

Abstract
Toll-like receptors (TLRs), a family of pattern recognition receptors, recognize and respond to conserved components of microbes and play a crucial role in both innate and adaptive immunity. In addition to binding exogenous ligands derived from pathogens, TLRs interact with endogenous molecules released from damaged tissues or dead cells and regulate many sterile inflammation processes. Putative endogenous TLR ligands include proteins and peptides, polysaccharides and proteoglycan, nucleic acids and phospholipids, which are cellular components, particularly extracellular matrix degradation products. Accumulating evidence demonstrates that endogenous ligand-mediated TLR signalling is involved in pathological conditions such as tissue injury, repair and regeneration; autoimmune diseases and tumorigenesis. The ability of TLRs to recognize endogenous stimulators appears to be essential to their function in regulating non-infectious inflammation. In this review, we summarize current knowledge of endogenous TLR ligands and discuss the biological significance of TLR signalling triggered by endogenous ligands in several sterile inflammation conditions.
Keywords: toll-like receptors, endogenous ligands, sterile inflammation, molecular medicine
Introduction
Toll-like receptors (TLRs) are a family of pattern recognition receptors (PRRs) responsible for recognizing conserved pathogen-associated molecular patterns (PAMPs). Engagement of TLRs initiates intracellular signalling pathways leading to the synthesis and secretion of various cytokines and chemokines by cells of the innate immune system. TLR-induced innate immune responses are also a prerequisite for the generation of most adaptive immune responses. TLRs therefore represent the first line of host defence against pathogens and play a pivotal role in both innate and adaptive immunity. In recent years, increasing evidence demonstrates that, under many pathological conditions, endogenous stimulators can activate TLR signalling to trigger a sterile inflammatory response. We review research progress on endogenous TLR ligands and their biological effects in non-infectious inflammation conditions such as tissue injury, tissue repair and regeneration, autoimmune disease and tumorigenesis.
TLR signalling
Eleven human TLRs and 13 mouse TLRs have been identified. Each TLR recognizes distinct PAMPs derived from various microorganisms including bacteria, viruses, protozoa and fungi [1]. TLRs are generally divided into three groups: those that recognize lipids and lipopeptides (TLR1, 2, 4 and 6), proteins (TLR5 and mouse TLR11) and nucleic acids (TLR3, 7, 8 and 9) [2]. TLR1 forms heterodimers with TLR2 (TLR1/2) and recognizes triacyl lipopeptides [3]. TLR2 in concert with TLR1 or TLR6 recognizes a wide variety of PAMPs, including peptidoglycan, lipopeptides and lipoproteins of gram+ bacteria, mycoplasma lipopeptides and fungal zymosan [4]. Heterodimerization of TLRs extends the spectrum of ligands recognized. TLR4, together with its extracellular components such as MD-2 and CD14, recognizes lipopolysaccharide (LPS), a constituent of cell walls of gram– bacteria [5]. In addition, studies in mice show that TLR4 recognizes not only bacterial motifs, but also viral motifs [6] and the plant product taxol [7]. TLR6 in association with TLR2 (TLR2/6) recognizes diacyl lipopeptides [8]. TLRs recognizing nucleic acids are localized in cytoplasmic compartments where they detect DNA and RNA derived from viruses and bacteria. TLR3 recognizes double-strand RNA [9], and TLR7 and TLR8 are responsive to the single-strand RNA [10] found during viral replication. TLR9 recognizes unmethylated deoxycytidyl-phosphate-deoxyguanosine (CpG) motifs commonly present in bacterial and viral genomes [11]. TLR5 recognizes bacterial flagellin [12] and TLR11 in mouse is responsive to a profilin-like molecule from the protozoan parasite Toxoplasma gondii [13]. Human TLR11 has been reported to be non-functional because of the presence of a stop codon in the gene [14]. TLR10 is able to homodimerize or heterodimerize with TLR1 and TLR2, but its ligand remains unknown [15].
Engagement of TLRs by PAMPs triggers intracellular signalling cascades through a set of toll/interleukin-1 receptor (TIR)-domain-containing adaptors, including myeloid differentiation primary response protein 88 (MyD88), TIR domain-containing adaptor protein (TIRAP/Mal), TIR domain-containing adaptor inducing interferon (IFN)-β (TRIF/TICAM1) and TRIF-related adaptor molecule (TRAM/TICAM2) (Fig. 1) [2]. Each TLR recruits a specific combination of adaptors to activate different transcription factors, giving rise to appropriate inflammatory responses. All TLRs, with exception of TLR3, share a common adaptor MyD88 to activate nuclear factor-κB (NF-κB) and activating protein-1 (AP-1) and induce the expression of various inflammatory cytokines through IL-1R-associated kinase (IRAK), tumour necrosis factor (TNF) receptor-associated factor-6 (TRAF6) and mitogen-activated protein (MAP) kinases (MyD88-dependent pathway) [2]. TIRAP mediates the activation of the MyD88-dependent pathway downstream of TLR2 and TLR4 [16, 17]. TRIF is recruited to TLR3 and TLR4 and mediates a MyD88-independent (TRIF-dependent) signalling pathway, leading to the activation of the late phase of NF-κB and IFN regulatory factor 3 (IRF3) and the subsequent production of type I IFN (IFN-α/β), IFN-inducible gene products and an immune regulatory response [18, 19]. TRAM selectively mediates the TRIF-dependent pathway downstream of TLR4, but not TLR3 [20].
Fig 1.
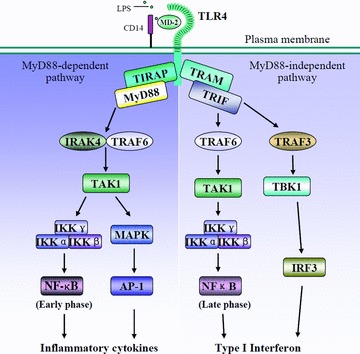
TLR4 signalling. TLR4 engagement initiates MyD88-dependent and independent signalling pathway and leads to production of inflammatory cytokines and type I IFN. AP-1, activating protein-1; CD14, cluster of differentiation 14; IKK, IκB kinase; IRAK, IL-1R-associated kinase; IRF, IFN regulatory factor; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MD-2, myeloid differentiation protein-2; MyD88, myeloid differentiation primary-response protein 88; TAK, TGF-β activated kinase; TBK, TANK binding kinase; TIRAP, Toll/interleukin-1 receptor domain-containing adaptor protein; TLR, toll-like receptor; TRAF, tumour necrosis factor receptor-associated factor; TRAM, TRIF-related adaptor molecule; TRIF, TIR-domain-containing adaptor inducing IFN-β; NF-κB, nuclear factor kappa B.
Endogenous TLR ligands
Recognition of microbial ligands fails to explain all functions of TLRs [21]. In addition to microbial PAMPs, an increasing number of endogenous stimulators are being reported as candidate ligands of TLRs (for review [22–25]). These endogenous molecules activate TLR signalling and induce sterile inflammatory responses in many pathological processes. Endogenous TLR ligands are a group of molecules derived from host tissues or cells, either components of cells or induced gene products in specific conditions. The majority are extracellular matrix components [22, 26] such as fibronectin [27], heparan sulphate [28], biglycan [29], fibrinogen [30], oligosaccharides of hyaluronan [31] and hyaluronan breakdown fragments [32–34]. Apart from previously identified protein ligands, for example high-mobility group box 1 (HMGB1) and heat shock proteins (HSP), it appears that more proteins such as tenascin-C, cardiac myosin and S100 proteins are implicated [35–38]. These so-called endogenous TLR ligands and their receptors are localized in different cellular compartments and cannot interact physiologically. In pathological conditions, endogenous ligands are either released passively from injured/inflamed tissues and dying cells or actively secreted by activated cells via a non-conventional lysosomal route [39]. Furthermore, it seems that even apoptotic cells, such as hypertrophic chondrocytes, may release some endogenous TLR ligands [40]. Endogenous TLR ligands can be categorized according to their properties (Table 1). Most are agonists of TLR4 and TLR2 (Table 2). Only a few bind to and stimulate other TLRs, but a single endogenous molecule, for example HMGB1, has the potential to interact with several TLRs [21, 41, 42].
Table 1.
Putative endogenous TLR ligands
| Property of ligands | Ligands and references |
|---|---|
| Proteins and peptides | β-defensin 2 [47, 48], fibrinogen [30], fibronectin [27], HMGB1 [21, 49, 50], HSP (HSP22, HSP60, HSP70, HSP72, endoplasmin and α-crystallin A chain) [51–60], human cardiac myosin [38], resistin [61], S100 proteins [35, 36], surfactant protein A [62] and tenascin-C [37] |
| Polysaccharides and proteoglycan | Biglycan [29], CD138 [23], heparan sulphate [28], oligosaccharides of hyaluronan [31] and hyaluronan breakdown fragments [32–34, 63] |
| Nucleic acids | DNA [64, 65], RNA [66, 67], mRNA [68, 69] and small interfering RNA (siRNA) [70] |
| Phospholipids | OxPAPC [71] |
| Small organic molecules | Monosodium urate crystals [72, 73] |
Table 2.
TLRs and their corresponding endogenous ligands
| TLRs | Ligands and references |
|---|---|
| TLR2 | Biglycan [29], endoplasmin [56], HMGB1 [74], HSP60 [52], HSP70 [53, 54], human cardiac myosin [38], hyaluronan [32, 63, 75] and monosodium urate crystals [72, 73] |
| TLR3 | mRNA [68, 69] |
| TLR4 | Biglycan [29], CD138 [23], α-crystallin A chain [57], β-defensin 2 [47, 48], endoplasmin [56], fibrinogen [30], fibronectin [27], heparan sulphate [28], HMGB1 [21, 41], HSP22 [57], HSP60 [51, 52, 60], HSP70 [53–55], HSP72 [58, 59], hyaluronan [32, 75, 76], monosodium urate crystals [72, 73], OxPAPC [71], resistin [61], S100 proteins [35, 36], surfactant protein A [62] and tenascin-C [37] |
| TLR7 | RNA [66, 67] and small interfering RNA (siRNA) [70] |
| TLR8 | Human cardiac myosin [38] and small interfering RNA (siRNA) [70] |
| TLR9 | DNA [64, 65] and HMGB1[42] |
Endogenous TLR ligands are often referred to as alarmins and serve as early warning signals to innate and adaptive immune systems. Matzinger [43, 44] has suggested that the activation of the innate immune system is not only based on the recognition of PAMPs but also relies on the presence of danger signals or danger-associated molecular patterns (DAMPs) released by injured cells. As tissue damage and cell lysis are often associated with infections and lead to the release of host molecules, recognition of DAMPs enables the immune system to not only sense an ongoing infection and recruit more immune cells, but also to initiate the repair of damaged tissue [45]. The endogenous alarmins and exogenous PAMPs are subgroups of the larger category of danger signals termed damage-associated molecular patterns (DAMPs) [46].
Biological significance of signalling triggered by endogenous TLR ligands
The inflammatory signals in some pathological situations, such as trauma and ischemia/reperfusion (I/R) injury, are activated even in the absence of infection. Evidence is accumulating that tissue damage is recognized at the cell level via receptor-mediated detection of endogenous molecules released by dead cells [46]. This is believed to be the major contribution of TLRs and inflammasome [77, 78]. Inflammasome is a large cytoplasmic molecular complex involved in the activation of inflammatory caspases resulting in the proteolytic activation of the pro-inflammatory cytokines IL-1β and IL-18 [78]. Cell disruption activates the inflammasome and initiates inflammatory responses. The inflammasome also senses extracellular danger signal ATP via purinoreceptor P2×7 [78]. Since an excellent review of the inflammasome has been published [78], it is not the focus of this review. TLR-mediated sterile inflammation, triggered by endogenous ligands, is involved in many pathological processes.
Ischemia and reperfusion injury
Ischemia/reperfusion injury is implicated in a broad array of pathological conditions such as myocardial infarction; cerebral stroke; and hepatic, renal and intestinal ischemia as well as following cardiovascular and transplant surgery [77]. The hallmark of these pathologies is extreme inflammation. Massive tissue injury with a large number of cells undergoing necrosis lead to the release of DAMPs from dead and dying cells resulting in the activation of TLRs and sterile inflammation (Fig. 2) [79]. Most of the evidence for the existence of endogenous TLR ligands is based on the study of I/R injury, the majority of endogenous ligands are likely to be involved in the activation of TLRs in I/R injury [80–82], but a strong necrosis-induced inflammatory response may, at least in part, be mediated by HMGB1 [83].
Fig 2.
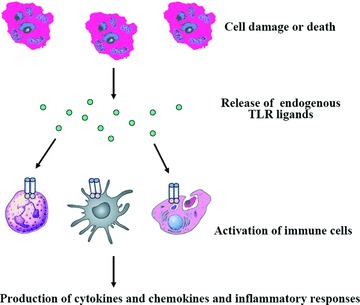
A proposed mechanism of inflammatory response in ischemia/reperfusion injury. Ischemia/reperfusion injury results in release of endogenous TLR ligands such as HMGB1, HSP and hyaluronan. Endogenous molecules bind to TLRs on DCs, macrophages and neutrophils that are recruited or infiltrated to injury sites and induce production of cytokines and chemokines and an inflammatory response.
The injury promoting roles of TLR4 are manifested in almost all organs as demonstrated by the protection of TLR4-deficient mice after hepatic, renal, cardiac and cerebral I/R [83–87]. Deficiency of TLR2, TLR4 or MyD88 leads to an attenuated myocardial inflammation, a smaller infarction size, better preserved ventricular function, and reduced ventricular remodelling after ischemic injury [88]. Much evidence points to endogenous TLR ligands as mediators of I/R-induced inflammation. The endotoxin blockade using the LPS-neutralizing agent (recombinant bactericidal/permeability-increasing protein) fails to protect mouse livers from warm I/R injury, and LPS-independent, heat-sensitive protein molecules contained in liver perfusates activate macrophages to produce TNF-α through the TLR4 pathway, providing evidence that endogenous TLR4 ligands are critical in the pathogenesis of liver I/R injury [89]. HMGB1 expression is rapidly up-regulated in hepatic and renal I/R injury and blockade of HMGB1 by a neutralizing antibody dramatically reduces hepatic injury in wild-type but not TLR4-deficient mice [83, 84]. HMGB1/TLR4 signalling induces generation of reactive oxygen species in hemorrhagic shock/resuscitation-activated neutrophils [90]. Tissue injury-induced neutrophil recruitment is reduced in mice treated with HMGB1 antibody [91] or HMGB1−/− cells in comparison to HMGB1+/+ cells [92]. Monocytes challenged with necrotic HMGB1−/− cells produce less TNF-α compared to those challenged with necrotic HMGB1+/+ cells [91]. In addition, HMGB1 stimulation enhances TLR4 expression in hepatic dendritic cells (DCs) in vitro and increasing numbers of hepatic DCs promote HMGB1-mediated I/R injury [93]. In adaptive immune cells, TLR4 expression is significantly increased on CD4+ T cells and CD8+ T cells after burn injury, which may be a mechanism for enhanced T-cell response late following burn injury [94].
Donor TLR4 and HMGB1 also contribute to graft inflammation and sterile injury following cold preservation and transplantation and may be associated with alloimmune responses after transplantation [93, 95, 96]. After cold I/R, grafts exhibit translocation of HMGB1 out of the nucleus of cardiac myocytes [96]. Administration of an anti-HMGB1 neutralizing antibody reduces TLR4-mediated cold I/R-induced inflammatory responses, suggesting that targeting TLR4 signalling may have value in preventing or treating post-ischemic acute organ injury after transplantation [96, 97].
TLR4 and/or TLR2 are responsible for the hyaluronan signalling [63, 75] and hyaluronan-induced inflammatory response [32, 98]. It has been reported that hyaluronan and biglycan are up-regulated in the I/R kidney, but their functional contribution to injury has not been characterized [84]. Hyaluronan is released during acute allograft rejection [75] and is released in higher levels in bronchial lavage fluid of patients with evidence of chronic lung rejection compared with lung transplant recipients who are free of this effect [81], suggesting that hyaluronan may mediate TLR activation in the setting of allograft rejection. It should be mentioned here that hyaluronan receptors CD44 and MD-2 are also important co-signalling molecules in the specific interaction between hyaluronan and TLR4 during sterile injury [34].
TLR3 has been shown to be an endogenous sensor of tissue necrosis during acute inflammatory events [69], and RNA escaping from damaged tissue or contained within endocytosed cells may serve as an endogenous ligand for TLR3 [68]. In experimental polymicrobial septic peritonitis and ischemic gut injury model, TLR3-deficient mice are protected from the lethal effects of sustained inflammation by a transient increase of inflammatory chemokine/cytokine [69]. Macrophages from TLR3-deficient mice respond normally to other TLR ligands but do not respond to RNA from necrotic neutrophils [69]. Anti-TLR3 antibody reduces generation of inflammatory chemokines of macrophages evoked by products of necrotic neutrophils, attenuates the tissue injury associated with gut ischemia and significantly decreases sepsis-induced mortality [69].
Tissue repair and regeneration
In addition to the injury-promoting role established in I/R injury [83–88], TLRs are crucial in the response to other tissue injuries and subsequent tissue repair and regeneration [22, 99–101]. TLR2/TLR4- and MyD88-deficient mice display an increased degree of bleomycin-induced lung injury [32] and dextran sulphate sodium (DSS)-induced intestinal epithelial injury [102, 103]. TLR signalling is required for the liver regeneration after partial hepatectomy [104] and the regeneration of intestinal epithelia following DSS-induced injury [102, 105]. Experimental hepatic fibrogenesis is significantly decreased in TLR4-deficient mice [106, 107]. A markedly slower healing of skin wounds has been observed in MyD88-deficient mice compared with wild-type, further suggesting a key role for TLR signalling in wound healing [108]. Interestingly, the R753Q polymorphism of the TLR2 gene has recently been found to be associated with severe ulcerative colitis, as the functional deficiency of the TLR2 variant leads to impaired wound healing [109].
TLR signalling in wound healing appears to be mediated by two classes of ligands: bacterial PAMPs and endogenous ligands [110]. It is believed that PAMPs from the commensal intestinal microbiota activate TLR signal pathways in the intestine to prevent epithelial injury as gut-sterilized mice display a similar increase in intestinal injury and DSS-induced death as MyD88-deficient mice [103]. The growth-promoting TLR ligand for liver restoration after hepatectomy may also derive from intestinal microbiota [110]. It is likely that this effect is dose dependent with lower concentrations mediating regeneration and higher concentrations mediating growth-suppressive responses [110]. A strong reduction of fibrogenesis in gut-sterilized mice implies a role of bacterial ligands from the intestine in promoting liver fibrogenesis [107]. Moreover, LPS induces a dose-dependent inhibition of keratinocyte migration and neutralizing antibodies to TLR4 and TLR2 relieve the migration inhibition, providing a possible explanation for the lack of healing found in ulcers [111]. Thus, detection of microbial patterns via epithelial TLRs directly regulates tissue homeostasis during the steady state and following injury [103, 105, 106, 112].
In sterile conditions, TLRs are likely to be activated by endogenous ligands that are released from necrotic cells [41, 91] or extracellular matrix components generated as a result of tissue injury [113]. Endogenous HMGB1 seems to be the predominant activator of TLR4 in I/R injury with a large number of cells undergoing necrosis [83]. In addition, it has been demonstrated that hyaluronan-mediated TLR signalling regulates tissue injury and repair [32, 76]. Hyaluronan fragments isolated from serum of mice with acute bleomycin-induced lung injury stimulate macrophage chemokine production, but this induction effect is completely abolished in TLR2/TLR4 double-deficient cells [32]. TLR2−/−/TLR4−/− mice are more susceptible than control mice to bleomycin-induced lung injury, and administration of hyaluronan-blocking peptide generates a phenotype that is notably similar to the TLR2-/TLR4- and MyD88-deficient state following acute lung injury, suggesting that the interaction of hyaluronan fragments with TLRs regulates lung injury and repair in vivo[32]. Hyaluronan accumulation is increased in injured skin tissue relative to normal skin and exogenous application of hyaluronan promotes wound repair, further supporting the potential involvement of hyaluronan in the wound-healing process [76]. It has also been observed that hyaluronan stimulates B cells, which infiltrate wounds to produce interleukin-6 and transforming growth factor-β through TLR4 [76].
Endogenous ligands and autoimmune diseases
Numerous reports have demonstrated that microbial PAMPs can trigger the onset of autoimmune diseases such as rheumatoid arthritis (RA) and experimental allergic encephalomyelitis (EAE), the animal model for multiple sclerosis [114, 115]. Whether endogenous TLR ligands contribute to the onset or perpetuation of these diseases is less clear. Data from mouse model studies indicate that endogenous TLR ligands may be involved in the pathogenesis of some autoimmune diseases (for review, see [24, 114, 116]).
Augmented TLR expression has been observed within the central nervous system (CNS) during EAE, even in the absence of apparent microbial involvement [117]. MyD88-deficient mice are EAE resistant and show no brain inflammation. This protection is due partly to TLR9 signalling [117]. These data indicate that both TLR9 and MyD88 are essential modulators of EAE and implicate an endogenous TLR9-dependent signal exerting an encephalitogenic effect [117]. As TLR9 is functionally expressed in microglia, it is likely that the ligand in the CNS is derived from cells that are damaged by the pathogenic effector T cells [118]. However, research results are inconsistent [119, 120]. It has been reported that TLR4- and TLR9-deficient mice exhibit more severe EAE symptoms than wild-type mice [119] and that stimulation of TLR3 with poly I:C induces IFN-β secretion and suppresses EAE [120].
Bacterial DNA and peptidoglycans have been detected in the joints of patients with RA and other arthritides, where they might enhance synovial inflammation [121]. However, recent evidence indicates that endogenous TLR ligand-mediated signalling plays an important role in RA. Several TLRs are expressed or even up-regulated in human synovial tissue from RA patients [122–124]. The inflammation resolves more quickly in TLR4-deficient experimental arthritic mice than in control mice [125] and blocking TLR4 in mice with collagen-induced arthritis leads to reduced disease severity [126]. Serum and synovial fluid from RA patients stimulate TLR4-expressing CHO cells to up-regulate CD25 [127] and RA synovial membrane culture supernatants are able to activate human macrophages via TLR signalling, suggesting the involvement of an endogenous ligand in the pathogenesis of RA [128]. Several putative endogenous TLR ligands have been associated with RA. HMGB1 is detectable in the synovial fluid of RA patients [129] and its expression is increased in synovial fibroblasts of RA patients compared to osteoarthritis patients, and this may stimulate the release of pro-inflammatory cytokines by fibroblasts [130]. RNA released from necrotic synovial fluid cells was recently shown to activate synovial fibroblasts via TLR3 [124]. Chromatin-IgG complexes activate B cells by dual engagement of antigen receptors and TLR9 to produce rheumatoid factor, a class of autoantibodies [64]. DNase II-deficient mice develop chronic polyarthritis resembling human RA due to excessive release of DNA following cell death, which may contribute to TLR9 activation [131]. Tenascin-C is an extracellular matrix glycoprotein specifically expressed in inflamed rheumatoid joints [37]. Tenascin-C-deficient mice show rapid resolution of acute joint inflammation and are protected from erosive arthritis [37]. Administration of exogenous tenascin-C promotes joint inflammation in vivo and induces cytokine synthesis in in vitro cultures in a TLR4-dependent manner, indicating that tenascin-C is an endogenous TLR activator essential for maintaining inflammation in arthritic joint disease [37].
It has been demonstrated that mammalian DNA and RNA, in the form of immune complexes, are potent self-antigens for TLR9 and TLR7, respectively, and induce IFN-α production by plasmacytoid pre-dendritic cells, which may promote systemic lupus erythematosus (SLE) [67, 132]. As TLR7 and TLR9 are located in cytoplasmic compartments, it is believed that these autoantigens gain access to them through a receptor-mediated delivery. One such access route is the B cell receptor (BCR)-mediated endocytosis, which is available not only for microorganisms but also for endogenous ligands that are liberated from damaged cells and recognized by an autoreactive BCR [114]. DNA and DNA-associated autoantigens activate autoreactive B cells via sequential engagement of the BCR and TLR9 [64, 65], whereas RNA and RNA-associated autoantigens react through BCR/TLR7 [66]. Autoimmune-prone mice, lacking the TLR adaptor protein MyD88, have markedly reduced autoantibody titres, further supporting the suggestion that TLRs play a key role in autoantibody responses in SLE [66].
Tumorigenesis and tumour progression
The study of the association of TLR signalling with tumours focuses primarily on the following three aspects. It has been found that TLR signal-mediated chronic inflammation is involved in tumorigenesis and that the activation of TLR signalling induces an antitumour T-cell response [41]. In addition, evidence indicates that TLRs are also expressed in tumour cells and tissues, and TLR signalling contributes to tumour progression and chemoresistance [133–135]. Moreover, it has been reported recently that dying tumour cells release endogenous TLR ligands [41, 74] that are involved in solid tumour progression [136, 137] and probably in leukemic cell growth [23].
HMGB1 is a major putative tumour-related TLR ligand. It has been revealed that the ability of chemotherapeutic agents to kill tumours is decreased in TLR4- and MyD88-deficient mice [41]. This is mainly attributed to HMGB1, released by chemotherapy-induced cell death, which activates TLR4 expressed by DCs and induces antitumour T-cell immunity [41]. Breast cancer patients with a TLR4 loss-of-function allele relapse more quickly after radiotherapy and chemotherapy than those carrying the normal TLR4 allele [41]. Mice with a mutated TLR4 develop more skin tumours than wild-type mice treated with mutagenic agent, suggesting that TLR4-dependent antitumour responses are important for inhibiting tumorigenesis [138]. HMGB1 released from dying tumour cells also activates TLR2 expressed on tumour-infiltrating myeloid DCs and mediates an effective anti-glioblastoma multiforme immune response [74]. TLR2 signalling can be specifically activated by supernatants from the drug-treated tumour cells and is blocked by a specific HMGB1 inhibitor glycyrrhizin or anti-HMGB1 antibodies [74]. Moreover, HMGB1 inhibitor or antibodies abolish therapeutic efficacy in the mouse model, highlighting the critical role of HMGB1-mediated TLR2 signalling to elicit tumour regression [74]. HMGB1 is released from melanoma, small cell lung carcinoma and glioma cells treated with radiation or temozolomide, and therapeutic efficacy is consistent with an increase in the level of circulating HMGB1 in an intracranial melanoma model [74]. In addition, HMGB1 is released by osteoclasts [139] and can stimulate adjacent multiple myeloma cells [23].
The extracellular matrix proteoglycan versican in Lewis lung carcinoma has been identified as a macrophage activator that acts through TLR2 and its co-receptors TLR6 and CD14 and strongly enhances Lewis lung carcinoma metastatic growth via TLR2/TLR6-mediated TNF-α secretion [140]. TLR4 serves as a functional receptor for serum amyloid A 3 in pre-metastatic lungs. Serum amyloid A 3 is induced by S100A8 and S100A9, stimulates NF-κB signalling and facilitates metastasis [141]. Small fragments of hyaluronan in melanoma might promote tumour invasiveness by inducing matrix metalloprotease- and cytokine expression in a TLR4-dependent manner [142]. In addition, heparan sulphate proteoglycan CD138 expressed on multiple myeloma cells can bind to TLR4 and act as an autocrine survival factor. Soluble CD138 and heparanase levels correlate with poor prognoses [23].
Other inflammatory responses mediated by endogenous TLR ligands
Several endogenous ligands are involved in the inflammatory responses of the respiratory tract. HSP72 is released and biologically active in the bronchoalveolar lavage fluid and can regulate airway epithelial cell cytokine expression and activate neutrophils via TLR4 [58, 59]. The endogenous oxidized phospholipid oxidized 1-palmitoyl-2-arachidonoyl-phosphatidylcholine (OxPAPC) has been demonstrated recently to induce acute lung injury and cytokine production by lung macrophages through TLR4-TRIF signalling [71]. TLR3 expression is increased in airway epithelial cells from patients with acute respiratory distress syndrome [143]. TLR3-deficiency or administration of anti-TLR3 antibody to wild-type mice protects these animals from hyperoxia-induced lung injury and inflammation in the absence of a viral pathogen, suggesting an important role of TLR3 signalling and the possible involvement of endogenous TLR3 ligands [143]. In addition, even mechanical ventilation significantly increases endogenous TLR4 ligands in bronchoalveolar lavage fluid and induces a TLR4-dependent inflammatory response in healthy mice [144].
Endogenous TLR stimulators have the potential to induce cardiac and vascular inflammation. Human cardiac myosin acts as an endogenous ligand to directly stimulate TLR2 and TLR8 and activate monocytes to release pro-inflammatory cytokines, which can promote chronic inflammation in the myocardium [38]. Minimally oxidized low-density lipoprotein (mmLDL) stimulates intracellular reactive oxygen species generation in macrophages in a TLR4-dependent manner, suggesting that mmLDL can serve as an endogenous TLR4 ligand inducing proatherogenic activation of macrophages. This suggests a causative role in the development of atherosclerosis [145]. It has been reported that oxidized LDL and amyloid-b can trigger inflammatory signalling through a heterodimer of TLR4 and TLR6 [146]. In addition, Mrp8 and Mrp14, two members of the S100 family of calcium-modulated proteins, broadly regulate vascular inflammation and contribute to the biological response to vascular injury by promoting leucocyte recruitment [36]. They have been revealed to be endogenous activators of TLR4, promoting lethal, endotoxin-induced shock [35].
Uric acid, the end-product of the cellular catabolism of purines, is a danger-associated molecule involved in gout, an acute arthritis caused by deposits of monosodium urate monohydrate (MSU) crystals in joints. TLR2, TLR4 and MyD88 are required for MSU crystals-induced cytokine expression by macrophages and required for mouse air pouch inflammation as well a response to MSU crystal. This indicates that specific TLRs expressed in innate immune cells recognize naked MSU crystals and regulate the course of gouty arthritis [72]. TLR2 expression is up-regulated in osteoarthritic cartilage chondrocytes and blockage of TLR2 on chondrocytes suppresses MSU crystal-induced release of nitric oxide, an inflammation mediator [73]. Moreover, engagement of CD14, a TLR2 and TLR4 co-receptor, mediates the inflammatory potential of MSU crystals [147]. Apart from involvement in gouty arthritis, uric acid released from injured cells constitutes a major endogenous danger signal that activates the NACHT (NAIP, CIITA, HET-E and TP-1) domain, leucine-rich repeat and pyrin domain-containing protein (NALP)3 inflammasome and initiates sterile inflammation in other organs, for example in lung injury inflammation and fibrosis [148].
Sterile inflammation in the injured nervous system can also be triggered by endogenous TLR signals (for review [149]). HSPs including HSP60 and HSP70 are locally expressed by injured axons [150] and released by CNS cells undergoing necrotic or apoptotic cell death [60]. HSP60 can activate TLR4 at the surface of microglia and mediate neuro-inflammation through a MyD88-dependent pathway [60]. Necrotic neuronal cells induce inflammatory Schwann cell activation via TLR2 and TLR3, which may be involved in Wallerian degeneration upon peripheral nerve injury [151]. Furthermore, TLR signalling may play a critical role in orchestrating the innate immune response leading to efficient and rapid clearance of inhibitory myelin debris and nerve regeneration [152].
Conclusions
Toll was first found to be a receptor involved in embryonic development of the fly [153]. TLRs, its mammalian homologues, were first discovered in 1997 [154] and were quickly demonstrated to be responsible for the recognition of microbial components [5]. In addition to binding to microbial PAMPs and involvement in host defence against pathogens, TLRs are involved in many sterile inflammation processes. The ability of TLRs to recognize endogenous mediators appears to be necessary for their ability to regulate sterile inflammation [110]. However, the biochemical evidence for the direct interaction of TLRs with endogenous stimulators is limited [35, 61]. Most evidence is provided from in vitro studies which have potential for contamination of bacterial TLR agonists, particularly those from commercial sources or recombinant products expressed in bacterial systems [25, 63]. For example, recombinant human HSP70 preparation has been reported to be contaminated by LPS [155]. Thus, in vivo= studies and biochemical evidence are essential for endogenous ligands to be widely accepted. In vivo studies suggest that matrix components biglycan and hyaluronan are endogenous TLR ligands [32, 63]. Although there is ongoing controversy concerning some putative endogenous ligands [110, 156, 157], accumulating evidence supports the suggestion that several endogenous TLR activators regulate some important non-infectious pathological events. Signalling mediated by endogenous ligands plays a pivotal role in sterile inflammation contributing to tissue injury and repair, autoimmune disease and tumorigenesis.
Despite rapid progress in understanding the broad roles of endogenous TLR ligands and their signalling, a number of questions remain unanswered. Initially, the specific contributions of endogenous TLR ligands to sterile inflammation need to be further evaluated, especially when the effects of microbial components cannot be eliminated. An interesting question is whether TLRs distinguish microbial ligands from endogenous ligands and respond differently. Many reported endogenous ligands (Table 2) have the potential to activate both TLR2 and TLR4. As far as we know, TLR2 and TLR4 recognize different microbial ligands. How do these receptors sense the same endogenous stimulators? Is it possible that they form a TLR2/4 heterodimer? As mentioned above, TLRs do not behave as singular molecular units, but form various heterodimers, which increases the versatility of the system. In addition to heterodimerization of TLR2 with TLR1 or TLR6 [3, 4, 8], a TLR4 and TLR6 heterodimer has recently been demonstrated to sense oxidized LDL and amyloid-β[146]. The scavenger receptor CD36, acting as a TLR4/TLR6 co-receptor, regulates TLR4/TLR6 complex formation and promotes sterile inflammation [146]. In addition, the endogenous stimulators for TLR2 or TLR4 vary greatly in their chemical properties. How can an individual TLR sense several agonists with different properties? It seems difficult to find a common moiety or structure among those molecules. It is more logical to postulate that different co-receptors or accessory molecules mediate interaction of an individual endogenous stimulator with a corresponding TLR. Then, the question is, which is the dominant endogenous activator for each TLR, if such exists? Moreover, in addition to interaction with TLRs, some endogenous molecules are able to stimulate other receptors, such as HMGB1 binding to the receptor for advanced glycation end products and hyaluronan acting on its receptor CD44. It is therefore necessary to estimate the respective contributions of individual receptors. There is no doubt that it is of great significance to identify the endogenous TLR ligands and elucidate their biological functions.
Acknowledgments
This study was funded by the Ph.D. Program Foundation of Ministry of Education of China (20090171110029) and grants from Yuexiu Science and Technology Bureau of Guangzhou, China (2005-WS-001, 2008-WS-003).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Takeuchi O, Sato S, Horiuchi T, et al. Cutting edge: role of toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169:10–4. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–51. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 5.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 6.Kurt-Jones EA, Popova L, Kwinn L, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 7.Kawasaki K, Akashi S, Shimazu R, et al. Mouse toll-like receptor 4.MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by Taxol. J Biol Chem. 2000;275:2251–4. doi: 10.1074/jbc.275.4.2251. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi O, Kawai T, Muhlradt PF, et al. Discrimination of bacterial lipoproteins by toll-like receptor 6. Int Immunol. 2001;13:933–40. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 9.Alexopoulou L, Holt AC, Medzhitov R, et al. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 10.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–9. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 11.Hemmi H, Takeuchi O, Kawai T, et al. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi F, Smith KD, Ozinsky A, et al. The innate immune response to bacterial flagellin is mediated by toll-like receptor 5. Nature. 2001;410:1099–103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 13.Yarovinsky F, Zhang D, Andersen JF, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–9. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 14.Zhang D, Zhang G, Hayden MS, et al. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–6. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- 15.Hasan U, Chaffois C, Gaillard C, et al. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol. 2005;174:2942–50. doi: 10.4049/jimmunol.174.5.2942. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto M, Sato S, Hemmi H, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324–9. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 17.Horng T, Barton GM, Flavell RA, et al. The adaptor molecule TIRAP provides signalling specificity for toll-like receptors. Nature. 2002;420:329–33. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto M, Sato S, Mori K, et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the toll-like receptor signaling. J Immunol. 2002;169:6668–72. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto M, Sato S, Hemmi H, et al. TRAM is specifically involved in the toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–50. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 21.Tang AH, Brunn GJ, Cascalho M, et al. Pivotal advance: endogenous pathway to SIRS, sepsis, and related conditions. J Leukoc Biol. 2007;82:282–5. doi: 10.1189/jlb.1206752. [DOI] [PubMed] [Google Scholar]
- 22.Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9:57–63. doi: 10.1038/nrc2541. [DOI] [PubMed] [Google Scholar]
- 23.Chiron D, Bekeredjian-Ding I, Pellat-Deceunynck C, et al. Toll-like receptors: lessons to learn from normal and malignant human B cells. Blood. 2008;112:2205–13. doi: 10.1182/blood-2008-02-140673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rifkin IR, Leadbetter EA, Busconi L, et al. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 25.Tsan MF, Gao B. Endogenous ligands of toll-like receptors. J Leukoc Biol. 2004;76:514–9. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- 26.Miyake K. Innate immune sensing of pathogens and danger signals by cell surface toll-like receptors. Semin Immunol. 2007;19:3–10. doi: 10.1016/j.smim.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Okamura Y, Watari M, Jerud ES, et al. The extra domain A of fibronectin activates toll-like receptor 4. J Biol Chem. 2001;276:10229–33. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 28.Johnson GB, Brunn GJ, Kodaira Y, et al. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by toll-like receptor 4. J Immunol. 2002;168:5233–9. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- 29.Schaefer L, Babelova A, Kiss E, et al. The matrix component biglycan is proinflammatory and signals through toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115:2223–33. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167:2887–94. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 31.Termeer C, Benedix F, Sleeman J, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 33.Taylor KR, Trowbridge JM, Rudisill JA, et al. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–84. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 34.Taylor KR, Yamasaki K, Radek KA, et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on toll-like receptor 4, CD44, and MD-2. J Biol Chem. 2007;282:18265–75. doi: 10.1074/jbc.M606352200. [DOI] [PubMed] [Google Scholar]
- 35.Vogl T, Tenbrock K, Ludwig S, et al. Mrp8 and Mrp14 are endogenous activators of toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–9. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 36.Croce K, Gao H, Wang Y, et al. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation. 2009;120:427–36. doi: 10.1161/CIRCULATIONAHA.108.814582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–80. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 38.Zhang P, Cox CJ, Alvarez KM, et al. Cutting edge: cardiac myosin activates innate immune responses through TLRs. J Immunol. 2009;183:27–31. doi: 10.4049/jimmunol.0800861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pollanen R, Sillat T, Pajarinen J, et al. Microbial antigens mediate HLA-B27 diseases via TLRs. J Autoimmun. 2009;32:172–7. doi: 10.1016/j.jaut.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 40.Taniguchi N, Yoshida K, Ito T, et al. Stage-specific secretion of HMGB1 in cartilage regulates endochondral ossification. Mol Cell Biol. 2007;27:5650–63. doi: 10.1128/MCB.00130-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 42.Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–96. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 43.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 44.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–78. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 45.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–35. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 46.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 47.Biragyn A, Ruffini PA, Leifer CA, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–9. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- 48.Biragyn A, Coscia M, Nagashima K, et al. Murine beta-defensin 2 promotes TLR-4/MyD88-mediated and NF-kappaB-dependent atypical death of APCs via activation of TNFR2. J Leukoc Biol. 2008;83:998–1008. doi: 10.1189/jlb.1007700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park JS, Gamboni-Robertson F, He Q, et al. High mobility group box 1 protein interacts with multiple toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–24. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 50.Park JS, Svetkauskaite D, He Q, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 51.Ohashi K, Burkart V, Flohe S, et al. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–61. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 52.Vabulas RM, Ahmad-Nejad P, Da Costa C, et al. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–9. doi: 10.1074/jbc.M103217200. [DOI] [PubMed] [Google Scholar]
- 53.Vabulas RM, Ahmad-Nejad P, Ghose S, et al. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–12. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 54.Asea A, Rehli M, Kabingu E, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–34. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 55.Dybdahl B, Wahba A, Lien E, et al. Inflammatory response after open heart surgery: release of heat-shock protein 70 and signaling through toll-like receptor-4. Circulation. 2002;105:685–90. doi: 10.1161/hc0602.103617. [DOI] [PubMed] [Google Scholar]
- 56.Vabulas RM, Braedel S, Hilf N, et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the toll-like receptor 2/4 pathway. J Biol Chem. 2002;277:20847–53. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- 57.Roelofs MF, Boelens WC, Joosten LA, et al. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J Immunol. 2006;176:7021–7. doi: 10.4049/jimmunol.176.11.7021. [DOI] [PubMed] [Google Scholar]
- 58.Chase MA, Wheeler DS, Lierl KM, et al. Hsp72 induces inflammation and regulates cytokine production in airway epithelium through a TLR4- and NF-kappaB-dependent mechanism. J Immunol. 2007;179:6318–24. doi: 10.4049/jimmunol.179.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wheeler DS, Chase MA, Senft AP, et al. Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via toll-like receptor (TLR)-4. Respir Res. 2009;10:31. doi: 10.1186/1465-9921-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lehnardt S, Schott E, Trimbuch T, et al. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. J Neurosci. 2008;28:2320–31. doi: 10.1523/JNEUROSCI.4760-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarkowski A, Bjersing J, Shestakov A, et al. Resistin competes with lipopolysaccharide for binding to toll-like receptor 4. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00899.x. Sep 14 [Epub ahead of print]; DOI: 10.1111/j.1582-4934.2009.00899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guillot L, Balloy V, McCormack FX, et al. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves toll-like receptor 4. J Immunol. 2002;168:5989–92. doi: 10.4049/jimmunol.168.12.5989. [DOI] [PubMed] [Google Scholar]
- 63.Scheibner KA, Lutz MA, Boodoo S, et al. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177:1272–81. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 64.Leadbetter EA, Rifkin IR, Hohlbaum AM, et al. Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature. 2002;416:603–7. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 65.Viglianti GA, Lau CM, Hanley TM, et al. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19:837–47. doi: 10.1016/s1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]
- 66.Lau CM, Broughton C, Tabor AS, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–7. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelly KM, Zhuang H, Nacionales DC, et al. “Endogenous adjuvant” activity of the RNA components of lupus autoantigens Sm/RNP and Ro 60. Arthritis Rheum. 2006;54:1557–67. doi: 10.1002/art.21819. [DOI] [PubMed] [Google Scholar]
- 68.Kariko K, Ni H, Capodici J, et al. mRNA is an endogenous ligand for toll-like receptor 3. J Biol Chem. 2004;279:12542–50. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- 69.Cavassani KA, Ishii M, Wen H, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–21. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sioud M. Innate sensing of self and non-self RNAs by toll-like receptors. Trends Mol Med. 2006;12:167–76. doi: 10.1016/j.molmed.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 71.Imai Y, Kuba K, Neely GG, et al. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–49. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu-Bryan R, Scott P, Sydlaske A, et al. Innate immunity conferred by toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 2005;52:2936–46. doi: 10.1002/art.21238. [DOI] [PubMed] [Google Scholar]
- 73.Liu-Bryan R, Pritzker K, Firestein GS, et al. TLR2 signaling in chondrocytes drives calcium pyrophosphate dihydrate and monosodium urate crystal-induced nitric oxide generation. J Immunol. 2005;174:5016–23. doi: 10.4049/jimmunol.174.8.5016. [DOI] [PubMed] [Google Scholar]
- 74.Curtin JF, Liu N, Candolfi M, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tesar BM, Jiang D, Liang J, et al. The role of hyaluronan degradation products as innate alloimmune agonists. Am J Transplant. 2006;6:2622–35. doi: 10.1111/j.1600-6143.2006.01537.x. [DOI] [PubMed] [Google Scholar]
- 76.Iwata Y, Yoshizaki A, Komura K, et al. CD19, a response regulator of B lymphocytes, regulates wound healing through hyaluronan-induced TLR4 signaling. Am J Pathol. 2009;175:649–60. doi: 10.2353/ajpath.2009.080355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Arumugam TV, Okun E, Tang SC, et al. Toll-like receptors in ischemia-reperfusion injury. Shock. 2009;32:4–16. doi: 10.1097/SHK.0b013e318193e333. [DOI] [PubMed] [Google Scholar]
- 78.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–65. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 79.Lotze MT, Zeh HJ, Rubartelli A, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 80.Beg AA. Endogenous ligands of toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol. 2002;23:509–12. doi: 10.1016/s1471-4906(02)02317-7. [DOI] [PubMed] [Google Scholar]
- 81.Shirali AC, Goldstein DR. Activation of the innate immune system by the endogenous ligand hyaluronan. Curr Opin Organ Transplant. 2008;13:20–5. doi: 10.1097/MOT.0b013e3282f3df04. [DOI] [PubMed] [Google Scholar]
- 82.Goldstein DR. The identity of innate immune activators in organ transplantation: origins from within or exterior to the host. Am J Transplant. 2007;7:1692–4. doi: 10.1111/j.1600-6143.2007.01851.x. [DOI] [PubMed] [Google Scholar]
- 83.Tsung A, Sahai R, Tanaka H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–43. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu H, Chen G, Wyburn KR, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–59. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang SC, Arumugam TV, Xu X, et al. Pivotal role for neuronal toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci USA. 2007;104:13798–803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oyama J, Blais C, Jr, Liu X, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–9. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 87.Takeishi Y, Kubota I. Role of toll-like receptor mediated signaling pathway in ischemic heart. Front Biosci. 2009;14:2553–8. doi: 10.2741/3397. [DOI] [PubMed] [Google Scholar]
- 88.Chao W. Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol. 2009;296:H1–12. doi: 10.1152/ajpheart.00995.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhai Y, Qiao B, Shen XD, et al. Evidence for the pivotal role of endogenous toll-like receptor 4 ligands in liver ischemia and reperfusion injury. Transplantation. 2008;85:1016–22. doi: 10.1097/TP.0b013e3181684248. [DOI] [PubMed] [Google Scholar]
- 90.Fan J, Li Y, Levy RM, et al. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J Immunol. 2007;178:6573–80. doi: 10.4049/jimmunol.178.10.6573. [DOI] [PubMed] [Google Scholar]
- 91.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 92.Chen CJ, Kono H, Golenbock D, et al. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 93.Tsung A, Zheng N, Jeyabalan G, et al. Increasing numbers of hepatic dendritic cells promote HMGB1-mediated ischemia-reperfusion injury. J Leukoc Biol. 2007;81:119–28. doi: 10.1189/jlb.0706468. [DOI] [PubMed] [Google Scholar]
- 94.Cairns B, Maile R, Barnes CM, et al. Increased toll-like receptor 4 expression on T cells may be a mechanism for enhanced T cell response late after burn injury. J trauma. 2006;61:293–8. doi: 10.1097/01.ta.0000228969.46633.bb. [DOI] [PubMed] [Google Scholar]
- 95.Kaczorowski DJ, Nakao A, Mollen KP, et al. Toll-like receptor 4 mediates the early inflammatory response after cold ischemia/reperfusion. Transplantation. 2007;84:1279–87. doi: 10.1097/01.tp.0000287597.87571.17. [DOI] [PubMed] [Google Scholar]
- 96.Kaczorowski DJ, Nakao A, Vallabhaneni R, et al. Mechanisms of toll-like receptor 4 (TLR4)-mediated inflammation after cold ischemia/reperfusion in the heart. Transplantation. 2009;87:1455–63. doi: 10.1097/TP.0b013e3181a36e5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kruger B, Krick S, Dhillon N, et al. Donor toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci USA. 2009;106:3390–5. doi: 10.1073/pnas.0810169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Noble PW, Jiang D. Matrix regulation of lung injury, inflammation, and repair: the role of innate immunity. Proc Am Thorac Soc. 2006;3:401–4. doi: 10.1513/pats.200604-097AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Larsen PH, Holm TH, Owens T. toll-like receptors in brain development and homeostasis. Sci STKE. 2007;2007:pe47. doi: 10.1126/stke.4022007pe47. [DOI] [PubMed] [Google Scholar]
- 100.Michelsen KS, Arditi M. Toll-like receptors and innate immunity in gut homeostasis and pathology. Curr Opin Hematol. 2007;14:48–54. doi: 10.1097/00062752-200701000-00010. [DOI] [PubMed] [Google Scholar]
- 101.Rakoff-Nahoum S, Medzhitov R. Role of toll-like receptors in tissue repair and tumorigenesis. Biochemistry. 2008;73:555–61. doi: 10.1134/s0006297908050088. [DOI] [PubMed] [Google Scholar]
- 102.Fukata M, Chen A, Klepper A, et al. Cox-2 is regulated by toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology. 2006;131:862–77. doi: 10.1053/j.gastro.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 104.Seki E, Tsutsui H, Iimuro Y, et al. Contribution of toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology. 2005;41:443–50. doi: 10.1002/hep.20603. [DOI] [PubMed] [Google Scholar]
- 105.Pull SL, Doherty JM, Mills JC, et al. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci USA. 2005;102:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–32. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 107.Isayama F, Hines IN, Kremer M, et al. LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1318–28. doi: 10.1152/ajpgi.00405.2005. [DOI] [PubMed] [Google Scholar]
- 108.Macedo L, Pinhal-Enfield G, Alshits V, et al. Wound healing is impaired in MyD88-deficient mice: a role for MyD88 in the regulation of wound healing by adenosine A2A receptors. Am J Pathol. 2007;171:1774–88. doi: 10.2353/ajpath.2007.061048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Podolsky DK, Gerken G, Eyking A, et al. Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology. 2009;137:209–20. doi: 10.1053/j.gastro.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kluwe J, Mencin A, Schwabe RF. Toll-like receptors, wound healing, and carcinogenesis. J Mol Med. 2009;87:125–38. doi: 10.1007/s00109-008-0426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Loryman C, Mansbridge J. Inhibition of keratinocyte migration by lipopolysaccharide. Wound Repair Regen. 2008;16:45–51. doi: 10.1111/j.1524-475X.2007.00290.x. [DOI] [PubMed] [Google Scholar]
- 112.Shaykhiev R, Behr J, Bals R. Microbial patterns signaling via toll-like receptors 2 and 5 contribute to epithelial repair, growth and survival. PloS one. 2008;3:e1393. doi: 10.1371/journal.pone.0001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiang D, Liang J, Li Y, Noble PW. The role of toll-like receptors in non-infectious lung injury. Cell Res. 2006;16:693–701. doi: 10.1038/sj.cr.7310085. [DOI] [PubMed] [Google Scholar]
- 114.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–35. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nichols FC, Housley WJ, O’Conor CA, et al. Unique lipids from a common human bacterium represent a new class of toll-like receptor 2 ligands capable of enhancing autoimmunity. Am J Pathol. 2009;175:2430–8. doi: 10.2353/ajpath.2009.090544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wagner H. Endogenous TLR ligands and autoimmunity. Adv Immunol. 2006;91:159–73. doi: 10.1016/S0065-2776(06)91004-9. [DOI] [PubMed] [Google Scholar]
- 117.Prinz M, Garbe F, Schmidt H, et al. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. 2006;116:456–64. doi: 10.1172/JCI26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Iliev AI, Stringaris AK, Nau R, et al. Neuronal injury mediated via stimulation of microglial toll-like receptor-9 (TLR9) FASEB J. 2004;18:412–4. doi: 10.1096/fj.03-0670fje. [DOI] [PubMed] [Google Scholar]
- 119.Marta M, Andersson A, Isaksson M, et al. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur J Immunol. 2008;38:565–75. doi: 10.1002/eji.200737187. [DOI] [PubMed] [Google Scholar]
- 120.Touil T, Fitzgerald D, Zhang GX, et al. Cutting Edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-beta. J Immunol. 2006;177:7505–9. doi: 10.4049/jimmunol.177.11.7505. [DOI] [PubMed] [Google Scholar]
- 121.Van Der Heijden IM, Wilbrink B, Tchetverikov I, et al. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. 2000;43:593–8. doi: 10.1002/1529-0131(200003)43:3<593::AID-ANR16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 122.Ospelt C, Brentano F, Rengel Y, et al. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 2008;58:3684–92. doi: 10.1002/art.24140. [DOI] [PubMed] [Google Scholar]
- 123.Radstake TR, Roelofs MF, Jenniskens YM, et al. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis Rheum. 2004;50:3856–65. doi: 10.1002/art.20678. [DOI] [PubMed] [Google Scholar]
- 124.Brentano F, Schorr O, Gay RE, et al. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via toll-like receptor 3. Arthritis Rheum. 2005;52:2656–65. doi: 10.1002/art.21273. [DOI] [PubMed] [Google Scholar]
- 125.Choe JY, Crain B, Wu SR, et al. Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by toll-like receptor 4 signaling. J Exp Med. 2003;197:537–42. doi: 10.1084/jem.20021850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Abdollahi-Roodsaz S, Joosten LA, Roelofs MF, et al. Inhibition of toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007;56:2957–67. doi: 10.1002/art.22848. [DOI] [PubMed] [Google Scholar]
- 127.Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, et al. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 2005;52:2313–22. doi: 10.1002/art.21278. [DOI] [PubMed] [Google Scholar]
- 128.Sacre SM, Andreakos E, Kiriakidis S, et al. The toll-like receptor adaptor proteins MyD88 and Mal/TIRAP contribute to the inflammatory and destructive processes in a human model of rheumatoid arthritis. Am J Pathol. 2007;170:518–25. doi: 10.2353/ajpath.2007.060657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kokkola R, Sundberg E, Ulfgren AK, et al. High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum. 2002;46:2598–603. doi: 10.1002/art.10540. [DOI] [PubMed] [Google Scholar]
- 130.Taniguchi N, Kawahara K, Yone K, et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003;48:971–81. doi: 10.1002/art.10859. [DOI] [PubMed] [Google Scholar]
- 131.Kawane K, Ohtani M, Miwa K, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443:998–1002. doi: 10.1038/nature05245. [DOI] [PubMed] [Google Scholar]
- 132.Barrat FJ, Meeker T, Gregorio J, et al. Nucleic acids of mammalian origin can act as endogenous ligands for toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–9. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huang B, Zhao J, Li H, et al. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005;65:5009–14. doi: 10.1158/0008-5472.CAN-05-0784. [DOI] [PubMed] [Google Scholar]
- 134.Yu L, Chen S. Toll-like receptors expressed in tumor cells: targets for therapy. Cancer Immunol Immunother. 2008;57:1271–8. doi: 10.1007/s00262-008-0459-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kelly MG, Alvero AB, Chen R, et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006;66:3859–68. doi: 10.1158/0008-5472.CAN-05-3948. [DOI] [PubMed] [Google Scholar]
- 136.Sanderson RD, Yang Y, Suva LJ, et al. Heparan sulfate proteoglycans and heparanase–partners in osteolytic tumor growth and metastasis. Matrix Biol. 2004;23:341–52. doi: 10.1016/j.matbio.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 137.Gotte M, Yip GW. Heparanase, hyaluronan, and CD44 in cancers: a breast carcinoma perspective. Cancer Res. 2006;66:10233–7. doi: 10.1158/0008-5472.CAN-06-1464. [DOI] [PubMed] [Google Scholar]
- 138.Yusuf N, Nasti TH, Long JA, et al. Protective role of toll-like receptor 4 during the initiation stage of cutaneous chemical carcinogenesis. Cancer Res. 2008;68:615–22. doi: 10.1158/0008-5472.CAN-07-5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Charoonpatrapong K, Shah R, Robling AG, et al. HMGB1 expression and release by bone cells. J Cell Physiol. 2006;207:480–90. doi: 10.1002/jcp.20577. [DOI] [PubMed] [Google Scholar]
- 140.Kim S, Takahashi H, Lin WW, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–6. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hiratsuka S, Watanabe A, Sakurai Y, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10:1349–55. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 142.Voelcker V, Gebhardt C, Averbeck M, et al. Hyaluronan fragments induce cytokine and metalloprotease upregulation in human melanoma cells in part by signalling via TLR4. Exp Dermatol. 2008;17:100–7. doi: 10.1111/j.1600-0625.2007.00638.x. [DOI] [PubMed] [Google Scholar]
- 143.Murray LA, Knight DA, McAlonan L, et al. Deleterious role of TLR3 during hyperoxia-induced acute lung injury. Am J Respir Crit Care Med. 2008;178:1227–37. doi: 10.1164/rccm.200807-1020OC. [DOI] [PubMed] [Google Scholar]
- 144.Vaneker M, Joosten LA, Heunks LM, et al. Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology. 2008;109:465–72. doi: 10.1097/ALN.0b013e318182aef1. [DOI] [PubMed] [Google Scholar]
- 145.Bae YS, Lee JH, Choi SH, et al. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ Res. 2009;104:210–8. doi: 10.1161/CIRCRESAHA.108.181040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2009;11:155–61. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Scott P, Ma H, Viriyakosol S, et al. Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J Immunol. 2006;177:6370–8. doi: 10.4049/jimmunol.177.9.6370. [DOI] [PubMed] [Google Scholar]
- 148.Gasse P, Riteau N, Charron S, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med. 2009;179:903–13. doi: 10.1164/rccm.200808-1274OC. [DOI] [PubMed] [Google Scholar]
- 149.Pineau I, Lacroix S. Endogenous signals initiating inflammation in the injured nervous system. Glia. 2009;57:351–61. doi: 10.1002/glia.20763. [DOI] [PubMed] [Google Scholar]
- 150.Willis D, Li KW, Zheng JQ, et al. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25:778–91. doi: 10.1523/JNEUROSCI.4235-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lee H, Jo EK, Choi SY, et al. Necrotic neuronal cells induce inflammatory Schwann cell activation via TLR2 and TLR3: implication in Wallerian degeneration. Biochem Biophys Res Commun. 2006;350:742–7. doi: 10.1016/j.bbrc.2006.09.108. [DOI] [PubMed] [Google Scholar]
- 152.Boivin A, Pineau I, Barrette B, et al. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J Neurosci. 2007;27:12565–76. doi: 10.1523/JNEUROSCI.3027-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Anderson KV, Bokla L, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell. 1985;42:791–8. doi: 10.1016/0092-8674(85)90275-2. [DOI] [PubMed] [Google Scholar]
- 154.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 155.Gao B, Tsan MF. Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J Biol Chem. 2003;278:174–9. doi: 10.1074/jbc.M208742200. [DOI] [PubMed] [Google Scholar]
- 156.Tsan MF, Baochong G. Pathogen-associated molecular pattern contamination as putative endogenous ligands of toll-like receptors. J Endotoxin Res. 2007;13:6–14. doi: 10.1177/0968051907078604. [DOI] [PubMed] [Google Scholar]
- 157.Zahringer U, Lindner B, Inamura S, et al. TLR2 – promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology. 2008;213:205–24. doi: 10.1016/j.imbio.2008.02.005. [DOI] [PubMed] [Google Scholar]