

Abstract
Brain energy disorders can be present in aged men and animals. To this respect, the mitochondrial and free radical theory of aging postulates that age-associated brain energy disorders are caused by an imbalance between pro- and anti-oxidants that can result in oxidative stress. Our study was designed to investigate brain energy metabolism and the activity of endogenous antioxidants during their lifespan in male Wistar rats. In vivo brain bioenergetics were measured using 31P nuclear magnetic resonance (NMR) spectroscopy and in vitro by polarographic analysis of mitochondrial oxidative phosphorylation. When compared to the young controls, a significant decrease of age-dependent mitochondrial respiration and adenosine-3-phosphate (ATP) production measured in vitro correlated with significant reduction of forward creatine kinase reaction (kfor) and with an increase in phosphocreatine (PCr)/ATP, PCr/Pi and PME/ATP ratio measured in vivo. The levels of enzymatic antioxidants catalase, GPx and GST significantly decreased in the brain tissue as well as in the peripheral blood of aged rats. We suppose that mitochondrial dysfunction and oxidative inactivation of endogenous enzymes may participate in age-related disorders of brain energy metabolism.
Keywords: aging, brain, 31P NMR, mitochondria, OXPHOS, antioxidants
Introduction
Mitochondrial and free radical theories of aging suggest that there is an imbalance between the activity of free radical generation and the endogenous radical scavenging system in brain disorders [1, 2] resulting in an impairment of ATP production [3, 4]. In this regard, mitochondria are not only intracellular generators of the reactive oxygen species (ROS) but also immediate targets of oxidative damage [5–8]. ROS are generated at sites of inflammation and injury. At low levels, they can function as signalling intermediates in the regulation of fundamental cell activities such as growth and adaptation responses. At higher concentrations, ROS can cause cell injury and death. This occurs during the aging process, where oxidative stress is incremented due to an accelerated generation of ROS and a gradual decline in cellular antioxidant defence mechanisms. Consequently, aging and the associated increase in oxidative stress are major risk factors for many neurodegenerative diseases [9]. Compared to other organs or tissues, the brain is more vulnerable to ROS-induced damage due to its high rate of oxygen consumption, high polyunsaturated lipid content and relative weariness of classic antioxidant enzymes [10, 11].
Organisms utilize antioxidant defence, including enzymes like superoxide dismutase (SOD), which converts O2.− into H2O2, catalase (CAT), that is responsible for the detoxification of H2O2 and GPx, that breaks down peroxides. The ATP metabolism is tightly coupled to the PCr metabolism via the enzyme system of creatine kinase (CK), which plays a central role in energy transfer in cells with high energy requirements and which is very susceptible to oxidative stress [12, 13]. Rate constant of CK reaction can be investigated by the magnetization transfer in vivo 31P magnetic resonance spectroscopy (MRS) experiment [14, 15]. The investigation of the kinetic parameter kfor using in vivo 31P MR spectroscopy magnetization transfer method contributes to better understanding of the underlying processes in age-dependent brain disorders. This technique can be used as a non-invasive in vivo biomarker for age-related neurodegenerative diseases as it can reveal energy metabolism impaired in brain tissue, which is not detectable by conventional MRS methods. By means of this technique we studied reaction kinetics of reversible exchange of the phosphate group in the reaction catalysed by CK in the brain of rats [9]. Moreover, mitochondrial oxidative phosphorylations (OXPHOS) as well as endogenous enzymatic antioxidant activities in the brain and peripheral blood of young, adult and aged rats were investigated in this study.
Materials and methods
Animals
Young (3 months old), adult (17 months old) and aged (36 months old), male Wistar rats supplied by Velaz (Prague, Czech Republic) were housed at 22 ± 2°C, 45% relative humidity, 12-hr light/dark photoperiodicity in air-conditioned rooms with free access to standard commercial laboratory pellets ST1 (Tp Dovo, Slovak Republic) and water ad libitum. All animals received human care in compliance with the Institutional Animal Ethic Committee and with the Guidelines of European Convention for the Protection of Vertebrate Animals Used for Experimental Purposes.
Methods
In vivo 31P magnetic resonance spectroscopy
Phosphorous saturation transfer experiments were performed on a 4.7 T 200 Mhz SISCO imaging spectrometer equipped with a horizontal magnet at 81 MHz. A three-turn 1.6-cm-diameter surfaře coil was positioned over the skull of an animal anesthetized by 0.8–1.0% of halothane. First, the static magnetic field was shimmed using proton signals which showed a typical line width of 20–35 Hz. Then, the phosphorous flip angle was adjusted to minimize the broad signal coming from the bone. The time of irradiation of γ-ATP resonance was varied from 0.3 to 1.6 sec., which resulted in an exponential decay of PCr signal to a steady-state value. The saturation was accomplished by an on-resonance series of 10 msec. DANTE pulses with interpulse delays of 400 msec. [16]. Either 96 or 128 transients were accumulated in the interleave mode with a repetition of 5 sec. [17].
In vitro mitochondrial respiration
The rats were euthanized with i.p. injection of Thiopental (Spofa, Czech Republic, 150 mg/kg b.w.). Removed brain tissue was placed on ice-cold isolation solution containing (in mmol/l) 225 manitol, 75 sucrose, 0.2 ethylenediaminetetraacetic acid (EDTA); pH 7.4. Tissue sample was minced and homogenized in the solution using a glass-teflon homogenizer. Brain mitochondria were isolated at 4°C by differential centrifugation [18]. Mitochondrial protein concentration was estimated by the method of Lowry [19] using bovine serum albumin as a standard. Respiratory chain function was measured in a respiratory buffer containing (in mmol/l) 12.5 HEPES, 3 KH2PO4, 122 KCl, 0.5 EDTA and 2% dextran; pH 7.2 at 30°C, by means of Oxygraph Gilson 5/6H (Madison, WI, USA) using Clark-type oxygen electrode. Sodium glutamate/malic acid (2.5 mmol/2.5 mmol) were used as a nicotinamide adenine dinucleotide (NAD) substrate for complex I and succinate (5 mmol) as a FAD substrate for complex II. To initiate state 3 respiratory activity, 500 nmol of ADP were added to the cuvette. When all the ADP was converted to ATP, state 4 respiration was measured. Parameters of OXPHOS, such as QO2(S3) [nAtO mg/prot/min.], i.e. oxygen consumption rate in presence of ADP (state S3); QO2S4 [nAtO mg/prot/min.], oxygen consumption rate without ADP (S4); RCR [S3/S4], respiratory control ratio; ADP:O (nmol ADP/nAtO], coefficient of OXPHOS and OPR [nmol ATP mg prot/min], OXPHOS rate, were determined, respectively.
Endogenous antioxidants
The enzymatic activity of SOD was determined using a RANSOD kit (Randox Labs, San Diego, CA, USA). CAT activity was determined as described by Aebi [20], glutathione peroxidase (GPx) activity according to Wendel [21], the ferric reducing ability of plasma (FRAP) according to Benzie [22] and ceruloplasmin (CPL) according to Schosinsky [23].
Statistical methods
The results were evaluated using ANOVA and Student’s t-test for unpaired data; the Sudent’s t-test was used to compare differences between groups. Calculated values were expressed as means ± S.E.M.. P-values were considered as a significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
Brain bioenergetic parameters were evaluated on young (3 months old), adult (17 months old) and aged (36 months old) male Wistar rats using in vivo 31P MRS and in vitro polarographic analysis of mitochondrial OXPHOS, respectively.
In vivo 31P magnetic resonance spectroscopy
Relative concentrations of phosphate metabolism were determined from integrals of their signal in 31P MR spectra using program MESTREC-C 1.5.1 (Table 1 and Fig. 1). Time-dependent 31P MRS saturation transfer was applied to determine the pseudo-first-order rate constant of forward CK reaction (kfor). While the analysis of steady-state 31P MR spectra revealed significant increase in the ratio of PCr/ATP (P < 0.001), PCr/Pi (P < 0.01) and PME/ATP (P < 0.05), we found significant (P < 0.001) decrease of forward kfor in aged rats as compared to the young group.
Table 1.
Brain bioenergetic measurements in young (3 months old) and aged (36 months old) rats by using 31P NMR spectroscopy
| Age | PCr/ATP | PCr/Pi | PME/ATP | kfor | pHi |
|---|---|---|---|---|---|
| 3 months | 1.94 ± 0.06 | 4.14 ± 0.42 | 0.77 ± 0.09 | 0.35 ± 0.02 | 7.18 ± 0.01 |
| 36 months | 2.12 ± 0.04*** | 5.64 ± 1.44** | 0.84 ± 0.07* | 0.30 ± 0.01*** | 7.16 ± 0.01* |
Data are presented as mean ± S.E.M., ***P < 0.001, **P < 0.01, *P < 0.05 versus 3 months.
Note: Adult (17 months old) rats were not measured due to disruptions in SISCO spectrometer.
Fig 1.
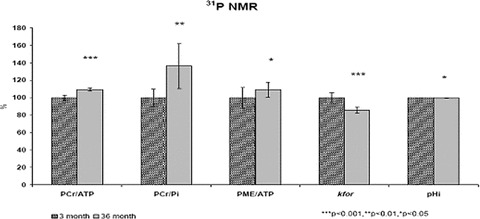
Changes of brain bioenergetics measured in vivo by 31P MRS in aged (36 months old) compared to young (3 months old) male Wistar rats. MRS, magnetic resonance spectroscopy; PCr/ATP, phosphocreatine to adenosine three phosphate ratio; PCr/Pi, PCr to inorganic phosphate ratio; PME/ATP, phosphate monoester to ATP ratio; kfor, forward rate constant of CK and pHi, intracellular pH. ***P < 0.001, **P < 0.01, *P < 0.05.
In vitro mitochondrial oxidative phosphorylation (Table 2 and Fig. 2)
Table 2.
Age-dependent mitochondrial OXPHOS activities in young (3 months), adult (17 months) and aged (36 months) male Wistar rats
| Parameters OXPHOS | Age | ||
|---|---|---|---|
| 3 months | 17 months | 36 months | |
| Complex I | |||
| RCI (S3.S4–1) | 3.63 ± 0.19 | 3.25 ± 0.12 | 2.86 ± 0.08*+ |
| ADP:O (nmol ADP.nAtO-1) | 2.67 ± 0.02 | 2.76 ± 0.02* | 2.63 ± 0.04+ |
| QO2S3 (nAtO.mg prot-1.min-1) | 76.73 ± 2.80 | 58.74 ± 2.89** | 48.68 ± 1.76***+ |
| QO2S4 (nAtO.mg prot-1.min-1) | 21.98 ± 1.52 | 18.16 ± 0.75 | 17.14 ± 0.80 |
| OPR (nmol ATP.mg prot-1.min-1) | 205.23 ± 6.08 | 163.97 ± 9.35** | 127.27 ± 5.26***++ |
| Complex II | |||
| RCI (S3.S4–1) | 1.66 ± 0.33 | 1.65 ± 0.06 | 1.64 ± 0.04 |
| ADP:O (nmol ADP.nAtO-1) | 1.69 ± 0.03 | 1.63 ± 0.04 | 1.72 ± 0.03 |
| QO2S3 (nAtO.mg prot-1.min-1) | 87.87 ± 4.36 | 68.20 ± 3.97** | 51.04 ± 2.72***++ |
| QO2S4 (nAtO.mg prot-1.min-1) | 53.02 ± 2.93 | 41.66 ± 2.75* | 31.33 ± 1.99***+ |
| OPR (nmol ATP.mg prot-1.min-1) | 205.23 ± 6.08 | 163.97 ± 9.35** | 127.27 ± 5.26***++ |
Fig 2.
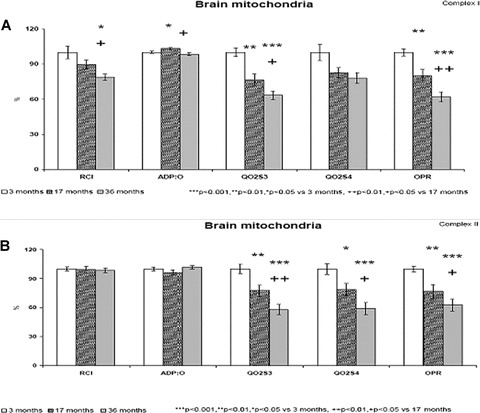
Percentual changes of in vitro measured OXPHOS parameters in brain mitochondria from young (3 months old), adult (17 months old) and aged (36 months old) male Wistar rats. (A) Complex I–sodium glutamate / malic acid NAD substrate, (B) Complex II–succinate FAD substrate (respectively, A and B). ***P < 0.001, **P < 0.01, *P < 0.05 versus 3 months old rats. ++P < 0.01, +P < 0.05 versus 17 months old rats.
2 days after 31P MR spectroscopy, the rats were killed and brains were removed for biochemical investigations. Polarographic analysis of OXPHOS was performed on mitochondria isolated from the forebrain of young (3 months old), adult (17 months old) and aged (36 months old) rats. State 3 respiration (QO2S3) was started by the addition of ADP to mitochondria respiring with substrates. When measured in complex I and in complex II, the ADP-stimulated state 3 respiration significantly decreased by 23% and by 22% in adult (P < 0.01), and by 37% and 42% (P < 0.001) in aged rats as compared with young rats, respectively. Respiratory control index (RCI), as indicator of mitochondrial membrane integrity was found to be significantly decreased by 21% (P < 0.05) only in the complex I of aged rats. There were no significant differences in state 4 respiration in complex I, however, in complex II state 4 respiration was decreased significantly by 21% in adult (P < 0.05) and by 41% (P < 0.001) in aged rats. Coefficient of OXPHOS (ADP:O) as an indicator of coupling oxidation and phosphorylation showed some changes in complex I, but not in complex II. Significant decrease in rates of OXPHOS and ATP production (OPR) by 20% and 23% (P < 0.01) in adult and by 38% and 37% (P < 0.001) in aged rats is suggested to be the consequence of age-related decrease in state 3 respiration.
Endogenous antioxidants (Table 3 and Fig. 3)
Table 3.
Brain and blood antioxidant measurements in adult (10 months old) and aged (36 months old) rats
| Age | ||
|---|---|---|
| 10 months | 36 months | |
| Brain | ||
| SOD (U.g-1) | 170.73 ± 21.43 | 160.63 ± 8.15 |
| CAT (U.ml-1) | 3.63 ± 0.42 | 4.59 ± 0.16 |
| GPX (U.g-1) | 0.84 ± 0.14 | 1.34 ± 0.16* |
| GST (U.g-1) | 2.69 ± 0.17 | 3.09 ± 0.06* |
| FRAP (mmol.g-1) | 4.64 ± 0.27 | 4.87 ± 0.06 |
| CPL (U.g-1) | 0.52 ± 0.04 | 0.07 ± 0.02*** |
| Blood | ||
| SOD e(U.ml-1) | 197.03 ± 11.06 | 179.83 ± 8.06 |
| CAT e(U.ml-1) | 1232.68 ± 143.68 | 3928.88 ± 155.88*** |
| GPXe(U.ml-1) | 8.83 ± 0.33 | 3.14 ± 0.03*** |
| GST e(U.ml-1) | 3.84 ± 0.36 | 5.70 ± 0.37** |
| FRAP p(mmol.ml-1) | 363.34 ± 32.18… | 315.50 ± 18.90 |
| CPL p (U.ml-1) | 0.14 ± 0.02 | 0.11 ± 0.01 |
Data are presented as mean ± S.E.M. ***P < 0.001, **P < 0.01, *P < 0.05.
Fig 3.
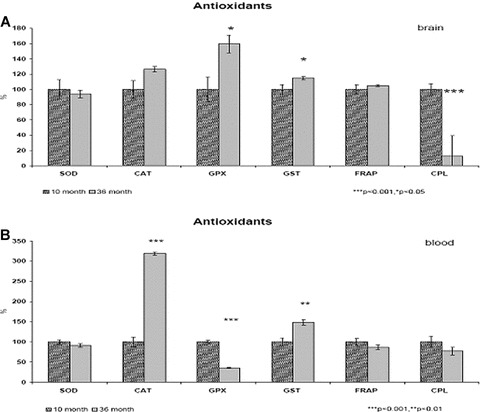
Percentual changes of endogenous antioxidants activity in brain tissue (A) and in peripheral blood (B) of aged (36 months old) compared to adult (10 months old) male Wistar rats. (A) (brain) ***P < 0.001, *P < 0.05, (B) (blood) ***P < 0.001, **P < 0.01.
The aim of the present study was to investigate the FRAP along with antioxidant enzyme activities and CPL. This analysis has revealed an significant (P < 0.05) increase of GPX and GST activity and significant (P < 0.001) decrease of CPL level in the brain as well as significant increase of CAT (P < 0.001) and GST (P < 0.01) activity and decrease of GPX (P < 0.001) activity in plasma of aged rats. As a result, antioxidant power (FRAP) of the brain and plasma was not disturbed in aged rats.
Discussion
It is well documented that age-related neurogenic dementia is a major risk factor in the lifespan of human beings and animals [24–27]. In aerobic cells, 90–95% of the total amount of ATP production comes from aerobic metabolism. The synthesis of ATP via the mitochondrial respiratory chain is the result of the electron transport coupled to OXPHOS [28]. During normal aging and due to the emergence of some neurodegenerative diseases like AD, damaged mitochondria are unable to maintain the energy demands of the cell [29–31]. This can lead to an increased production of free radicals, which induces the interruption of OXPHOS and results in decreased levels of ATP [32]. Excitotoxicity, advanced age, oxidative stress, hypertension, mitochondrial dysfunction, free radical-induced oxidative damage have all been implicated in the pathogenesis of several different neurodegenerative diseases in addition to AD, and include Huntington’s disease, Parkinson’s disease, amyotrophic lateral sclerosis and Schizophrenia [33]. The main radical produced by mitochondria is the superoxide anion; intra mitochondrial antioxidant systems scavenge this radical to avoid oxidative damage, which can lead to impaired ATP production [4, 24, 30, 32, 34, 35].
Neurons are very sensible to oxidative stress due to their high demand for energy. Most of the energy is derived from the metabolism of glucose to carbon dioxide and water through the glycolysis and mitochondrial OXPHOS [36]. The ATP production and utilization is fundamental in cerebral bioenergetics [37–39]. The brain ATP metabolism is mainly controlled by ATPase and CK reactions of PCR< = >ATP< = >Pi chemical exchange systems [40, 41]. In our 31P MRS study, performed on young (3 months) and aged (36 months) rats, we found significantly increased ratios of PCr to ATP (P < 0.001), PCr to Pi (P < 0.01) and PME to ATP (P < 0.05).
The kinetics of ATP metabolism and the associated chemical exchange rates should be more sensitive to brain energy states than the steady-state ATP concentration [42, 43]. CK plays a fundamental role in cellular energetics of the brain. The forward rate constant (kfor) of the CK can be used as an indicator of brain metabolic changes. In our study, kfor was found to be significantly (P < 0.001) diminished in the brain of aged rats compared to young rats. We assume that kfor of CK in the brain could serve as a sensitive early in vivo MRS biomarker of brain disorders. The results of 31P MRS correspond with the results of in vitro mitochondrial OXPHOS assessed polarographically in the brain of rats. These results support the hypothesis that impairment of mitochondrial respiration may be a causal factor of the aging process, and that such impairment may be result from increased H2O2 production in vivo[18]. Our findings suggest that the CK reaction could play a key role in the energetic system of young and aged rat brains.
Progressive age-related significant decrease of oxygen uptake (QO2S3) and ATP synthesis (OPR) describe the condition of mitochondrial dysfunction. Age-related cerebral disorders are usually accompanied by oxidative stress [44] and perturbation of the cellular oxidant/antioxidant balance [45, 46]. The brain has relatively low levels of antioxidants [47, 48]. SOD-2 is a highly inducible mitochondrial protein synthesized within the cytosol [49, 50]. These consequences have been strongly implicated in the pathogenesis of human as well as animal models of neurodegenerative diseases [51–54], particularly AD [4, 24, 29, 30, 34, 35]. In our study, no significant differences in SOD expression were detectable between old (10 months) and aged (36 months) in rat brain homogenate and in peripheral blood (see Table 3 and Fig. 3). The activity of GPX significantly increased (P < 0.05) in the brain and decreased (P < 0.001) in peripheral blood. The activity of GST significantly increased in brain (P < 0.05) as well as in peripheral blood (P < 0.01). The activity of CAT significantly increased (P < 0.001) in blood only. However, age-related changes in brain oxidant enzyme activities remain controversial [55].
The high energy demand and lack of endogenous fuel reserves make the brain highly dependent upon continuous blood supply. Cerebrovascular lesions observed in aging are believed to be responsible for cognitive impairment [30, 31, 56] due to chronic hypoperfusion and suboptimal delivery of energy substrates [57, 58]. These observations demonstrate the importance of studying vascular alterations during aging. Clinical symptoms of vascular dementia are manifested when the amount of neuron loss has reached a threshold around 50–70%[59]. Our observations suggest that the mitochondria found in neurons, as well as in all of the brain cellular compartments (glia and vessels wall cells), are a primary target of brain damage due to their high energy demand and susceptibility to oxidation, leading to energy failure, and resulting in cognitive impairment and memory decline. Moreover, mounting evidence indicates that targeting mitochondria with antioxidants is a powerful form of treatment capable of restoring cell integrity and eliminating damage in the brain, resulting in significantly restored cognitive function and spatial memory. Therefore, in vivo 31P MRS and magnetic resonance imaging measurements are suitable to detect the disturbances early in the preclinical stage. Expanding feature research towards this avenue will open the doors for more and better treatment strategies developed towards aged associated neruodegenerative and cerebrovascular diseases.
Acknowledgments
This study was supported by the Slovak Research and Development Agency, project APVV-21–022004.
Conflicts of interest
All authors have no conflicts of interest.
References
- 1.Dogru-Abbasoglu S, Tamer-Toptani S, Ugurnal B, et al. Lipid peroxidation and antioxidant enzymes in livers and brains of aged rats. Mech Ageing Dev. 1997;98:177–80. doi: 10.1016/s0047-6374(97)00082-1. [DOI] [PubMed] [Google Scholar]
- 2.Evans PH. Free radicals in brain metabolism and pathology. Br Med Bull. 1993;49:577–87. doi: 10.1093/oxfordjournals.bmb.a072632. [DOI] [PubMed] [Google Scholar]
- 3.Bowling AC, Mutisya EM, Walker LC, et al. Age-dependent impairment of mitochondrial function in primate brain. J Neurochem. 1993;60:1964–7. doi: 10.1111/j.1471-4159.1993.tb13430.x. [DOI] [PubMed] [Google Scholar]
- 4.Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab. 1999;19:351–69. doi: 10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 6.Harman D. The biologic clock: the mitochondria. J Am Geriatr Soc. 1972;20:145–7. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 7.Kwong LK, Sohal RS. Substrate and site specificity of hydrogen peroxide generation in mouse mitochondria. Arch Biochem Biophys. 1998;350:118–26. doi: 10.1006/abbi.1997.0489. [DOI] [PubMed] [Google Scholar]
- 8.Miquel J, Economos AC, Fleming J, et al. Mitochondrial role in cell aging. Exp Gerontol. 1980;15:575–91. doi: 10.1016/0531-5565(80)90010-8. [DOI] [PubMed] [Google Scholar]
- 9.Linseman DA. Targeting oxidative stress for neuroprotection. Antioxidants Redox Signal. 2008;11:421–24. doi: 10.1089/ars.2008.2236. [DOI] [PubMed] [Google Scholar]
- 10.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–95. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 11.Aliev G, Obrenovich ME, Reddy VP, et al. Antioxidant therapy in Alzheimer’s disease: theory and practice. Mini Rev Med Chem. 2008;8:1395–406. doi: 10.2174/138955708786369582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta. 1998;1366:211–23. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- 13.Matthews RT, Yang L, Jenkins BG, et al. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. J Neurosci. 1998;18:156–63. doi: 10.1523/JNEUROSCI.18-01-00156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corbett RJ, Laptook AR. Age-related changes in swine brain creatine kinase-catalyzed 31P exchange measured in vivo using 31P NMR magnetization transfer. J Cereb Blood Flow Metab. 1994;14:1070–7. doi: 10.1038/jcbfm.1994.140. [DOI] [PubMed] [Google Scholar]
- 15.Sauter A, Rudin M. Determination of creatine kinase kinetic parameters in rat brain by NMR magnetization transfer. Correlation with brain function. J Biol Chem. 1993;268:13166–71. [PubMed] [Google Scholar]
- 16.Clark JF, Harris GI, Dillon PF. Multisite saturation transfer using DANTE and continuous wave. Magn Reson Med. 1991;17:274–8. doi: 10.1002/mrm.1910170130. [DOI] [PubMed] [Google Scholar]
- 17.Forsen F, Hoffman R. Study of moderately rapid chemical exchange reactions by means of nuclear magnetic double resonance. J Chem Phys. 1963;39:2892–901. [Google Scholar]
- 18.Sarma JS, Ikeda S, Fischer R, et al. Biochemical and contractile properties of heart muscle after prolonged alcohol administration. J Mol Cell Cardiol. 1976;8:951–72. doi: 10.1016/0022-2828(76)90077-8. [DOI] [PubMed] [Google Scholar]
- 19.Lowry OH, Rosebrough NJ, Farr AL, et al. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 20.Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–6. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 21.Wendel A. Glutathione peroxidase. Methods Enzymol. 1981;77:325–33. doi: 10.1016/s0076-6879(81)77046-0. [DOI] [PubMed] [Google Scholar]
- 22.Benzie IF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem. 1996;239:70–6. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- 23.Schosinsky KH, Lehmann HP, Beeler MF. Measurement of ceruloplasmin from its oxidase activity in serum by use of o-dianisidine dihydrochloride. Clin Chem. 1974;20:1556–63. [PubMed] [Google Scholar]
- 24.Aliev G, Gasimov E, Obrenovich ME, et al. Atherosclerotic lesions and mitochondria DNA deletions in brain microvessels: implication in the pathogenesis of Alzheimer’s disease. Vasc Health Risk Manag. 2008;4:721–30. doi: 10.2147/vhrm.s2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aliev G, Palacios H, Walrafen B, et al. Brain mitochondria as a primary target in the development of treatment strategies for Alzheimer disease. Int J Biochim Cell Biol. 2009 doi: 10.1016/j.biocel.2009.03.015. (in press). doi: 10.1016/j.biocel.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 26.Kunz WS, Kuznetsov AV, Clark JF, et al. Metabolic consequences of the cytochrome c oxidase deficiency in brain of copper-deficient Mo(vbr) mice. J Neurochem. 1999;72:1580–5. doi: 10.1046/j.1471-4159.1999.721580.x. [DOI] [PubMed] [Google Scholar]
- 27.Raz N, Rodrigue KM, Acker JD. Hypertension and the brain: vulnerability of the prefrontal regions and executive functions. Behav Neurosci. 2003;117:1169–80. doi: 10.1037/0735-7044.117.6.1169. [DOI] [PubMed] [Google Scholar]
- 28.Acuna-Castroviejo D, Martin M, Macias M, et al. Melatonin, mitochondria, and cellular bioenergetics. J Pineal Res. 2001;30:65–74. doi: 10.1034/j.1600-079x.2001.300201.x. [DOI] [PubMed] [Google Scholar]
- 29.Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–23. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aliev G, Smith MA, De La Torre JC, et al. Mitochondria as a primary target for vascular hypoperfusion and oxidative stress in Alzheimer’s disease. Mitochondrion. 2004;4:649–63. doi: 10.1016/j.mito.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 31.Aliyev A, Chen SG, Seyidova D, et al. Mitochondria DNA deletions in atherosclerotic hypoperfused brain microvessels as a primary target for the development of Alzheimer’s disease. J Neurol Sci. 2005:229–230. doi: 10.1016/j.jns.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 32.Schulz JB, Matthews RT, Klockgether T, et al. The role of mitochondrial dysfunction and neuronal nitric oxide in animal models of neurodegenerative diseases. Mol Cell Biochem. 1997;174:193–7. [PubMed] [Google Scholar]
- 33.Rao AV, Balachandran B. Role of oxidative stress and antioxidants in neurodegenerative diseases. Nutritional neuroscience. 2002;5:291–309. doi: 10.1080/1028415021000033767. [DOI] [PubMed] [Google Scholar]
- 34.Castellani R, Hirai K, Aliev G, et al. Role of mitochondrial dysfunction in Alzheimer’s disease. J Neurosci Res. 2002;70:357–60. doi: 10.1002/jnr.10389. [DOI] [PubMed] [Google Scholar]
- 35.Aliev G, Liu J, Shenk JC, et al. Neuronal mitochondrial amelioration by feeding acetyl-L-carnitine and lipoic acid to aged rats. J Cell Mol Med. 2009;13:320–33. doi: 10.1111/j.1582-4934.2008.00324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erecinska M, Silver IA. ATP and brain function. J Cereb Blood Flow Metab. 1989;9:2–19. doi: 10.1038/jcbfm.1989.2. [DOI] [PubMed] [Google Scholar]
- 37.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–45. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- 38.Kemp GJ. Non-invasive methods for studying brain energy metabolism: what they show and what it means. Dev Neurosci. 2000;22:418–28. doi: 10.1159/000017471. [DOI] [PubMed] [Google Scholar]
- 39.Raichle ME, Mintun MA. Brain work and brain imaging. Annu Rev Neurosci. 2006;29:449–76. doi: 10.1146/annurev.neuro.29.051605.112819. [DOI] [PubMed] [Google Scholar]
- 40.Du F, Zhu XH, Qiao H, et al. Efficient in vivo 31P magnetization transfer approach for noninvasively determining multiple kinetic parameters and metabolic fluxes of ATP metabolism in the human brain. Magn Reson Med. 2007;57:103–14. doi: 10.1002/mrm.21107. [DOI] [PubMed] [Google Scholar]
- 41.Lei H, Ugurbil K, Chen W. Measurement of unidirectional Pi to ATP flux in human visual cortex at 7 T by using in vivo 31P magnetic resonance spectroscopy. Proc Natl Acad Sci USA. 2003;100:14409–14. doi: 10.1073/pnas.2332656100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Degani H, Alger JR, Shulman RG, et al. 31P magnetization transfer studies of creatine kinase kinetics in living rabbit brain. Magn Reson Med. 1987;5:1–12. doi: 10.1002/mrm.1910050102. [DOI] [PubMed] [Google Scholar]
- 43.Ugurbil K. Magnetization transfer measurements of creatine kinase and ATPase rates in intact hearts. Circulation. 1985;72:IV94–6. [PubMed] [Google Scholar]
- 44.Long J, Gao F, Tong L, et al. Mitochondrial decay in the brains of old rats: ameliorating effect of alpha-lipoic acid and acetyl-L-carnitine. Neurochem Res. 2009;34:755–63. doi: 10.1007/s11064-008-9850-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–23. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 46.Halliwell B. Antioxidant defence mechanisms: from the beginning to the end (of the beginning) Free Radic Res. 1999;31:261–72. doi: 10.1080/10715769900300841. [DOI] [PubMed] [Google Scholar]
- 47.Cassarino DS, Bennett JP., Jr An evaluation of the role of mitochondria in neurodegenerative diseases: mitochondrial mutations and oxidative pathology, protective nuclear responses, and cell death in neurodegeneration. Brain Res Brain Res Rev. 1999;29:1–25. doi: 10.1016/s0165-0173(98)00046-0. [DOI] [PubMed] [Google Scholar]
- 48.Otterbein LE, Choi AM. Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1029–37. doi: 10.1152/ajplung.2000.279.6.L1029. [DOI] [PubMed] [Google Scholar]
- 49.Akai F, Maeda M, Suzuki K, et al. Immunocytochemical localization of manganese superoxide dismutase (Mn-SOD) in the hippocampus of the rat. Neurosci Lett. 1990;115:19–23. doi: 10.1016/0304-3940(90)90510-g. [DOI] [PubMed] [Google Scholar]
- 50.Manganaro F, Chopra VS, Mydlarski MB, et al. Redox perturbations in cysteamine-stressed astroglia: implications for inclusion formation and gliosis in the aging brain. Free Radic Biol Med. 1995;19:823–35. doi: 10.1016/0891-5849(95)02008-x. [DOI] [PubMed] [Google Scholar]
- 51.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–81. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 52.Beckman KB, Ames BN. Mitochondrial aging: open questions. Ann N Y Acad Sci. 1998;854:118–27. doi: 10.1111/j.1749-6632.1998.tb09897.x. [DOI] [PubMed] [Google Scholar]
- 53.Beckman KB, Ames BN. Endogenous oxidative damage of mtDNA. Mutat Res. 1999;424:51–8. doi: 10.1016/s0027-5107(99)00007-x. [DOI] [PubMed] [Google Scholar]
- 54.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–8. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 55.Serrano F, Klann E. Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res Rev. 2004;3:431–43. doi: 10.1016/j.arr.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 56.Sarti C, Pantoni L, Bartolini L, et al. Cognitive impairment and chronic cerebral hypoperfusion: what can be learned from experimental models. J Neurol Sci. 2002:203–204. doi: 10.1016/s0022-510x(02)00302-7. [DOI] [PubMed] [Google Scholar]
- 57.De La Torre JC. Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer’s pathogenesis. Neurobiol Aging. 2000;21:331–42. doi: 10.1016/s0197-4580(00)00111-1. [DOI] [PubMed] [Google Scholar]
- 58.De La Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke. 2002;33:1152–62. doi: 10.1161/01.str.0000014421.15948.67. [DOI] [PubMed] [Google Scholar]
- 59.Mueller SG, Schuff N, Weiner MW. Evaluation of treatment effects in Alzheimer’s and other neurodegenerative diseases by MRI and MRS. NMR Biomed. 2006;19:655–68. doi: 10.1002/nbm.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]