

Abstract
Purpose.
Expansion of the intronic CTG18.1 triplet repeat locus within TCF4 contributes significant risk to the development of Fuchs' endothelial corneal dystrophy (FECD) in Eurasian populations, but the mechanisms by which the expanded repeats result in degeneration of the endothelium have been hitherto unknown. The purpose of this study was to examine FECD endothelial samples for the presence of RNA nuclear foci, the hallmark of toxic RNA, as well as evidence of haploinsufficiency of TCF4.
Methods.
Using fluorescence in situ hybridization, we examined for the presence of nuclear RNA foci containing expanded CUG transcripts in corneal endothelial samples from FECD subjects with CTG18.1 expansion. We also examined for any changes in expression levels of TCF4 by quantitative real-time PCR.
Results.
Numerous discrete nuclear RNA foci were identified in endothelial samples of FECD subjects (n = 8) harboring the CTG18.1 expansion, but not in controls lacking the expansion (n = 5) (P = 7.8 × 10−4). Percentage of cells with foci in expansion-positive endothelial samples ranged from 33% to 88%. RNA foci were absent in endothelial samples from an FECD subject without CTG18.1 expansion and a subject with endothelial dysfunction without FECD. Expression of the constitutive TCF4 exon encoding the basic helix-loop-helix domain was unaltered with CTG18.1 expansion.
Conclusions.
Our findings suggest that the RNA nuclear foci are pathognomonic for CTG18.1 expansion-mediated endothelial disease. The RNA nuclear foci have been previously found only in rare neurodegenerative disorders caused by repeat expansions. Our detection of abundant ribonuclear foci in FECD implicates a role for toxic RNA in this common disease.
Keywords: Fuchs' endothelial corneal dystrophy, TCF4, CTG18.1, RNA nuclear foci, triplet repeat expansion
CTG18.1 triplet repeat expansion in TCF4 confers significant risk to the development of Fuchs' dystrophy in Eurasian populations. The presence of nuclear RNA foci containing expanded CUG transcripts in corneal endothelial samples with CTG18.1 expansion implicate toxic RNA in Fuchs' dystrophy.
Introduction
Fuchs' endothelial corneal dystrophy (FECD; MIM 136800) is an age-related degenerative disorder resulting in corneal edema and loss of vision. This bilateral, progressive disorder occurring in 5% of Caucasians older than 40 years in the United States is the most common genetic disorder of the corneal endothelium.1 Forty percent of the 66,305 keratoplasty (corneal transplant) procedures performed in the United States in 2013 were for FECD and other related cases of endothelial dysfunction.2 Although studies on the prevalence of FECD worldwide are limited, the disorder is thought to be more common in Eurasian populations, with its corneal manifestations documented in 11% of females and 7% of males in Reykjavik, Iceland,3 8.5 % of Singapore Chinese,4 and 5.5% of Japanese.4
In FECD, the corneal endothelium, the inner terminally differentiated monolayer responsible for maintenance of stromal dehydration, undergoes accelerated senescence and apoptosis.5–10 Descemet membrane, the basement membrane of the endothelium, becomes diffusely thickened and develops focal excrescences called guttae that are clinically visible with slit lamp biomicroscopy.11 In the early stages of the disease process, the normal hexagonal morphology of the endothelium is lost and replaced by cellular polymorphism and polymegathism (variation in cell size) as an indicator of premature senescence.6 As the guttae become confluent centrally, there is progressive loss of central endothelial cell density, resulting in corneal stromal edema, and scarring, resulting in loss of vision.
Late-onset FECD is a complex genetic disease with locus heterogeneity. Although the disorder has been described as an autosomal dominant trait,12 incomplete penetrance and phenocopies within large pedigrees are not uncommon.13,14 Rare heterozygous mutations in COL8A2 (MIM 120252) cause an early-onset corneal endothelial dystrophy.15 Although SLC4A11 (MIM 610206), TCF8 (MIM 189909), LOXHD1 (MIM 613267), and AGBL1 (MIM 615523) have been implicated in adult-onset FECD, they are responsible for only a minority of cases.14,16–22
Expanded trinucleotide repeats at the CTG18.1 locus in the third intron of transcription factor 4, TCF4 (MIM 602272) (aliases: E2-2, SEF2, SEF2-1B, or ITF2), were recently identified to be strongly associated with adult-onset FECD23,24 after both traditional linkage studies13 and genome-wide association studies25 highlighted the same vicinal region on chromosome 18 (MIM 613267). The TCF4 is a conserved class I basic helix-loop-helix (bHLH) transcription factor that binds to the canonical E-box sequence CANNTG of promoters of target genes.26,27 The CTG18.1 locus was initially discovered in 1997 by the Repeat Expansion Detection assay with expanded alleles of greater than 37 CTG repeats shown to be unstable and present in 3% of subjects in Caucasian pedigrees without any known associated phenotype.28 Defying the norm for common variants, each copy of the expanded CTG18.1 allele of more than 40 CTG triplet repeats confers significant risk for the development of FECD with an odds ratio (OR) of 32.3 in Caucasians.24 In a study of 29 Caucasian FECD pedigrees, we showed that the expanded CTG18.1 allele cosegregates with the trait with complete penetrance in 52% of the families and with incomplete penetrance in an additional 10% of the remaining families examined.24 After we performed transethnic haplotype analysis and replication of the association with an OR of 66.5 for each expanded CTG18.1 allele in Singapore Chinese, we concluded that the repeat expansion is a shared, common variant predisposing susceptibility for FECD in Eurasian populations.29 Additional corroborating genetic evidence that the triplet repeat expansion is an FECD susceptibility allele rather than a tagged polymorphism in linkage disequilibrium with another functional variant is the recent report of the association of the triplet expansion in an FECD cohort from India30 with a different genetic background from Caucasians or Chinese, and the failure of targeted next-generation sequencing of 465 kb of this region, including TCF4 exons, introns, and flanking regions, in a Caucasian cohort to reveal any other variant that was a better predictor of disease than the CTG18.1 repeat expansion.31
However, the mechanisms by which the expanded CTG repeats contribute to the degeneration of the corneal endothelium have been hitherto unknown but may include RNA-mediated toxicity, haploinsufficiency of TCF4, or a combination of both mechanisms. A role for RNA gain of function in disease was initially reported for expanded CUG transcripts in myotonic dystrophy (DM1; MIM 160900).32 Subsequently, RNA toxicity has been shown to play a central role in numerous other neurodegenerative and neuromuscular diseases caused by simple repeat expansions.33,34 A hallmark of these nucleotide repeat expansion disorders is the accumulation of the mutant expanded transcripts into nuclear RNA foci that were first identified in DM1.35,36 Subsequently, RNA nuclear foci have been found to play a central role in numerous other degenerative disorders, including myotonic dystrophy 2 (DM2; MIM 602668), fragile X-associated tremor ataxia syndrome (MIM 300623), Huntington's disease-like 2 (MIM 606438), spinocerebellar ataxia (SCA) type 8 (MIM 608768), SCA type 10 (MIM 603516), SCA 31 (MIM 117210), and most recently amyotrophic lateral sclerosis/frontotemporal degeneration (MIM 105550) caused by expanded hexanucleotide repeats in the C9orf72 gene (MIM 614260).33,34,37–43 The RNA transcripts with expanded repeats form stable hairpin structures, interact and sequester RNA binding proteins, and trigger molecular pathways resulting in neurodegeneration.34 The RNA foci have deleterious effects on the host cells, generally resulting in aberrant splicing and stimulation of apoptosis.42,44
In this current study, we identify discrete nuclear RNA foci containing expanded CUG transcripts in corneal endothelial samples from FECD subjects with CTG18.1 expansion and absent in control samples lacking the triplet expansion. No changes in expression levels of TCF4 were identified between FECD and control endothelial samples using quantitative-PCR. The RNA nuclear foci, a hallmark of toxic RNA, have been previously found only in rare neurodegenerative disorders caused by repeat expansions. Our detection of ribonuclear foci in FECD implicates a role for RNA gain of function in this common disease.
Methods
Subjects
The study was approved by the University of Texas (UT) Southwestern Medical Center Institutional Review Board (IRB) and conducted in adherence to the tenets of the Declaration of Helsinki. All study subjects underwent a complete ophthalmic examination, including slit lamp biomicroscopy, Cell Check XL specular microscopy (Konan Medical, Irvine, CA, USA), and funduscopy by a cornea fellowship-trained ophthalmologist. Subjects with visually significant FECD with a severity grade of 5 or 6 on the modified Krachmer scale (Grade 5 is ≥5 mm central confluent guttae without edema; Grade 6 is ≥5 mm central confluent guttae with edema)45 or other related endothelial disorders undergoing endothelial keratoplasty were enrolled after written informed consent. The diseased central 8 mm endothelium Descemet membrane monolayer removed at the time of endothelial keratoplasty was either immediately fixed in a 4% phosphate-buffered formaldehyde and equilibrated in a 30% sucrose solution for cytoprotection before freezing in Tissue-Tek Optimal Cutting Tissue compound (Sakura, Torrance, CA, USA) for FISH studies or alternatively snap frozen in liquid nitrogen for gene expression studies. Genomic DNA was extracted from leukocytes of peripheral blood samples from each study subject using Autogen Flexigene (Qiagen, Valencia, CA, USA). For controls, we obtained corneal endothelial samples from postmortem donor corneas preserved in Optisol GS corneal storage media (Bausch & Lomb, Rochester, NY, USA) from the eye bank of Transplant Services at UT Southwestern. The donor corneal endothelium had been screened with slit lamp biomicroscopy and Cellchek EB-10 specular microscopy (Konan Medical) by certified eye bank technicians. Endothelium Descemet membrane monolayers from donor corneas were micro-dissected as previously described46 and stored similarly as the FECD endothelial samples. The DNA from the remaining donor corneal tissue layers was extracted with Trizol reagent (Life Technologies, Carlsbad, CA, USA) per the manufacturer's protocol.
CTG18.1 Triplet Repeat Genotyping
The CTG18.1 triplet repeat polymorphism was genotyped using a combination of short-tandem repeat analysis, triplet repeat primed PCR (TP-PCR), and Southern blot analysis.23,24 Short-tandem repeat analysis was performed on DNA samples with PCR by using a forward primer (5′-AATCCAAACCGCCTTCCAAGT-3′) labeled fluorescently with 6-carboxyfluorescein at the 5′ end in combination with reverse primer (5′-CAAAACTTCCGAAAGCCATTTCT-3′). Polymerase chain reaction products were examined with the ABI 3730XL DNA analyzer (Applied Biosystems, Foster City, CA). Triplet repeat primed PCR assay was performed by using P1 (5′-AATCCAAACCGCCTTCCAAGT-3′), a fluorescent primer designed to a region upstream from the CTG18.1 allele. The companion reverse primer P4 (5′-TACGCATCCCAGTTTGAGACGCAGCAGCAGCAGCAG-3′) is composed of five units of the CTG repeat and a 5′ tail to serve as an anchor for a second reverse primer P3 (5′-TACGCATCCCAGTTTGAGACG-3′). The TP-PCR amplicons were analyzed on the ABI 3730XL DNA analyzer to resolve zygosity and detect the presence of an expanded allele based on characteristic electropherogram tracings.24 Southern blot analysis was performed to determine length of large CTG repeat expansions. Genomic DNA extracted from peripheral leukocytes or donor corneal tissue was digested with EcoRI restriction endonuclease. Two microns of digested genomic DNA was run onto a 1.2% agarose gel overnight at low voltage with digoxigenin (DIG)-labeled Molecular Weight Marker VII (Roche, Mannheim, Germany) as a size standard. After downward capillary gel transfer to a positively charged nylon membrane, the membrane was hybridized with 2 μL/mL of 513bp of PCR fragment labeled with DIG in DIG Easy Hyb buffer (Roche). Forward primer (5′-GTTTTGCCAGGAAACGTAGC-3′) and reverse primer (5′-TTCATTAGATGGCCAAGCAG-3′) were used to synthesize the DIG-labeled probes. After hybridization, the membrane was washed twice in 2× SSC, 0.1% SDS at room temperature and then twice in 0.5× SSC, 0.1% SDS at 65°C. Then, the membrane was exposed to X-ray film after applying DIG Luminescent Detection Kit (Roche).
FISH
The investigators performing the FISH were blinded to underlying CTG18.1 genotype of the donor endothelial samples. Optimal cutting tissue solution–preserved FECD and donor endothelial samples were treated with 100 to 150 μL 70% ethanol and incubated overnight at 4°C to permeabilize the cells. The samples were rehydrated for 5 minutes at room temperature with 2× SSC and 50% formamide in PBS. The samples were then hybridized overnight at 37°C with chemically synthesized (CAG)6CA-5′ Texas red-labeled 2-O-methyl RNA 20-mers probe (8 μL at 20 ng/μL) (Integrated DNA Technologies, Coralville, IA, USA) in Vysis LSI/WCP Hybridization Buffer (72 μL) (Abbott, Abbott Park, IL, USA) for a final probe concentration of 2 ng/μL. The samples were washed twice for 30 minutes in 150 μL 2× SSC and 50% formamide (Sigma-Aldrich Corp., St. Louis, MO, USA) at 37°C and once again in 150 μL 1× PBS. The tissue samples were stained with 100 μL 200 nM 4′,6-diamidino-2-phenylindole (DAPI) (Southern Biotech, Birmingham, AL, USA) for 1 hour and washed once in 150 μL 1× PBS. The samples were mounted with Fluoromount G (Southern Biotech) and examined with the Leica DMI 4000B fluorescence microscope (Leica Microsystems, Wetzlar, Germany) equipped with a Hamamatsu Flash 4.0 digital camera (Hamamatsu, Hamamatsu City, Japan).
Nuclease Treatment
Endothelial tissue from an FECD subject with an expansion of 91 CTG repeats was divided into three portions. One was processed for RNA-FISH as described above. The other two portions were treated with either RNase I or DNase I separately for 1 hour in a moist chamber at 37°C. The RNase I treatment consisted of 200 U/mL in 1× RNase I reaction buffer (Promega, Madison, WI, USA). The DNase I treatment consisted of 200 U/mL in 1× DNase I buffer (Roche, Mannheim, Germany). After treatment, cells were washed with PBS and processed for RNA-FISH.
Quantitative Analysis of Foci
Quantitative analysis of foci images was performed by an individual blinded to the tissue source, diagnosis, and CTG18.1 genotype of the samples. Three representative images from each sample were analyzed. Using the MetaMorph Microscopy Automation and Image Analysis Software (Molecular Devices, Sunnyvale, CA, USA), foci from the FISH channel and nuclei from the DAPI channel were each manually counted. The percentage of cells with nuclear foci and the number of foci per nucleus in each image were also calculated. Next, foci were segmented from the background and further processed using Metamorph's Integrated Morphometry Analysis function, which measured the area of each focus.
TCF4 Expression Studies
Total RNA from the cornea endothelium was extracted by RNeasy Micro Kit (Qiagen) with on-column DNase digestion. Total RNA was converted to cDNA using random primers and VILO Superscript III Reverse Transcriptase (Invitrogen Life Technologies, Carlsbad, CA, USA) followed by digestion with RNaseH to free the cDNA strand. Polymerase chain reaction was performed with 12.5 ng RNA equivalent of cDNA, 500 ng each of forward and reverse primer, and 1 U Taq DNA polymerase (5 Prime, Gaithersburg, MD, USA) in a 10-μL reaction volume containing 5 μL diluted cDNA and 5 μL reaction mixture including TCF4 exon-specific primer pairs. Polymerase chain reaction products were analyzed on ethidium bromide–stained 1.5% agarose gel. For the real-time PCR (qPCR) studies, the control corneal endothelial tissue from donor corneas and FECD endothelial specimens were homogenized gently and total RNA was extracted by RNeasy Micro Kit (Qiagen) with on-column DNase digestion. The cDNA was made with cDNA synthesis kit (Life Technologies) by random primers and oligo dT combination and Superscript III reverse transcriptase. The SYBR-Green–based real-time PCR amplification was performed in technical duplicates for each sample and gene on 96-well reaction plates with CFX real-time PCR system (Bio-Rad, Hercules, CA, USA). All the primers were optimized by semiquantitative reverse transcription PCR (RT-PCR) to confirm amplification of a single PCR product of right size. Minus RT control and nontemplate controls containing H2O substituted for template cDNA were run in duplicates on every reaction plate. Reactions were prepared in a total volume of 20 μL containing 8 μL cDNA, 2 μL mixed 10 μM primer (500 nM each; Life Technologies), and 10 μL iTaq Universal SYBR−Green supermix (Bio-Rad). The cycling parameters were as follows: initial denaturation at 95°C for 3 minutes, followed by 39 cycles of denaturation at 95°C for 10 seconds, combined primer annealing/elongation at 60°C for 40 seconds, and final elongation at 95°C for 10 seconds. This cycle was followed by a melting curve analysis, ranging from 55°C to 95°C, with temperature increasing by steps of 0.5°C at every 5 seconds. The comparative Ct method was used for normalizing target gene transcript to glyceraldehyde 3-phosphate dehydrogenase transcript. Fold changes in gene expression were represented by 2−ΔΔCt method and plotted.
Results
First, we examined for the presence of CUG repeat RNA nuclear foci in FECD endothelial samples using FISH. The corneal endothelial tissue examined by FISH included eight FECD endothelial samples from subjects carrying the CTG18.1 expansion, one FECD endothelial sample from a subject without the CTG18.1 expansion, one endothelial sample from a subject with endothelium dysfunction without guttae or the CTG18.1 expansion, and seven control endothelial samples (Table 1). The mean age of the subjects with FECD was 69.8 years and the mean age of the donors was 62.9 years (P = 0.26).
Table 1.
Corneal Endothelial Samples Assessed for RNA Foci by FISH
|
Endothelium ID |
Diagnosis |
CTG18.1 Genotype |
Age |
Sex |
Race |
Death to Preservation, h |
Endothelium Findings by Slit Lamp/Specular Microscopy |
Percentage of Cells With RNA Nuclear Foci, % |
No. of Foci per Nucleus, Mean ± SD |
Maximum Foci Size, μm2 |
| CA025 | FECD | 16, 79 | 72 | F | Caucasian | NA | Grade 6 guttae | 40 | 0.51 ± 0.26 | 0.43 |
| CA041 | FECD | 26, 1300 | 68 | F | Caucasian | NA | Grade 6 guttae | 46 | 0.53 ± 0.09 | 0.47 |
| CA042 | FECD | 17, 91 | 70 | M | Caucasian | NA | Grade 6 guttae | 33 | 0.39 ± 0.32 | 0.64 |
| CA044 | FECD | 17, 91 | 58 | M | Caucasian | NA | Grade 6 guttae | 88 | 1.55 ± 0.10 | 2.59 |
| VVM573 | FECD | 12, 120 | 50 | F | Caucasian | NA | Grade 6 guttae | 38 | 0.45 ± 0.16 | 0.81 |
| CA036 | FECD | 25, 150 | 76 | M | Caucasian | NA | Grade 6 guttae | 82 | 1.29 ± 0.10 | 1.45 |
| CA037 | FECD | 17, 120 | 89 | F | Caucasian | NA | Grade 6 guttae | 59 | 1.26 ± 0.27 | 1.23 |
| CA038 | FECD | 12, 1300 | 74 | M | Caucasian | NA | Grade 6 guttae | 38 | 0.43 ± 0.16 | 0.64 |
| CA035 | FECD | 12, 17 | 71 | M | Caucasian | NA | Grade 5 guttae | No foci detected | NA | NA |
| CA028 | Corneal edema | 12, 12 | 86 | F | Caucasian | NA | No guttae | No foci detected | NA | NA |
| 14-1117 | Control | NA | 71 | F | Caucasian | 13.8 | Normal (ECD = 3247) | No foci detected | NA | NA |
| 14-0852 | Control | 17, 17 | 38 | M | Hispanic | 12.9 | Normal (ECD = 3030) | No foci detected | NA | NA |
| 14-0874 | Control | 12, 14 | 56 | M | Caucasian | 11.9 | Moderate diffuse polymorphism (ECD = 2331) | No foci detected | NA | NA |
| 14-1674 | Control | 17, 28 | 72 | M | Hispanic | 7.9 | NA | No foci detected | NA | NA |
| 14-1658 | Control | 12, 12 | 71 | F | Asian | 1.7 | Normal (ECD = 3115) | No foci detected | NA | NA |
| 14-0832 | Control with CTG18.1 Expansion | 17, 100 | 63 | F | Caucasian | 6.5 | Moderate diffuse polymorphism and polymegathism (ECD = 2146) | 37 | 0.43 ± 0.05 | 0.81 |
| 14-0842 | Control with CTG18.1 expansion | 18, 87 | 69 | F | Caucasian | 8.2 | Severe diffuse polymorphism and polymegathism (ECD = 1597) | 53 | 0.68 ± 0.09 | 0.89 |
Control, control endothelial tissue from donor cornea from eye bank; CTG18.1 Genotype, CTG repeat number; F, female; M: male; Grade 5 guttae, modified FECD grade proposed by Krachmer with >5 mm confluent guttae without edema; Grade 6 guttae, >5 mm confluent guttae with edema; ECD, central endothelial cell density (cells/mm2); Death to Preservation, interval in hours between death of donor and preservation of cornea by eye bank.
Abundant discrete, punctate nuclear RNA foci of variable size were identified in all eight endothelial samples of FECD subjects with the CTG18.1 repeat expansion and absent in all five control endothelial samples without the expansion (P = 7.8 × 10−4 by Fisher's exact test) (Fig. 1; Table 1). The RNA foci were absent in endothelial samples from an FECD subject without the CTG18.1 expansion and a subject with endothelial dysfunction without FECD (Fig. 2). We detected nuclear RNA foci in two control endothelial samples (Figs. 1C, 1D). We subsequently genotyped these samples to discover that they both harbor the CTG18.1 repeat expansion. Specular microscopic findings of the corneal endothelium from these two controls revealed cellular polymorphism and polymegathism (Fig. 1D).
Figure 1.

Nuclear RNA foci accumulate in corneal endothelial cells with CTG18.1 triplet repeat expansion in TCF4. (A) Fluorescence in situ hybridization with a (CAG)6CA-5′ Texas red-labeled 2-O-methyl RNA 20-mers probe (Integrated DNA Technologies) on endothelial keratoplasty samples of FECD subjects with CTG18.1 triplet repeat expansion revealed punctate, nuclear CUG repeat RNA foci (red). These foci were absent in control tissue without the expansion. Affected FECD subjects shown here had CTG18.1 expansion with 1300 CTG repeats or 91 repeats, whereas the control endothelial sample had two short alleles with 12 and 14 CTG repeats at the locus. DNA was stained with DAPI (blue). Scale bar: 25 μm. (B) Preoperative in vivo specular microscopy of the central endothelium of the FECD subjects corresponding to endothelial samples examined by FISH in (A) revealed typical findings for the disorder, including increased cellular polymorphism (with loss of normal hexagonal pattern) and polymegathism (variation in cell size) with focal dark spots corresponding to corneal guttae (arrows) (left) and large dark areas corresponding to confluence of the corneal guttae and marked loss of endothelial cell density and grotesque morphology of remaining cells (right). Scale bar: 100 μm. (C) Fluorescence in situ hybridization on endothelial samples from two control corneas revealed punctate, nuclear RNA foci (red) and subsequent genotyping of the donor corneal tissue revealed expansion of CTG18.1 triplet repeat. The tissue harbored expanded alleles with either 100 CTG repeats or 87 CTG repeats. DNA was stained with DAPI (blue). Scale bar: 25 μm. (D) Postmortem specular microscopy of the central endothelium of corresponding donor corneas with nuclear RNA foci revealed either moderate diffuse polymorphism and polymegathism (left) or severe diffuse polymorphism and polymegathism (right). The presence of foci seen in these endothelial samples with morphological changes typical for the early stages of FECD indicate that the foci are present and may play a critical role early in the disease course. Scale bar: 100 μm. (E) Quantification of the percentage of endothelial cells with foci detected by RNA-FISH as in (A). Gray bars indicate eight endothelial keratoplasty samples from FECD subjects with CTG18.1 expansion and the two white bars indicate the donor cornea endothelial samples with the triplet repeat expansion. Bars represent SE. The number of CTG repeats at this locus is shown for each sample below the corresponding bar.
Figure 2.
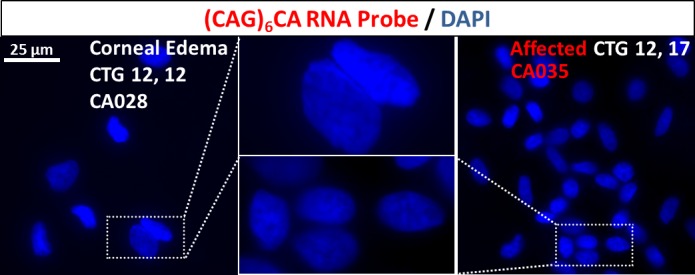
The RNA nuclear foci are specific for CTG18.1 expansion-mediated endothelial dysfunction. The RNA foci were absent in the endothelial sample from a subject with endothelial dysfunction without FECD that had two short alleles of 12 CTG repeats (left). Note the paucicellularity of the endothelium that is characteristic of endothelial specimens with corneal decompensation. The RNA foci were absent in an endothelial sample from an FECD subject without CTG18.1 repeat expansion with 12 and 17 repeats at locus (right).
Next, we quantified the number of nuclear RNA foci in the endothelial samples with the CTG18.1 repeat expansion. The average percentage of cells with nuclear RNA foci in the eight CTG18.1 expansion-positive FECD endothelial samples ranged from 33% to 88% (Fig. 1E; Table 1). The number of RNA foci ranged from zero to five per nucleus in the FECD samples, with a range of 0.39 to 1.55 average foci per nucleus (Table 1). The maximum size of foci observed in the eight FECD endothelial samples ranged from 0.43 to 2.59 μm2 (Table 1). No correlations between CTG18.1 allele length and percentage of cells with nuclear RNA foci were found.
The morphology of the observed punctate nuclear RNA foci in endothelial samples with CTG18.1 repeat expansion was spheroidal. The nuclear RNA foci in FECD endothelial samples were found to be sensitive to degradation when treated with RNase I but not DNase I treatment (Fig. 3).
Figure 3.

Foci are sensitive to degradation with RNase. The nuclear foci were sensitive to degradation with RNase I but not DNase I treatment. Note chromatin digestion with DNase and loss of nuclear borders in middle panel. The DNA was stained with DAPI (blue). These results are consistent with these foci being composed primarily of RNA. Scale bar: 10 μm.
Using reverse transcription PCR (RT-PCR), TCF4 transcripts were detected in adult corneal endothelial samples derived from donor corneas (Fig. 4); TCF4 is a large gene with more than 40 tissue-specific transcripts with a variable number of internal exons that encode for numerous protein isoforms with at least 18 different N-termini,47,48 making expression studies challenging especially given the limited number of cells from FECD endothelial keratoplasty specimens. Therefore, we examined for any changes in expression by real-time PCR (qPCR) of the constitutive exon encoding the bHLH domain present in all TCF4 protein isoforms but found no significant alteration in FECD endothelial samples with CTG18.1 expansion compared with control endothelial samples (Fig. 4C).
Figure 4.
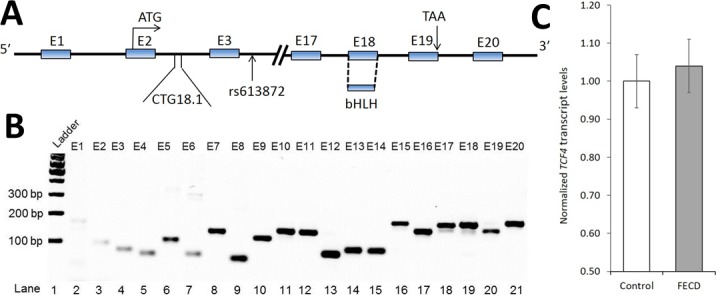
Expression of TCF4. (A) Gene schematic diagram of TCF4 showing 20 exons and relevant elements including intronic CTG18.1 locus and single nucleotide polymorphism rs613872 (NCBI Accession # NM_001083962.1). The CTG18.1 repeat locus reaches the threshold for instability with more than 37 CTG repeats. The gene regulatory basic helix-loop-helix is present in constitutive exon 18. (B) Semiquantitative RT-PCR expression of all exons of TCF4 in control corneal endothelial tissue. Control endothelial tissue was from an 18-year-old donor with two short alleles with 14 and 17 CTG repeats at locus. Similar expression results were seen using control endothelial samples from a 60-year-old donor as well as a 66-year-old donor (data not shown). (C) Bar diagram depicting qPCR analysis of total TCF4 mRNA (control, n = 5; FECD, n = 5). Demographic information of the FECD subjects and control corneas is shown in Table 2. The mean age of the FECD subjects was 63.0 years and the mean age of the donors was 60.6 years (P = 0.70). The qPCR analysis of endothelial samples from patients with FECD compared with control endothelial samples reveals no significant fold change in total TCF4 mRNA level. Bars represent SE. Details of primers are included in Supplementary Table S1.
Table 2.
Corneal Endothelial Samples Assessed for TCF4 Expression by qPCR
|
ID |
Diagnosis |
CTG18.1 Genotype |
Age |
Sex |
Race |
Death to Preservation, h |
| VVM220 | FECD | 28, 91 | 62 | M | Caucasian | NA |
| VVM358 | FECD | 24, 88 | 63 | F | Caucasian | NA |
| VVM434 | FECD | 12, 70 | 69 | M | Caucasian | NA |
| VVM463 | FECD | 12, 120 | 64 | F | Caucasian | NA |
| VVM552 | FECD | 15, 91 | 57 | F | Caucasian | NA |
| 12-1319 | Control | NA | 69 | M | Caucasian | 10.2 |
| 13-0395 | Control | 12, 25 | 70 | M | Caucasian | 14.8 |
| 13-0759 | Control | 12, 12 | 46 | F | Caucasian | 17.7 |
| 13-1212 | Control | 12, 12 | 70 | F | Caucasian | 8.5 |
| 14-0564 | Control | 22, 26 | 48 | F | Caucasian | 7.2 |
Discussion
Expansions of the intronic CTG repeat locus within TCF4 (CTG18.1) contribute significant risk to the development of FECD in Eurasian populations.24,29,31 Association studies of late-onset FECD performed by our group using case (unrelated familial and idiopathic singleton cases)/control comparisons in Caucasian and Singapore Chinese cohorts indicate that one copy of the expanded allele increases the odds of the carrier being affected with FECD by greater than 30-fold.24,29 The mechanisms by which the expanded CTG repeats at the CTG18.1 locus in TCF4 contribute to the degeneration of the corneal endothelium have been hitherto unknown but may include an RNA gain-of-function model, haploinsufficiency of TCF4, or a combination of both mechanisms. Our data suggest that rather than haploinsufficiency of TCF4, toxic RNA is the primary mechanism of disease of FECD with CTG18.1 triplet repeat expansion mediated by CUG repeat RNA foci.
Using FISH, we identify discrete nuclear RNA foci containing expanded CUG transcripts in all corneal endothelial samples from FECD subjects with CTG18.1 expansion but not in control subjects lacking the expansion. The RNA foci also were absent in endothelial samples from an FECD subject without CTG18.1 expansion and a subject with endothelial dysfunction without FECD. This evidence supports that the foci are not nonspecific markers of endothelial dysfunction but rather a pathognomonic finding of CTG18.1 expansion-mediated endothelial disease.
The presence of ribonuclear foci in two control endothelial samples with CTG18.1 expansion and moderate morphological changes (Figs. 1C, 1D) suggests that the foci also may be present early in the FECD disease course in asymptomatic individuals with expanded repeats and subclinical disease.
The morphology of the observed punctate nuclear RNA foci in endothelial samples with CTG18.1 repeat expansion was spheroidal and similar to the CUG foci in DM1 rather than the rod-shaped foci in DM2 or the large, patchy foci seen in CGG or CAG repeat expansion expressing cells.34 Additionally, the foci in FECD were sensitive to degradation when treated with RNase, which is consistent with these foci being composed primarily of RNA (Fig. 3).
A higher percentage of cells with CUG RNA nuclear foci and number of foci per nucleus have been reported in DM1 muscle biopsy specimens with longer CTG mutations in the 3′ untranslated region of the DMPK gene (MIM 60537).49 Our FECD endothelial sample cohort was inadequately powered to see such a trend with CTG18.1 allele length but larger studies are certainly warranted to explore any relationships between quantitative measures of the nuclear RNA foci and CTG18.1 mutation length, as well as phenotypic characteristics of the FECD subjects.
We report the detection of foci in 33% to 88% of endothelial cells from FECD subjects with the expansion. In a study by Borderie et al.,8 2.65% of endothelial cells of keratoplasty specimens from FECD patients had evidence of apoptosis compared with 0.23% in the control group, which is compatible with the slow progressive decline in endothelial cell counts over decades in these patients. Mutant expanded CUG RNA foci may be necessary and sufficient to cause FECD via sequestration of critical RNA binding proteins. Sequestration of RNA binding proteins over a long period of time may trigger apoptosis as in SCA 10 and/or result in splicing misregulation of downstream effector genes as thought to be the primary disease mechanism in DM1 and DM2.42,44
Our qPCR expression data show no significant alteration of TCF4 expression levels in FECD endothelial samples of subjects with intronic CTG18.1 triplet repeat expansion. Other intronic repeat expansion disorders may give some insight to the significance of our findings. Friedreich's ataxia (MIM 229300) is an autosomal recessive disorder caused by large GAA repeat expansions in the first intron in FXN (MIM 606829) that markedly hinder the transcription of that gene.42 Although there is alteration of ZNF9 (MIM 116955) expression in DM2 caused by intronic CCTG tetranucleotide repeat expansions, the primary mechanism of disease is thought to be splicing dysregulation as a consequence of toxic CCUG RNA transcripts aggregating as nuclear foci and sequestration of the RNA binding protein MBNL1.44 In the autosomal dominant disorder SCA 10 caused by intronic ATTCT repeat expansions in ATXN10 (MIM 611150), expression levels of ATXN10 are unaltered.42 Toxic RNA is the primary mechanism of disease in SCA 10 in which expanded intronic AUUCU repeats aggregate as nuclear foci to trigger apoptosis.42 Based on our qPCR results, the intronic triplet repeat expansion in TCF4 does not appear to affect expression levels of the gene, but further studies are warranted. Haploinsufficiency of TCF4 is also unlikely to underlie the pathogenesis of CTG18.1 expansion-mediated FECD based on the discovery of the genetic basis of Pitt-Hopkins syndrome (MIM 610954). This disorder, consisting of episodic hyperventilation and apnea, mental and motor retardation, hypotonia, and seizures, results primarily from de novo heterozygous splice-site, frameshift, nonsense, or missense mutations in TCF4 or large deletions involving the entire or part of the gene, resulting in haploinsufficiency.48
In the current article, we report the presence of abundant nuclear RNA foci in FECD with CTG18.1 triplet repeat expansion in TCF4. We propose an RNA gain-of-function model in which mutant expanded CUG transcripts are stabilized through their interaction with RNA binding proteins to form nuclear inclusions triggering corneal endothelium-specific aberrant splicing and/or apoptosis. Nuclear RNA foci have previously been reported as the hallmark of toxic RNA only in rare neurodegenerative disorders caused by repeat expansions. The presence of ribonuclear foci in FECD suggests a role for toxic RNA in this common disease.
While this manuscript was under review, Du et al.50 also reported the presence of RNA foci in the endothelial tissue of FECD subjects. They showed some evidence of the splicing regulator muscleblind-like (MBNL1) colocalizing in the foci and differential splicing of genes known to be sensitive to MBNL1 sequestration.50 Results from both independent studies highlight the central role that CUG RNA nuclear foci play in the pathogenesis of FECD with CTG18.1 triplet repeat expansion in TCF4.
Acknowledgments
The authors thank the patients for their participation in this study. R. Wayne Bowman, MD, C. Bradley Bowman, MD, and Walter Beebe, MD, for their assistance in the recruitment of patients. The authors acknowledge the study coordinator efforts of Emily Linsenbardt and Mike Molai. We thank Charles A. Thornton, MD, for the generous gift of the RNA probe. We thank Helen H. Hobbs, MD, for helpful discussions on project and manuscript.
Supported by Grants R01EY022161 (VVM) and P30EY020799 from the National Eye Institute, National Institutes of Health, Bethesda, Maryland, United States, and an unrestricted grant from Research to Prevent Blindness, New York, New York, United States. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure: V.V. Mootha, None; I. Hussain, None; K. Cunnusamy, None; E. Graham, None; X. Gong, None; S. Neelam, None; C. Xing, None; R. Kittler, None; W.M. Petroll, None
References
- 1. Lorenzetti DW, Uotila MH, Parikh N, Kaufman HE. Central cornea guttata. Incidence in the general population. Am J Ophthalmol. 1967; 64: 1155–1158. [PubMed] [Google Scholar]
- 2. Eye Bank Association of America. 2013 Eye Banking Statistical Report. Washington: Eye Bank Association of America; 2013. Available at: http://www.restoresight.org/wpcontent/uploads/2014/04/2013_Statistical_Report-FINAL.pdf. Accessed December 8, 2014. [Google Scholar]
- 3. Zoega GM, Fujisawa A, Sasaki H, et al. Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study. Ophthalmology. 2006; 113: 565–569. [DOI] [PubMed] [Google Scholar]
- 4. Kitagawa K, Kojima M, Sasaki H, et al. Prevalence of primary cornea guttata and morphology of corneal endothelium in aging Japanese and Singaporean subjects. Ophthalmic Res. 2002; 34: 135–138. [DOI] [PubMed] [Google Scholar]
- 5. Chi HH, Teng CC, Katzin HM. Histopathology of primary endothelial-epithelial dystrophy of the cornea. Am J Ophthalmol. 1958; 45: 518–535. [DOI] [PubMed] [Google Scholar]
- 6. Laing RA, Leibowitz HM, Oak SS, Chang R, Berrospi AR, Theodore J. Endothelial mosaic in Fuchs' dystrophy. A qualitative evaluation with the specular microscope. Arch Ophthalmol. 1981; 99: 80–83. [DOI] [PubMed] [Google Scholar]
- 7. Bigar F. Specular microscopy of the corneal endothelium. Optical solutions and clinical results. Dev Ophthalmol. 1982; 6: 1–94. [PubMed] [Google Scholar]
- 8. Borderie VM, Baudrimont M, Vallee A, Ereau TL, Gray F, Laroche L. Corneal endothelial cell apoptosis in patients with Fuchs' dystrophy. Invest Ophthalmol Vis Sci. 2000; 41: 2501–2505. [PubMed] [Google Scholar]
- 9. Li QJ, Ashraf MF, Shen DF, et al. The role of apoptosis in the pathogenesis of Fuchs endothelial dystrophy of the cornea. Arch Ophthalmol. 2001; 119: 1597–1604. [DOI] [PubMed] [Google Scholar]
- 10. Matthaei M, Zhu AY, Kallay L, Eberhart CG, Cursiefen C, Jun AS. Transcript profile of cellular senescence-related genes in Fuchs endothelial corneal dystrophy. Exp Eye Res. 2014; 129: 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hogan MJ, Wood I, Fine M. Fuchs' endothelial dystrophy of the cornea. 29th Sanford Gifford Memorial lecture. Am J Ophthalmol. 1974; 78: 363–383. [DOI] [PubMed] [Google Scholar]
- 12. Magovern M, Beauchamp GR, McTigue JW, Fine BS, Baumiller RC. Inheritance of Fuchs' combined dystrophy. Ophthalmology. 1979; 86: 1897–1923. [DOI] [PubMed] [Google Scholar]
- 13. Sundin OH, Broman KW, Chang HH, Vito EC, Stark WJ, Gottsch JD. A common locus for late-onset Fuchs corneal dystrophy maps to 18q21.2-q21.32. Invest Ophthalmol Vis Sci. 2006; 47: 3919–3926. [DOI] [PubMed] [Google Scholar]
- 14. Riazuddin SA, Vasanth S, Katsanis N, Gottsch JD. Mutations in AGBL1 cause dominant late-onset Fuchs corneal dystrophy and alter protein-protein interaction with TCF4. Am J Hum Genet. 2013; 93: 758–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Biswas S, Munier FL, Yardley J, et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2001; 10: 2415–2423. [DOI] [PubMed] [Google Scholar]
- 16. Zhang C, Bell WR, Sundin OH, et al. Immunohistochemistry and electron microscopy of early-onset Fuchs corneal dystrophy in three cases with the same L450W COL8A2 mutation. Trans Am Ophthalmol Soc. 2006; 104: 85–97. [PMC free article] [PubMed] [Google Scholar]
- 17. Vithana EN, Morgan P, Sundaresan P, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet. 2006; 38: 755–757. [DOI] [PubMed] [Google Scholar]
- 18. Vithana EN, Morgan PE, Ramprasad V, et al. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet. 2008; 17: 656–666. [DOI] [PubMed] [Google Scholar]
- 19. Riazuddin SA, Zaghloul NA, Al-Saif A, et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am J Hum Genet. 2010; 86: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krafchak CM, Pawar H, Moroi SE, et al. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet. 2005; 77: 694–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mehta JS, Vithana EN, Tan DT, et al. Analysis of the posterior polymorphous corneal dystrophy 3 gene, TCF8, in late-onset Fuchs endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2008; 49: 184–188. [DOI] [PubMed] [Google Scholar]
- 22. Riazuddin SA, Parker DS, McGlumphy EJ, et al. Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs corneal dystrophy. Am J Hum Genet. 2012; 90: 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wieben ED, Aleff RA, Tosakulwong N, et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One. 2012; 7: e49083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mootha VV, Gong X, Ku HC, Xing C. Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs' endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2014; 55: 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baratz KH, Tosakulwong N, Ryu E, et al. E2-2 protein and Fuchs's corneal dystrophy. N Engl J Med. 2010; 363: 1016–1024. [DOI] [PubMed] [Google Scholar]
- 26. Murre C, Bain G, van Dijk MA, et al. Structure and function of helix-loop-helix proteins. Biochim Biophys Acta. 1994; 1218: 129–135. [DOI] [PubMed] [Google Scholar]
- 27. Ephrussi A, Church GM, Tonegawa S, Gilbert W. B lineage–specific interactions of an immunoglobulin enhancer with cellular factors in vivo. Science. 1985; 227: 134–140. [DOI] [PubMed] [Google Scholar]
- 28. Breschel TS, McInnis MG, Margolis RL, et al. A novel, heritable, expanding CTG repeat in an intron of the SEF2-1 gene on chromosome 18q21.1. Hum Mol Genet. 1997; 6: 1855–1863. [DOI] [PubMed] [Google Scholar]
- 29. Xing C, Gong X, Hussain I, et al. Transethnic replication of association of CTG18.1 Repeat expansion of TCF4 gene with Fuchs' corneal dystrophy in Chinese implies common causal variant. Invest Ophthalmol Vis Sci. 2014; 55: 7073–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nanda GG, Padhy B, Samal S, Das S, Alone DP. Genetic association of TCF4 intronic polymorphisms, CTG18.1 and rs17089887 with Fuchs' endothelial corneal dystrophy in Indian population. Invest Ophthalmol Vis Sci. 2014; 55: 7674–7680. [DOI] [PubMed] [Google Scholar]
- 31. Wieben ED, Aleff RA, Eckloff BW, et al. Comprehensive assessment of genetic variants within TCF4 in Fuchs' endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2014; 55: 6101–6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mankodi A, Logigian E, Callahan L, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000; 289: 1769–1773. [DOI] [PubMed] [Google Scholar]
- 33. Li LB, Bonini NM. Roles of trinucleotide-repeat RNA in neurological disease and degeneration. Trends Neurosci. 2010; 33: 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wojciechowska M, Krzyzosiak WJ. Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet. 2011; 20: 3811–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol. 1995; 128: 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miller JW, Urbinati CR, Teng-Umnuay P, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000; 19: 4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lagier-Tourenne C, Baughn M, Rigo F, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013; 110: E4530–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sellier C, Rau F, Liu Y, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010; 29: 1248–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rudnicki DD, Holmes SE, Lin MW, Thornton CA, Ross CA, Margolis RL. Huntington's disease–like 2 is associated with CUG repeat-containing RNA foci. Ann Neurol. 2007; 61: 272–282. [DOI] [PubMed] [Google Scholar]
- 40. Daughters RS, Tuttle DL, Gao W, et al. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009; 5: e1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sato N, Amino T, Kobayashi K, et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009; 85: 544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. White MC, Gao R, Xu W, et al. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 2010; 6: e1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Margolis JM, Schoser BG, Moseley ML, Day JW, Ranum LP. DM2 intronic expansions: evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum Mol Genet. 2006; 15: 1808–1815. [DOI] [PubMed] [Google Scholar]
- 44. Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007; 117: 3952–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Krachmer JH, Purcell JJ Jr, Young CW, Bucher KD. Corneal endothelial dystrophy. A study of 64 families. Arch Ophthalmol. 1978; 96: 2036–2039. [DOI] [PubMed] [Google Scholar]
- 46. Kruse FE, Laaser K, Cursiefen C, et al. A stepwise approach to donor preparation and insertion increases safety and outcome of Descemet membrane endothelial keratoplasty. Cornea. 2011; 30: 580–587. [DOI] [PubMed] [Google Scholar]
- 47. Sepp M, Kannike K, Eesmaa A, Urb M, Timmusk T. Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5′ exon usage and splicing. PLoS One. 2011; 6: e22138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sepp M, Pruunsild P, Timmusk T. Pitt-Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum Mol Genet. 2012; 21: 2873–2888. [DOI] [PubMed] [Google Scholar]
- 49. Botta A, Rinaldi F, Catalli C, et al. The CTG repeat expansion size correlates with the splicing defects observed in muscles from myotonic dystrophy type 1 patients. J Med Genet. 2008; 45: 639–646. [DOI] [PubMed] [Google Scholar]
- 50. Du J, Aleff RA, Soragni E, et al. RNA toxicity and missplicing in the common eye disease fuchs endothelial corneal dystrophy. J Biol Chem. 2015; 290: 5979–5990. [DOI] [PMC free article] [PubMed] [Google Scholar]