

Abstract
Exosomes (EXO) derived from tumour cells have been used to stimulate antitumour immune responses, but only resulting in prophylatic immunity. Tumour-derived heat shock protein 70 (HSP70) molecules are molecular chaperones with a broad repertoire of tumour antigen peptides capable of stimulating dendritic cell (DC) maturation and T-cell immune responses. To enhance EXO-based antitumour immunity, we generated an engineered myeloma cell line J558HSP expressing endogenous P1A tumour antigen and transgenic form of membrane-bound HSP70 and heat-shocked J558HS expressing cytoplasmic HSP70, and purified EXOHSP and EXOHS from J558HSP and J558HS tumour cell culture supernatants by ultracentrifugation. We found that EXOHSP were able to more efficiently stimulate maturation of DCs with up-regulation of Iab, CD40, CD80 and inflammatory cytokines than EXOHS after overnight incubation of immature bone-marrow-derived DCs (5 × 106 cells) with EXO (100 μg), respectively. We also i.v. immunized BALB/c mice with EXO (30 μg/mouse) and assessed P1A-specific T-cell responses after immunization. We demonstrate that EXOHSP are able to stimulate type 1 CD4+ helper T (Th1) cell responses, and more efficient P1A-specific CD8+ cytotoxic T lymphocyte (CTL) responses and antitumour immunity than EXOHS. In addition, we further elucidate that EXOHSP-stimulated antitumour immunity is mediated by both P1A-specific CD8+ CTL and non-P1A-specific natural killer (NK) responses. Therefore, membrane-bound HSP70-expressing tumour cell-released EXO may represent a more effective EXO-based vaccine in induction of antitumour immunity.
Keywords: tumour exosome, membrane-bound HSP70, DC maturation, CTL, NK, antitumour immunity
Introduction
One general characteristic of tumour cells is releasing or shedding membrane vesicular, nowadays, called exosomes (EXO), which was initially described by Taylor et al. 25 years ago [1]. Recently, tumour derived EXO have attracted much attention as a source of tumour antigens (Ag) for vaccines [2–4]. EXO are small (∼100 nm in diameter), membrane-bound vesicles of the endocytic pathway that are externalized by a variety of cell types. They are formed by the fusion of multivesicular bodies with the plasma membrane, followed by exocytosis [5, 6]. Such EXO display a discrete set of proteins involved in antigen presentation, that is, major histocompatibility complex class I and II (MHC-I and MHC-II), costimulatory (CD80,CD86) and tetraspan molecules (CD63, CD82) and are selectively enriched in molecules potentially involved in effector cell targeting, that is, CD11b, lactadherin and CD9 molecules [7, 8]. These tumour-derived EXO isolated from malignant effusions can transfer tumour Ags to dendritic cells (DCs) and induce tumour-specific cytotoxic T lymphocyte (CTL) responses and antitumour immunity [9–11]. It has been reported that EXO need the host DC as an adjuvant for stimulation of CD8+ CTL responses [12, 13]. Zitvogel et al. first demonstrated eradication of tumours by EXO vaccination in animal models [10]. Subsequently, EXO-based vaccines have been confirmed to stimulate CD8+ CTL responses and induce antitumour immunity [9, 14–16]. However, its efficiency is less effective because it only induces either prophylatic antitumour immunity in animal models or very weak antitumour immune responses in clinical trials [17, 18].
Heat shock protein (HSP) molecules are stress-induced molecular chaperones that function to facilitate presentation of endogenous antigenic peptides [19] leading to potent adjuvant effect on stimulation of DC maturation and enhanced CD8+ CTL responses [20]. Tumour-derived HSP have thus been used as adjuvant in cancer vaccines [21]. It has been demonstrated that enhanced expression of cytoplasmic HSP in EXO derived from heat-shocked tumour cells induced more efficient CD8+ CTL responses and antitumour immunity than EXO derived from untreated tumour cells [22, 23]. It has also been shown that expression of membrane-bound exosomal HSP stimulated cytolytic activity of natural killer (NK) cells [24]. Within HSP family, HSP70, the peptides of which can be quickly loaded onto MHC I and II complexes of DCs, can exhibit potent adjuvant effect in stimulation of the host immune responses and antitumour immunity [25–27].
In this study, we compared the efficiency of stimulation of T-cell responses and antitumour immunity between membrane-bound HSP70-expressing EXO derived from HSP70-engineered tumour cells engineered and cytoplasmic HSP70-expressing EXO derived from heat-shocked tumour cells. We first transfected a myeloma cell line J558 expressing its tumour Ag P1A [28] with pcDNAHSP70 vector expressing membrane-bound HSP70 and the control vector pcDNAneo without HSP70 expression, respectively [28]. We also incubated J558 tumour cells at 42°C for 1 hr for heat shock treatment to generate heat-shocked J558 (J558HS) tumour cells. We then purified EXOHSP, EXOneo and EXOHS from transfected J558HSP and J558neo and heat-shocked J558HS cell culture supernatants, respectively. To assess the antitumour immunity derived from EXO vaccination, we immunized wild-type BALB/c mice with membrane-bound HSP70-expressing EXOHSP or control EXOneo or cytoplasmic HSP70-expressing EXOHS. We demonstrate that EXOHSP vaccination is able to more efficiently induce DC maturation leading to stimulation of type 1 helper CD4+ T (Th1) and more efficient CD8+ T-cell responses and immunity against J558 tumour cells than EXPHS and EXOneo vaccination. We also elucidate that the antitumour immunity resulted from EXOHSP vaccination is mediated by both CD8+ CTL and NK cells.
Materials and methods
Reagents, cell lines and animals
The myeloma cell line J558 expressing tumour antigen P1A was obtained from American Type Culture Collection (ATCC; Rockville, MD, USA). Biotin-labelled or fluorenscein isothiocyanate (FITC)-labelled antibodies (Abs) specific for H-2Kd (SF-1.1), Iad (39–10-8), CD11c (HL3), CD40 (3/23), CD54 (3E2) and CD80 (16–10A1) as well as FITC-conjugated avidin were all obtained from BD Pharmingen, Inc. (Mississauga, Ontario, Canada). The Abs specific for LAMP-1 (1D4B) and AIP1 (49/AIP1) were obtained from BD Biosciences (Mississauga, Ontario, Canada). The anti-galectin Ab (M3/38) was obtained from BioLegend (San Diego, CA, USA). The anti-HSP70 Ab (BRM-22) was obtained from SIGMA (Oakville, Ontario, Canada). The anti-P1A Ab [28] was obtained from Dr. Yang Liu, The Ohio State University Medical Center (Columbus, OH, USA). The anti-CD8 and anti-NK Abs were purified from ascites of hybridoma cell line 3.155 and PK136 obtained from ATCC. The P1A peptide (EILPYLGWLVFA) of J558 tumour cells and an irrelevant peptide (YPHFMPTNL) of mouse cytomegalovirus [28] both specific for H-2Ld were synthesized by Multiple Peptide Systems (San Diego, CA, USA). Cytokine ELISA kits were obtained from R&D Systems (Minneapolis, MN, USA). Female wild-type BALB/c mice were obtained from the Jackson Laboratory (Bar Harbor, MA, USA). Mice were treated according to animal care committee guidelines of the University of Saskatchewan.
Generation of engineered J558HSP cell line
Twenty million J558 tumour cells were resuspended in 0.7 ml phosphate-buffered saline (PBS) and mixed with 0.3 ml PBS containing 10 μg pcDNA-HSP70 DNA expressing membrane-bound inducible HSP70 or the control pcDNAneo DNA [29]. Tumour cells were transfected with these DNA using a Bio-Rad gene pulser (Bio-Rad Lab, Mississauga, Ontario, Canada) with parameters of 250 V and 125 μF capacitance as previously described [29]. Transfected cells were selected for growth in medium containing G418 (4 mg/ml). The selected J558HSP and the control J558neo clones maintained in culture medium containing 10% foetal calf serum (FCS) and G418 (0.5 mg/ml) were subjected to flow cytometric analysis and production of tumour-released EXO.
Purification of tumour cell-released EXO
J558 tumour cells were exposed to 42°C in a water bath for 1 hr for heat shock treatment to induce HSP70 expression [30] and termed J558HS. EXO were isolated from tumour cells as described previously [16, 31]. Briefly, the supernatants of J558HS cells cultured for 4 hrs in FCS-free AIM-V medium to avoid contamination of FCS-derived EXO and the supernatants of J558HSP or J558neo cells cultured overnight in AIM-V medium containing G418 (0.5 mg/ml) were subjected to four successive centrifugations at 300 ×g for 5 min. to remove cells, 1200 ×g for 20 min. and 10,000 ×g for 30 min. to remove cellular debris and 100,000 ×g for 1 hr to pellet EXO. The EXO pellets were washed twice in a large volume of PBS and recovered by centrifugation at 100,000 ×g for 1 hr. The amount of exosomal proteins recovered was measured using Bradford assay (Bio-Rad, Richmond, CA, USA). EXO derived from J558HSP, J558neo and J558HS tumour cells were termed as EXOHSP, EXOneo and EXOHS, respectively.
Tumour cell lysate preparation
Live J558 tumour cell lysates were prepared as previously described [32]. Briefly, tumour cells were lysed using extraction buffer containing 125 mM tris(hydroxymethyl)aminomethane, 0.05% sodium dodecyl sulphate and 10%β-mercaptoethanol. Cell extracts were harvested. To remove cellular debris, cell extracts were centrifuged at 1000 ×g for 5 min. The supernatant containing tumour cell lysate protein was freezed at –80°C until use.
Electron microscopy
EXO were fixed in 4% paraformaldehyde. The pellets were then loaded onto carbon-coated formvar grids. After incubation in a moist atmosphere for 20 min., the samples were washed twice in PBS and then fixed for 5 min. in 1% glutaraldehyde. After three washes, the EXO sample-loaded grids were stained for 10 min. with saturated aqueous uranyl. EXO samples were then examined with a Zeiss EM10C (Carl Zeiss Canada Ltd., Montreal, Canada) electron microscope at 60 kV.
Western blot analysis
EXO samples (30 μg/each) were loaded onto 12.5% acrylamide gels (SDS-PAGE) and transferred onto nitrocellulose membrane (Millipore, Bedford, MA, USA). The membrane was blocked by incubation for 2 hrs at room temperature with OYSSEY blocking buffer (LI-COR Bioscience, Lincoln, NE, USA), and immunoblotted with various antibodies at 4°C for overnight. After three washes with PBS, the membrane was further incubated with goat anti-rabbit/antimouse IRDyeR800CW and scanned using ODYSSEY instrument according to manufacturer’s instruction (LI-COR Bioscience).
Flow cytometric analysis of tumour cells and tumour cell-released EXO
Tumour cells and tumour cell-derived EXO were analysed by flow cytometry as previously described [13, 33]. Briefly, J558, J558neo and J558HSP tumour cells (1 × 106 cells) were stained with a panel of biotin-labelled Abs and followed with FITC-avidin. To check apoptosis formation, J558, irradiated (9000 rad) J558, engineered J558HSP and heat-shocked J559HS tumour cells were stained with FITC-annexin V (BD Pharmingen, Inc.) on ice for 30 min., washed with PBS for two times, and then analysed by flow cytometry. EXO such as EXOHSP, EXOneo and EXOHS (30 μg/300 μl PBS) were incubated with a panel of FITC-Abs on ice for 30 min., and then directly analysed by flow cytometry.
Stimulation of immature DCs
The generation of bone marrow derived immature DCs from wild-type BALB/c mice in presence of GM-CSF (1 ng/ml) has been previously described [34]. To assess stimulatory effect, immature DCs (5 × 106 cells) were incubated with EXOHSP or EXOneo or EXOHS (100 μg) for overnight, then stained with a panel of Abs, and analysed by flow cytometry.
T-cell proliferation assays
BALB/c mice were i.v. immunized with EXOneo, EXOHS and EXOHSP (30 μg/mouse), respectively. The mice injected with PBS as control. In in vitro T-cell proliferation assay, 6 days after immunization, CD4+ T cells were purified from splenocytes of the immunized mice by passage through nylon wool columns followed with CD8-microbeads for removal of CD8+ T cells [35]. CD4+ T cells (0.2 × 106 cells/well) and its two-fold dilutions were then incubated with irradiated (4000 rad) J558 tumour cells at a cell ratio of 4:1 in presence of interleukin (IL)-2 (10 U/ml) in 96-well U-bottom plate for 3 days [36]. For measurement of T-cell proliferation, 3H-thymidine incorporation was assessed by liquid scintillation counting. For assessment of cytokine secretion, T-cell supernatants were harvested for measurement of IL-4 and interferon (IFN)-γ secretion by cytokine ELISA kits [35]. In in vitro mixed T lymphocyte reaction assay, irradiated (4000 rad) EXOHSP- or EXOneo- or EXOHS-DCs (1 × 105 cells per well) and its two-fold dilutions were co-cultured in 96-well plates with a constant number of allogeneic C57BL/6 naïve T cells (2 × 105 cells per well). After incubation for 3 days, T-cell proliferation was measured by adding 1 μCi 3H-thymidine (1 mCi/ml, Amersham, Baie D'Urfe, Canada) to each well. After incubation for overnight, the levels of 3H-thymidine incorporation into cellular DNA were determined by liquid scintillation counting [37]. In in vivo T-cell proliferation assay, 6 days after immunization, the tail blood samples or the splenocytes were harvested and stained with PE-conjugated H-2Ld/P1A peptide tetramer (ProImmune, Inc., Springfield, VA, USA) and FITC-conjugated anti-CD8 (Beckman Coulter, Mississauga, Ontario, Canada) for 30 min. at room temperature. The erythrocytes were then lysed using lysis/fixed buffer (Beckman Coulter). The cells were then analysed by flow cytometry [35].
Cytotoxicity assay
The in vivo cytotoxicity assay was performed as previously described [35]. C57BL/6 splenocytes were harvested from naive mouse spleens and incubated with either high (3.0 μM, CFSEhigh) or low (0.6 μM, CFSElow) concentrations of CFSE, to generate differentially labelled target cells. The CFSEhigh cells were pulsed with P1A peptide, whereas the CFSElow cells were pulsed with an irrelevant control peptide and served as internal control. These peptide-pulsed target cells were i.v. injected at 1:1 ratio into the above mice 6 days after immunization. Sixteen hours later, the spleens of immunized mice were removed and residual CFSEhigh and CFSElow target cells remaining in the recipients’ spleens were analysed by flow cytometry [35]. In NK cytotoxicity assay, spleen T cells of mice i.v. immunized with EXOHSP (30 μg/mouse) at different times (2, 4 and 6 days) after immunization were prepared by passing splenocytes through nylon wool columns, and red cells were then lysed using 0.84% ammonium chloride. These T cells were used as the source of effector NK cells. (51Cr)-chromate-labelled J558 tumour cells were used as target cells [38]. Ten thousand labelled target cells per well were mixed with effector cells at various effector/target cell ratios in triplicate in a 96-well plate with V bottom and were incubated for 6 hrs. Percentage of specific lysis was calculated as: 100 × ([experimental CPM – spontaneous CPM]/[maximal CPM – spontaneous CPM]). Spontaneous CPM of target cells was released in absence of effector cells, whereas maximal CPM was released by adding 1% Triton X-100 to wells in the experiment.
Animal studies
To examine whether EXOHSP or DCs with uptake of EXO can induce protective antitumour immunity, wild-type BALB/c mice (n = 8) were s.c. injected with EXOneo, EXOHS and EXOHSP (30 μg/mouse) or DCs with uptake of EXOneo, EXOHS and EXOHSP (1 × 106 cells per mouse), respectively. The immunized mice were s.c. challenged with 0.5 × 106 J558 tumour cells 6 days later. We previously demonstrated that i.p. injection of anti-CD8 (3.155) and anti-NK (PK136) Ab (0.5 mg/mouse) leads to in vivo depletion of more than 95% CD8+ T and NK cells in mouse spleens by flow cytometric analysis [38]. To assess whether CD8+ T or NK cells were involved in EXO-induced antitumour immunity, BALB/c mice (n = 8) were i.p. injected with anti-CD8 or anti-NK Ab (0.5 mg/mouse) 1 day before immunization, and the treatment was repeated every 3 days for a total of three time [38]. The Ab-treated mice were first immunized by s.c. injection of EXOneo, EXOHS and EXOHSP (30 μg/mouse), respectively, and then s.c. challenged with 0.5 × 106 J558 tumour cells 6 days after the immunization. The tumour growth was monitored daily. For humanitarian reasons, all mice with tumours that achieved a size of 1.5 cm in diameter were killed. Log-rank test and Graphpad Prism software (Graphpad Software, Inc., San Diego, CA, USA) were used to compare the mouse survival data [39].
Results
Phenotypical characterization of engineered and heat-shocked J558 tumour cells
The original J558 tumour cells expressed H-2Kd, CD54 and P1A molecules, but not Iad and HSP70 (Fig. 1A). In addition to the above molecules expressed on J558 tumour cells, the engineered J558HSP cells transfected with pcDNAHSP70 displayed cellular surface HSP70 expression, whereas the heat-shocked J558HS tumour cells did not express membrane-bound HSP70. The control J558neo tumour cells transfected with the control vector pcDNA expressed a similar pattern of cellular surface molecules as the original J558 (data not shown). The above molecules expressed on J558HSP were stable since a long-term culturing J558HSP cell line had a similar phenotype as the originally generated one (data not shown). Heat-shocked J558HS cells were generated by culturing J558 tumour cells at 42°C for 1 hr [22, 23]. As shown in Fig. 1(B), the original J558, transfected J558HSP and heat-shocked J558SH tumour cells were mostly live cells without staining of annexin V (early apoptosis marker) [22, 23], whereas irradiated J558 tumour cells (95%) became apoptosis.
Fig 1.

Phenotypic analysis of J558HSP-released EXO. (A) J558HSP, J558HS and J558 tumour cells were stained with a panel of Abs (solid lines) or isotype-matched irrelevant Abs (dotted lines), and analysed by flow cytometry. (B) J558HSP, J558HS and J558 tumour cells as well as irradiated J558 tumour cells were stained with FITC-annexin V (solid lines) or FITC anti-rat IgG (dotted lines) and analysed by flow cytometry. (C) Electron micrograph of EXO. The image shows small cup-shaped vesicles of 60–100 nm in diameter, which are representative of EXO released by tumour cells. Bar, 100 nm. (D) Western blot analysis of tumour cell-released EXO and tumour cell lysates was performed with a panel of antibodies. (E) Flow cytometric analysis of EXO. EXO were grouped for analysis of expression of surface molecules. (F) EXOneo, EXOHS and EXOHSP released from J558neo, J558HS and J558HSP tumour cells were stained with a panel of FITC antibodies (solid lines) or isotype-matched irrelevant FITC antibodies (dotted lines), and analysed by flow cytometry. One representative experiment of two is displayed.
Phenotypical characterization of J558 tumour cell-released EXO
EXO derived from J558, J558neo and J558HSP tumour cells were purified from tumour cell culture supernatants by differential ultracentrifugation. The yields of J558-released EXO, J558neo-released EXOneo and J558HSP-released EXOHSP were 1.0 μg/106 cells/24 hrs, 2.6 mg/106 cells/24 hrs and 2.5 μg/106 cells/24 hrs, respectively, indicating that tumour cells with DNA transfection secrete more EXO than the original tumour cells. The yield of J558HS-released EXOHS was 0.94 μg/106 cells/24 hrs similar to J558-released EXO. These EXO were then subjected to electron microscopic, Western blot and flow cytometric analysis. As shown in Fig. 1(C), EXO derived from either J558neo or J558HSP or J558HS had typical exosomal characteristic of ‘saucer’ or round shape with a diameter between 50 and 90 nm [40]. We also confirmed that EXO-associated proteins including LAMP-1, Alix/AIP1 and H-2Kd were abundant by Western blot analysis (Fig. 1D) [41], whereas galectin, a protein found in the endoplasmic reticulum [42] was only detectable in tumour cell lysates but absent in the EXO samples, indicating that there is no contamination of other vesicles in EXO preparation. Both EXOHS and EXPHSP contained HSP70 by Western blot analysis (Fig. 1D). EXO were then stained with a panel of FITC-Abs and analysed by flow cytometry. As shown in Fig. 1(E), EXO (50–90 nm in diameter) were detectable by flow cytometry even though they were much smaller than microbeads (5 μm in diameter). J558HSP-derived EXOHSP, J558neo-derived EXOneo and J558HS-released EXOHS all displayed expression of molecules (H-2Kd, CD54 and P1A), but in much less extent than J558HSP, J558HS and J558neo tumour cells (Fig. 1F). In addition, EXOHSP did, but EXOHS did not display surface HSP70 expression by flow cytometric analysis, indicating that EXOHSP and EXOHS express membrane-bound and cytoplasmic HSP70, respectively.
Immature DC uptake J558 tumour cell-released EXO
We previously established a protocol for in vitro generation of immature DCs by culturing bone marrow cells in low amount of GM-CSF, but no IL-4 [34] and demonstrated that immature DCs can uptake mature DC-released EXO [39]. To assess the phenotype of immature bone marrow-derived DCs generated in presence of low amount of GM-CSF, we performed flow cytometric analysis. As shown in Fig. 2(A), DCs cultured in presence of low GM-CSF (1 ng/ml) expressed CD11c and CD54, but no Iad, CD40 and CD80 [41], indicating that they are immature DCs. To assess uptake of EXO, immature DCs were incubated with CFSE-labelled J558 tumour cell-released EXO (EXOCFSE) for different times and then analysed by flow cytometry and confocal fluorescence microscopic analysis. As shown in Fig. 2(B), immature DCs displayed CFSE expression after incubation with EXOCFSE, indicating that immature DC uptake J558 tumour cell-released EXO. As shown in Fig. 2(C) and (D), the amount of immature DCs with acquired CFSE increased with the time of incubation with EXOCFSE and reached a maximal level after a 4-hr incubation.
Fig 2.

Immature DCs uptake J558-released EXO. (A) Flow cytometric analysis of immature DCs with a panel of antibodies (solid lines) or isotype-matched irrelevant antibodies (dotted lines). (B) Immature DCs (5 × 106 cells) were incubated with J558 tumour cell-released EXO (dotted line) or CFSE-labelled J558 tumour cell-released EXOCFSE (100 mg) (solid line) for 4 hrs and analysed by flow cytometry. (C and D) Immature DCs (5 × 106 cells) were incubated with CFSE-labelled J558 tumour cell-released EXOCFSE (100 mg) (solid line) for different times and analysed by (C) flow cytometry and (D) confocal fluorescence microscopy under differential interference contrast. One representative experiment of two is displayed.
EXOHSP efficiently stimulate DC maturation
It has been demonstrated that HSP70 can stimulate DC maturation [20]. To assess whether EXOHSP were also able to stimulate DC maturation, we analysed immature DCs cultured with EXOHSP expressing membrane-bound HSP70 or EXOHS expressing cytoplasmic HSP70 or EXOneo without HSP70 expression. EXOneo-stimulated DCs still showed a similar pattern of the expression of the above molecules on immature DCs (Fig. 3A), indicating that EXOneo does not modulate DC maturation. Interestingly, we found that EXOHS- and EXOHSP-stimulated DCs up-regulated expression of Iad, CD40 and CD80, indicating that both EXOHS and EXOHSP are able to stimulate DC maturation. However, the up-regulation of the above molecules stimulated by the later was more than the former, indicating that EXOHSP is a stronger stimulator for DC maturation than EXOHS. In addition, EXOHSP-stimulated mature DCs also secreted more amount of inflammatory cytokines such as IL-1β (1.8 ng/ml/106 cells/24 hrs), IL-12 (1.1 ng/ml/106 cells/24 hrs), IFN-γ (0.8 ng/ml/106 cells/24 hrs) and tumour necrosis factor (TNF)-α (0.9 ng/ml/106 cells/24 hrs) (Fig. 3B) than EXOHS-stimulated DCs. Since EXOHSP harboured the above immune molecules, they may have potent effect in stimulation of T-cell immune responses [43]. We first assessed whether DCs with uptake of EXO by incubation of immature DCs with EXO for overnight stimulate allogeneic T-cell proliferation in a mixed T lymphocyte reaction assay. As shown in Fig. 3C, DCs with uptake of EXOHSP (DC + EXOHSP) stimulated the strongest allogeneic T-cell proliferation than DCs with uptake of EXOneo (DC + EXOneo) and EXOHS (DC + EXOHS) (P < 0.05). We then assessed whether DCs with uptake of EXO by incubation of immature DCs with EXO for overnight induce P1A-specific antitumour immunity. As shown in Fig. 3(D), DCs with uptake of EXOneo (DC + EXOneo), EXOHS (DC + EXOHS) and EXOHSP (DC + EXOHSP) were able to stimulate P1A-specific antitumour immunity to protect one of eight, three of eight and eight of eight mice from tumour growth after the immunized mice were challenged with J558 tumour cells, respectively, indicating that EXOHSP-treated DCs are the most immunogenic among these three types of DCs.
Fig 3.

J558HSP-released EXOHSP stimulate DC maturation. (A) Flow cytometric analysis of EXO-treated DCs. Immature DCs with or without incubation of EXOneo, EXOHS and EXOHSP were stained with a panel of antibodies (solid lines) or isotype-matched irrelevant antibodies (dotted lines), and analysed by flow cytometry. Mean fluorescence intensity of solid line/mean fluorescence intensity of dotted line was presented in each panel. (B) Cytokine secretion. Culture supernatants of DCs alone (DC), DC with EXOHS (DC + EXOHS) and DC with EXOHSP (DC + EXOHSP) were tested using cytokine ELISA kits. Values represent the mean of triplicates from three experiments. (C) Mixed T lymphocyte reaction assay. Irradiated DCs including DC 1 EXOHS and DC 1 EXOHSP and their two-fold dilutions were co-cultured in 96-well plates with a constant number of allogeneic C57BL/6 naïve T cells. After 3 days, T-cell proliferation was measured by adding 1 μCi 3H-thymidine to each well in an overnight 3H-thymidine uptake assay. The levels of 3H-thymidine incorporation into cellular DNA were determined by liquid scintillation counting. (D) BALB/c mice were s.c. vaccinated with irradiated (4000 rad) DC 1 EXOneo, DC 1 EXOHS and DC 1 EXOHSP (0.5 × 106 cells per mouse). 6 days after immunization, the immunized mice were s.c. inoculated with J558 tumour cells (0.5 × 106 cells per mouse). Animal mortality and tumour growth were monitored daily for up to 60 days. *, P < 0.05 versus cohorts in EXOHSP group (log-rank test). One representative experiment of two is displayed.
EXOHSP stimulate type 1 CD4+ T-cell responses
To assess whether EXOHSP can stimulate CD4+ T-cell responses, we performed in vitro T-cell proliferation assay by using CD4+ T cells derived from EXO-immunized mouse spleens. As shown in Fig. 4(A), EXOHSP immunization could mount a more efficient CD4+ T-cell proliferation compared to that of EXOHS-immunized group (P < 0.05). To assess the type of CD4+ T-cell response, we measured the cytokine secretion of CD4+ T cells derived from EXOHSP-immunized mice by ELISA. CD4+ T cells derived from EXOHSP immunized mice secreted IL-2 (1.4 ng/ml/106 cells/24 hrs) and IFN-γ (1.1 ng/ml/106 cells/24 hrs), but not IL-4 (Fig. 4B), indicating that EXOHSP stimulate type 1 CD4+ helper T (Th1) cell responses.
Fig 4.
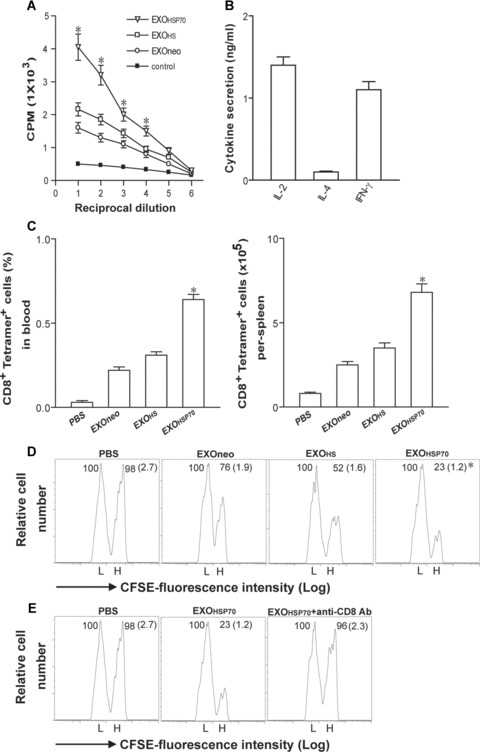
J558HSP-released EXOHSP stimulate T-cell responses. (A) A T-cell proliferation assay. Splenic CD4+ T cells from EXOneo- or EXOHS- or EXOHSP-immunized mice were incubated with irradiated J558 tumour cells for 3 days. 3H-thymidine incorporation was assessed by liquid scintillation counting. *, P < 0.05 versus cohorts in EXOneo or EXOHS group (Student’s t-test). (B) Cytokine secretion. The culture supernatants of CD4+ T cells derived from EXOHSP-immunized mice were tested using cytokine ELISA kits. Values represent the mean of triplicates from three experiments. (C) Tetramer staining assay. 6 days after immunization of mice with EXOneo, EXOHS and EXOHSP, the tail blood and splenocyte samples were taken from the immunized mice, stained with PE-H-2Ld/P1A peptide tetramer and FITC anti-CD8 Ab and analysed by flow cytomery. The value in each panel represents the percentage of tetramer+ CD8+ T cells versus the total blood CD8+ T-cell population or the total tetramer+ CD8+ T cells per spleen. *, P < 0.05 versus cohorts in EXOneo or EXOHS group (Student’s t-test). (D and E) In in vivo cytotoxicity assay. BALB/c splenocytes were harvested from naive mouse spleens and incubated with either high (3.0 μM, CFSEhigh) or low (0.6 μM, CFSElow) concentrations of CFSE, to generate differentially labelled target cells. The CFSEhigh cells were pulsed with P1A peptide, whereas the CFSElow cells were pulsed with the control peptide and served as internal controls. These peptide-pulsed target cells were i.v. injected at 1:1 ratio into (D) the above immunized mice 6 days after immunization of EXOneo, EXOHS and EXOHSP, respectively, or into (E) the mice immunized with EXOHSP but also treated with anti-CD8 Ab to deplete CD8+ T cells. 16 hrs later, the spleens of immunized mice were removed and the percentages of the residual P1A-specific CFSEhigh (H) and control CFSElow (L) target cells remaining in the recipients’ spleens were analysed by flow cytometry. The value in each panel represents the percentage of CFSEhigh versus CFSElow target cells remaining in the spleen. The value in parenthesis represents the standard deviation. *, P < 0.05 versus cohorts in EXOneo or EXOHS group (Student’s t-test). One representative experiment of three is shown.
EXOHSP stimulate stronger CD8+ effector CTL responses than EXOHS
To assess the stimulatory effect of EXOHSP on in vivo CD8+ T-cell responses, we performed PE-H-2Ld/P1A peptide tetramer staining assay [43]. As shown in Fig. 4(C), PBS-treatment group did not stimulate any P1A-specific (tetramer+) CD8+ T-cell responses. However, EXOHSP immunization was able to induce 0.64% tetramer+ CD8+ T cells of the total CD8+ T-cell population, whereas EXOHS and EXOneo immunization only stimulated 0.34% and 0.22% tetramer+ CD8+ T cells of the total CD8+ T-cell population at day 6 after immunization (P < 0.05), indicating that EXOHSP expressing membrane-bound HSP70 can activate more efficient P1A-specific CD8+ T-cell responses in vivo than EXOHS expressing cytoplasmic HSP70 and EXOneo without HSP70 expression, respectively. To confirm that P1A-specific (tetramer+) CD8+ T cells detected in peripheral blood represent the systemic condition, we also measured the tetramer+CD8+ T cells in mouse spleens. The pattern of P1A-specific CD8+ T cells in different groups of mouse spleens was similar to that seen in mouse blood samples. To assess whether EXO can also stimulate CD8+ T-cell differentiation into CTL effectors in vivo, we adoptively transferred P1A peptide-pulsed splenocytes that had been strongly labelled with CFSE (CFSEhigh) as well as the control peptide-pulsed splenocytes that had been weakly labelled with CFSE (CFSElow) into the recipient mice that had been vaccinated with EXOHSP, EXOHS and EXOneo, respectively. We then assessed loss of P1A-specific CFSEhigh target cells in the recipient mice, which represent the killing activity of P1A-specific effector CD8+ CTLs in the recipient mice. As shown in Fig. 4(D), little CFSEhigh target cells loss (2%) was observed in mice immunized with PBS. As expected, there was a substantial loss (24% and 48%) of CFSEhigh cells in mice immunized with EXOneo and EXOHS, indicating that EXOHS and EXOneo can stimulate CD8+ T-cell differentiation into CTL effectors in vivo. However, there was a more substantial loss (77%) of CFSEhigh cells in mice immunized with EXOHSP (P < 0.05), indicating that EXOHSP stimulate stronger CD8+ T-cell differentiation into CTL effectors than EXOHS and EXOneo, respectively. To confirm that loss of CFSEhigh cells in mice immunized with EXOHSP is due to P1A-specific effector CD8+ CTLs in the recipient mice, we depleted CD8+ T cells using neutralizing anti-CD8 antibody treatment before adoptive transfer of P1A peptide-pulsed splenocytes. As shown in Fig. 4(E), little CFSEhigh target cells loss (4%) was seen in EXOHSP-immunized mice with CD8+ T-cell depletion, confirming that EXOHSP efficiently stimulates P1A-specific effector CD8+ CTL responses.
EXOHSP induce stronger antitumour immunity than EXOHS
To investigate the antitumour immunity derived from EXOHSP vaccination, BALB/c mice were s.c. immunized with EXOHSP, EXOneo, EXOHS and PBS, respectively. 6 days after the immunization, the immunized mice were s.c. challenged with J558 tumour cells. As shown in Fig. 5(A), all the mice injected with PBS died of tumour within 14 days after tumour cell challenge. EXOHS and EXOneo vaccine protected four of eight (50%) and two of eight (25%) mice from tumour growth, respectively, and the rest of four or six tumour-bearing mice had significantly delayed tumour growth compared to the control group of mice treated with PBS (P < 0.05). However, EXOHSP immunization protected all eight of eight (100%) mice from tumour growth, indicating that EXOHSP expressing membrane-bound HSP70 can also induce more efficient antitumour immunity than EXOHS expressing cytoplasmic HSP70 and the control EXOneo without HSP70 expression.
Fig 5.

Animal studies. (A) BALB/c mice were s.c. vaccinated with EXOneo, EXOHS and EXOHSP or PBS. 6 days after immunization, the immunized mice were s.c. inoculated with J558 tumour cells. Animal mortality and tumour growth were monitored daily for up to 60 days. *, P < 0.05 versus cohorts in EXOHSP group (log-rank test). (B) Kinetic tetramer staining assay. 2 to 8 days after immunization of mice with EXOHSP, the tail blood samples were taken from the immunized mice, stained with PE-H-2Ld/P1A peptide tetramer and FITC anti-CD8 Ab and analysed by flow cytometry. The value in each panel represents the percentage of tetramer+ CD8+ T cells versus the total blood CD8+ T-cell population. (C) Kinetic study of NK activity. 2 to 8 days after immunization of mice with EXOHSP, the mouse spleen T cells were tested for NK killing activity to J558 tumour cells. BALB/c mice with treatment of anti-CD8 or anti-NK Ab were s.c. vaccinated with either (D) EXOneo or (E) EXOHS (F) EXOHSP. 6 days after immunization, the immunized mice were s.c. inoculated with J558 tumour cells. Animal mortality and tumour growth were monitored daily for up to 6 weeks. One representative experiment of three is shown.
The EXOHSP-induced antitumour immunity is mediated by CD8+ CTL and NK cells
Since CD8+ T cells have been defined as effector T cells with cytotoxic activity to tumour or virally infected cells [44] and the membrane-bound HSP70 molecules are identified as a target structure for cytolytic attack mediated by NK cells [45], we assessed whether CD8+ T and NK cells are involved in EXOHSP-induced antitumour immunity. We first performed a kinetic study on CD8+ CTL and NK cell killing activity derived from EXOHSP vaccination. The killing activity of NK derived from naïve BALB/c mice was 8% (data not shown). As shown in Fig. 5(B) and (C), P1A-specific CD8+ CTL responses (0.64%) and non-P1A-specific NK killing activity (38%) was peaked at day 6 and day 2 after vaccination. At day 6 after immunization, NK killing activity was still 20%, indicating that both CD8+ CTL and NK cells may be involved in the antitumour immunity seen at day 6 after EXOHSP vaccination (Fig. 5A). We then treated BALB/c mice with either anti-CD8 or anti-NK Ab, immunized the mice with EXOneo or EXOHS or EXOHSP and then challenged the immunized mice with J558 tumour cells. We found that the EXOneo-, EXOHS- and EXOHSP-immunized mice with treatment of anti-CD8 Ab completely lost the protective antitumour immunity, since all (eight of eight) immunized mice died of tumours within these three mouse groups with treatment of anti-CD8 Ab (Fig. 5D, E and F), indicating that EXOneo-, EXOHS- and EXOHSP-induced antitumour immunities are mainly mediated by CD8+ T cells. When mice were treated with anti-NK Ab, 25% (two of eight) of EXOneo-immunized mice were protected against tumour cell challenge (Fig. 5D), indicating that NK cell responses are not involved in EXOneo-induced antitumour immunity. However, 38% (three of eight) of EXOHS-immunized mice were tumour free after treatment of anti-NK Ab, compared to six of eight (76%) immune protection in EXOHSP-immunized mice with anti-NK Ab treatment (Fig. 5E and F), indicating that (i) NK cell responses are involved in both EXOHS- and EXOHSP-induced antitumour immunity and (ii) EXOHSP-vaccination stimulates a stronger NK activity than EXOHS-vaccination.
Discussion
In recent years, EXO research has attracted more attention by the finding that human and mouse tumour cells constitutively release membrane vesicles, similar to DC-derived EXO in their morphology, density and expression of certain membrane markers [10]. To enhance immunogenicity of tumour cell-released EXO, tumour cells have been engineered to express various immunologically related genes for use in EXO production. For example, Xiu et al. genetically engineered tumour cells to express superantigen Staphylococcal enterotoxin A [46]. Chen et al. generated IL-2-expressing EXO released from transgene IL-2-engineered tumour cells [47]. They demonstrated that all these modified EXO were able to stimulate more efficient T-cell responses and antitumour immunity. Furthermore, EXO derived from IL-18 transgene engineered human tumour cells expressing carcinoembryonic Ag were able to more efficiently stimulate carcinoembryonic Ag specific CTL responses [36]. In addition, EXO derived from heat-shocked tumour cells displayed enhanced expression of HSP by Western blotting analysis induced DC maturation and more efficient CD8+ CTL responses than EXO derived from untreated tumour cells [22, 48]. However, whether these enhanced HSP are cytoplasmic soluble ones or membrane-bound cellular surface ones is unclear. It has been reported that DCs secrete pro-inflammatory cytokines in responses to soluble HSP protein through a CD14-dependent signalling pathway [23, 49], whereas membrane-bound HSP70 molecules are identified as a target structure for cytolytic attack mediated by NK cells [45]. In this study, we generated HSP70-expressing EXOHSP derived from engineered J558HSP tumour cell line expressing membrane-bound transgene HSP70 and assessed the efficiency of tumour-specific CD8+ CTL responses induced by EXOHSP. For the first time, we demonstrated that EXOHSP expressing membrane-bound HSP70 were able to stimulate tumour-specific CD4+ Th1 cell responses possibly due to IL-12 secretion of EXOHSP-activated mature DCs. In addition, we also demonstrated that EXOHSP expressing membrane-bound HSP70 were able to stimulate more efficient tumour-specific CD8+ CTL responses and antitumour immunity than EXOHS expressing cytoplasmic HSP70 and EXOneo derived from the control J558neo tumour cells without HSP70 expression. Furthermore, we elucidated that EXOHSP-induced antitumour immunity is mediated by both CD8+ T and NK cells, indicating that EXOHSP vaccination can not only stimulate the P1A tumour Ag-specific T cell, but also the non-P1A-specific NK cell responses.
It has been previously reported that EXO may need the host DCs as an adjuvant for induction of immune responses based only upon in vitro experiments [12, 50]. We have recently demonstrated that DC- or tumour cell-derived immunogenic EXO exhibiting efficient T-cell responses completely lost their stimulatory effects in induction of CD8+ T responses in diphtheria toxin (DT)-treated DT receptor transgenic mice with the host DC deficiency [13], indicating that EXO need the host DCs for delivery of their stimulatory effect to CD8+ CTL responses in vivo. In this study, we demonstrated that EXOHSP expressing membrane-bound HSP70 were able to stimulate maturation of DCs with up-regulation of Iab, costimulatory molecules (CD40 and CD80) and inflammatory cytokines (IL-1β, TNF-α, IFN-γ and IL-12). Therefore, it is conceivable that the enhanced CD8+ CTL responses and antitumour immunity induced by EXOHSP may be derived from the adjuvant effect of membrane-bound expression of HSP70. This is because HSP70 can trigger a danger signal by binding to DC receptors such as Toll-like receptor 4 [49, 51] and CD14 [23] through NF-κB to up-regulate costimulatory molecules of DCs and induce DC maturation and secretion of pro-inflammatory cytokines [52] leading to stimulation of both CD8+ CTL and NK cell responses. Thus, the dual role of HSP70 as antigenic peptide chaperone [53] and danger signal [52] thus makes them especially critical in EXO-based antitumour vaccine.
Taken together, our data showed that EXO derived from tumour cells engineered to express membrane-bound HSP70 can induce DC maturation and stimulate CD4+ Th1, CD8+ CTL and NK cell responses leading to more efficient antitumour immunity. Therefore, membrane-bound HSP70-expressing EXO may represent a more effective EXO-based vaccine in induction of antitumour immunity.
Acknowledgments
This research work was supported by research grants from Canadian Institutes of Health Research (MOP 79415 and 89713). We appreciated Mark Boyd for help in flow cytometry.
References
- 1.Taylor DD, Homesley HD, Doellgast GJ. Binding of specific peroxidase-labeled antibody to placental-type phosphatase on tumor-derived membrane fragments. Cancer Res. 1980;40:4064–9. [PubMed] [Google Scholar]
- 2.Andre F, Schartz NE, Chaput N, et al. Tumor-derived exosomes: a new source of tumor rejection antigens. Vaccine. 2002;20:A28–31. doi: 10.1016/s0264-410x(02)00384-5. [DOI] [PubMed] [Google Scholar]
- 3.Kim JV, Latouche JB, Riviere I, et al. The ABCs of artificial antigen presentation. Nat Biotechnol. 2004;22:403–10. doi: 10.1038/nbt955. [DOI] [PubMed] [Google Scholar]
- 4.Chaput N, Schartz NE, Andre F, et al. Exosomes for immunotherapy of cancer. Adv Exp Med Biol. 2003;532:215–21. doi: 10.1007/978-1-4615-0081-0_17. [DOI] [PubMed] [Google Scholar]
- 5.Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–79. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 6.Denzer K, Kleijmeer MJ, Heijnen HF, et al. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci. 2000;113:3365–74. doi: 10.1242/jcs.113.19.3365. [DOI] [PubMed] [Google Scholar]
- 7.Thery C, Regnault A, Garin J, et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol. 1999;147:599–610. doi: 10.1083/jcb.147.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Escola JM, Kleijmeer MJ, Stoorvogel W, et al. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem. 1998;273:20121–7. doi: 10.1074/jbc.273.32.20121. [DOI] [PubMed] [Google Scholar]
- 9.Andre F, Schartz NE, Movassagh M, et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet. 2002;360:295–305. doi: 10.1016/S0140-6736(02)09552-1. [DOI] [PubMed] [Google Scholar]
- 10.Wolfers J, Lozier A, Raposo G, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7:297–303. doi: 10.1038/85438. [DOI] [PubMed] [Google Scholar]
- 11.Altieri SL, Khan AN, Tomasi TB. Exosomes from plasmacytoma cells as a tumor vaccine. J Immunother. 2004;27:282–8. doi: 10.1097/00002371-200407000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Vincent-Schneider H, Stumptner-Cuvelette P, Lankar D, et al. Exosomes bearing HLA-DR1 molecules need dendritic cells to efficiently stimulate specific T cells. Int Immunol. 2002;14:713–22. doi: 10.1093/intimm/dxf048. [DOI] [PubMed] [Google Scholar]
- 13.Hao S, Bai O, Yuan J, et al. Dendritic cell-derived exosomes stimulate stronger CD81 CTL responses and antitumor immunity than tumor cell-derived exosomes. Cell Mol Immunol. 2006;3:205–11. [PubMed] [Google Scholar]
- 14.Hsu DH, Paz P, Villaflor G, et al. Exosomes as a tumor vaccine: enhancing potency through direct loading of antigenic peptides. J Immunother. 2003;26:440–50. doi: 10.1097/00002371-200309000-00007. [DOI] [PubMed] [Google Scholar]
- 15.Chaput N, Schartz NE, Andre F, et al. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol. 2004;172:2137–46. doi: 10.4049/jimmunol.172.4.2137. [DOI] [PubMed] [Google Scholar]
- 16.Cho JA, Yeo DJ, Son HY, et al. Exosomes: a new delivery system for tumor antigens in cancer immunotherapy. Int J Cancer. 2005;114:613–22. doi: 10.1002/ijc.20757. [DOI] [PubMed] [Google Scholar]
- 17.Escudier B, Dorval T, Chaput N, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med. 2005;3:10. doi: 10.1186/1479-5876-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morse MA, Garst J, Osada T, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005;3:9. doi: 10.1186/1479-5876-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Eden W, Van Der Zee R, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol. 2005;5:318–30. doi: 10.1038/nri1593. [DOI] [PubMed] [Google Scholar]
- 20.Zeng Y, Feng H, Graner MW, et al. Tumor-derived, chaperone-rich cell lysate activates dendritic cells and elicits potent antitumor immunity. Blood. 2003;101:4485–91. doi: 10.1182/blood-2002-10-3108. [DOI] [PubMed] [Google Scholar]
- 21.Srivastava PK, Menoret A, Basu S, et al. Heat shock proteins come of age: primitive functions acquire new roles in an adaptive world. Immunity. 1998;8:657–65. doi: 10.1016/s1074-7613(00)80570-1. [DOI] [PubMed] [Google Scholar]
- 22.Chen W, Wang J, Shao C, et al. Efficient induction of antitumor T cell immunity by exosomes derived from heat-shocked lymphoma cells. Eur J Immunol. 2006;36:1598–607. doi: 10.1002/eji.200535501. [DOI] [PubMed] [Google Scholar]
- 23.Asea A, Kraeft SK, Kurt-Jones EA, et al. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–42. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 24.Gastpar R, Gehrmann M, Bausero MA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005;65:5238–47. doi: 10.1158/0008-5472.CAN-04-3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold-Schild D, Hanau D, Spehner D, et al. Cutting edge: receptor-mediated endocytosis of heat shock proteins by professional antigen-presenting cells. J Immunol. 1999;162:3757–60. [PubMed] [Google Scholar]
- 26.Moroi Y, Mayhew M, Trcka J, et al. Induction of cellular immunity by immunization with novel hybrid peptides complexed to heat shock protein 70. Proc Natl Acad Sci USA. 2000;97:3485–90. doi: 10.1073/pnas.070550797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castelli C, Ciupitu AM, Rini F, et al. Human heat shock protein 70 peptide complexes specifically activate antimelanoma T cells. Cancer Res. 2001;61:222–7. [PubMed] [Google Scholar]
- 28.Bai XF, Liu JQ, Joshi PS, et al. Different lineages of P1A-expressing cancer cells use divergent modes of immune evasion for T-cell adoptive therapy. Cancer Res. 2006;66:8241–9. doi: 10.1158/0008-5472.CAN-06-0279. [DOI] [PubMed] [Google Scholar]
- 29.Chan T, Chen Z, Hao S, et al. Enhanced T-cell immunity induced by dendritic cells with phagocytosis of heat shock protein 70 gene-transfected tumor cells in early phase of apoptosis. Cancer Gene Ther. 2007;14:409–20. doi: 10.1038/sj.cgt.7701025. [DOI] [PubMed] [Google Scholar]
- 30.Feng H, Zeng Y, Whitesell L, et al. Stressed apoptotic tumor cells express heat shock proteins and elicit tumor-specific immunity. Blood. 2001;97:3505–12. doi: 10.1182/blood.v97.11.3505. [DOI] [PubMed] [Google Scholar]
- 31.Thery C, Duban L, Segura E, et al. Indirect activation of naive CD41 T cells by dendritic cell-derived exosomes. Nat Immunol. 2002;3:1156–62. doi: 10.1038/ni854. [DOI] [PubMed] [Google Scholar]
- 32.Campbell I, Magliocco A, Moyana T, et al. Adenovirus-mediated p16INK4 gene transfer significantly suppresses human breast cancer growth. Cancer Gene Ther. 2000;7:1270–8. doi: 10.1038/sj.cgt.7700226. [DOI] [PubMed] [Google Scholar]
- 33.Clayton A, Court J, Navabi H, et al. Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation and flow cytometry. J Immunol Methods. 2001;247:163–74. doi: 10.1016/s0022-1759(00)00321-5. [DOI] [PubMed] [Google Scholar]
- 34.Chen Z, Dehm S, Bonham K, et al. DNA array and biological characterization of the impact of the maturation status of mouse dendritic cells on their phenotype and antitumor vaccination efficacy. Cell Immunol. 2001;214:60–71. doi: 10.1006/cimm.2001.1883. [DOI] [PubMed] [Google Scholar]
- 35.Xiang J, Huang H, Liu Y. A new dynamic model of CD81 T effector cell responses via CD41 T helper-antigen-presenting cells. J Immunol. 2005;174:7497–505. doi: 10.4049/jimmunol.174.12.7497. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, Xiu F, Cai Z, et al. Increased induction of antitumor response by exosomes derived from interleukin-2 gene-modified tumor cells. J Cancer Res Clin Oncol. 2007;133:389–99. doi: 10.1007/s00432-006-0184-7. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Zhang W, Chan T, et al. Engineered fusion hybrid vaccine of IL-4 gene-modified myeloma and relative mature dendritic cells enhances antitumor immunity. Leuk Res. 2002;26:757–63. doi: 10.1016/s0145-2126(02)00002-4. [DOI] [PubMed] [Google Scholar]
- 38.Xiang J, Chen Z, Huang H, et al. Regression of engineered myeloma cells secreting interferon-gamma-inducing factor is mediated by both CD4(+)/CD8(+) T and natural killer cells. Leuk Res. 2001;25:909–15. doi: 10.1016/s0145-2126(01)00052-2. [DOI] [PubMed] [Google Scholar]
- 39.Hao S, Bai O, Li F, et al. Mature dendritic cells pulsed with exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumour immunity. Immunology. 2007;120:90–102. doi: 10.1111/j.1365-2567.2006.02483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehmann BD, Paine MS, Brooks AM, et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008;68:7864–71. doi: 10.1158/0008-5472.CAN-07-6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luketic L, Delanghe J, Sobol PT, et al. Antigen presentation by exosomes released from peptide-pulsed dendritic cells is not suppressed by the presence of active CTL. J Immunol. 2007;179:5024–32. doi: 10.4049/jimmunol.179.8.5024. [DOI] [PubMed] [Google Scholar]
- 42.Fevrier B, Raposo G. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol. 2004;16:415–21. doi: 10.1016/j.ceb.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Hwang I, Shen X, Sprent J. Direct stimulation of naive T cells by membrane vesicles from antigen-presenting cells: distinct roles for CD54 and B7 molecules. Proc Natl Acad Sci USA. 2003;100:6670–5. doi: 10.1073/pnas.1131852100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiedemann A, Depoil D, Faroudi M, et al. Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc Natl Acad Sci USA. 2006;103:10985–90. doi: 10.1073/pnas.0600651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Multhoff G, Botzler C, Jennen L, et al. Heat shock protein 72 on tumor cells: a recognition structure for natural killer cells. J Immunol. 1997;158:4341–50. [PubMed] [Google Scholar]
- 46.Xiu F, Cai Z, Yang Y, et al. Surface anchorage of superantigen SEA promotes induction of specific antitumor immune response by tumor-derived exosomes. J Mol Med. 2007;85:511–21. doi: 10.1007/s00109-006-0154-1. [DOI] [PubMed] [Google Scholar]
- 47.Dai S, Zhou X, Wang B, et al. Enhanced induction of dendritic cell maturation and HLA-A*0201-restricted CEA-specific CD8(+) CTL response by exosomes derived from IL-18 gene-modified CEA-positive tumor cells. J Mol Med. 2006;84:1067–76. doi: 10.1007/s00109-006-0102-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai S, Wan T, Wang B, et al. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin Cancer Res. 2005;11:7554–63. doi: 10.1158/1078-0432.CCR-05-0810. [DOI] [PubMed] [Google Scholar]
- 49.Asea A, Rehli M, Kabingu E, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–34. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 50.Andre F, Chaput N, Schartz NE, et al. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J Immunol. 2004;172:2126–36. doi: 10.4049/jimmunol.172.4.2126. [DOI] [PubMed] [Google Scholar]
- 51.Vabulas RM, Braedel S, Hilf N, et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem. 2002;277:20847–53. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- 52.Basu S, Binder RJ, Suto R, et al. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12:1539–46. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- 53.Chandawarkar RY, Wagh MS, Srivastava PK. The dual nature of specific immunological activity of tumor-derived gp96 preparations. J Exp Med. 1999;189:1437–42. doi: 10.1084/jem.189.9.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]