

Abstract
Increasing evidence has shown that chemokine receptors may form functional dimers with unique pharmacological profiles. A common practice to characterize such G protein-coupled receptor dimerization processes is to apply bivalent ligands as chemical probes which can interact with both receptors simultaneously. Currently, two chemokine receptor dimers have been studied by applying bivalent compounds: the CXCR4-CXCR4 homodimer and the CCR5-MOR heterodimer. These bivalent compounds have revealed how dimerization influences receptor function and may lead to novel therapeutics. Future design of bivalent ligands for chemokine receptor dimers may be aided with the recently available CXCR4 homodimer, and CCR5 monomer crystal structures by more accurately simulating chemokine receptors and their dimers.
Keywords: bivalent ligand, CCR5, chemokine receptor, CXCR4, dimerization, GPCR, MOR
1. Introduction
All chemokine receptors are G protein-coupled receptors (GPCR), which have seven transmembrane helixes (TM) and couple to heterotrimeric G proteins. The GPCR superfamily of proteins has approximately 791 genes encoding for the six different receptor subtypes [1]. Currently, all 19 known chemokine receptors belong to the class A (rhodopsin-like) family of GPCRs and are classified into four main subfamilies based upon which chemokines they bind: CC, CXC, XC, and CX3C receptors [2–4]. Many of the chemokine receptors are promiscuous and bind to several chemokines within their family and allow for tailored chemokine response and redundancy [2,4].
Originally, it was postulated that GPCRs functioned in a monomeric fashion and that there was a general stoichiometry of 1:1 for the receptor ligand interaction [5,6]. However, increasing evidence has begun to support the possibility that they may act in dimeric, or even oligomeric assemblies [7–9]. One of the first observations of dimerization in rhodopsin-like GPCRs was seen in β-adrenergic receptors; it was seen that binding of one ligand decreased the binding of a second one [10]. This type of “cross-talk,” or better known as negative cooperativity, occurs when a dimer bound ligand either inhibits the binding, or signaling of a second ligand to the dimer pair [8,9].
One of the earliest methods for elucidating dimer pairs was to use co-immunoprecipitation (co-IP) techniques. First used for the β2-adrenergic receptor, two populations of the receptor were engineered to either have an influenza hemaglutinin (HA) or a myc-epitope tag incorporated into the receptor [11]. These two receptor subtypes were then co-expressed, and using an anti-myc-epitope antibody, immunoprecipitation was performed. If only monomers were present, then only the myc-epitope tagged β2-adrenergic receptor should show up on a Western blot analysis due to the selectivity of the anti-myc-epitope antibody. However, it was found that the HA tagged β2-adrenergic receptor was present (by co-staining with an anti-HA antibody) along with the myc-epitope tagged β2-adrenergic receptor [10]. Therefore, the two receptor populations had to be directly interacting with each other to both be isolated using co-immunoprecipitation, and thus showed that the β2-adrenergic receptor was able to homodimerize [10]. This technique has subsequently been used as a preliminary technique to study the homo and heterodimerization of numerous GPCRs [7–9].
Another important technique for GPCR dimerization/oligomerization detection is Fröster Resonance Energy Transfer (FRET). Both bioluminescence and fluorescence (BRET and FRET respectively) have been used in this technique. For FRET detection, the two receptors suspected of dimerizing are tagged with two different fluorescent proteins: i.e., a cyan fluorescent protein (CFP) and a yellow fluorescent protein (YFP). It is essential for the FRET that the excited state of one fluorescent protein (donor chromophore) can transfer energy at a very specific distance to an acceptor chromophore and permit it to emit its unique excitation wavelength. In order for FRET to take place, the two chromophores (and associated proteins) must be in close proximity (10 to 100 Å) [11]. Therefore, excitation of the CFP at ~436 nm would give only one emission wavelength at ~480 nm if no dimerization was present since the two GPCRs are not in close proximity. If the receptors dimerized, exciting the CFP would yield both the emission wavelength at ~480 nm (for CFP) and an additional emission wavelength at ~535 nm, which would correspond to the excitation/emission from the YFP on the other GPCR in close proximity. This technique can also be coupled with a bioluminescent luciferase enzyme instead of the CFP to excite the YFP through BRET [12]. The combination of co-immunoprecipitation and FRET/BRET has led to a network of GPCR homodimers and heterodimers being discovered [7].
In addition to the biochemical techniques, direct observation of GPCR dimers and oligomers has been obtained using both GPCR crystallization and atomic-force microscopy techniques [13–15]. Using atomic-force microscopy, oligomer formations of rhodopsin were able to be observed, giving the first direct visualization of GPCR oligomization [15]. Additionally, both chemokine receptor CXCR4 (CXCR4) and the mu opioid receptor (MOR) were observed to form dimers within their crystal lattice (Fig. 1a and 1b). While these observed dimers might partially be due to an artifact of the crystallization process, it did lend credence to GPCR dimerization [13,14].
Fig. (1).

Observed GPCR dimer construct from crystal structures. a) Crystal structure of the CXCR4 dimer with a TM5/TM6 interface (PDB code 3ODU) [14]. b) Crystal structure of the MOR dimer with a TM5/TM6 interface (PDB code 4DKL) [13].
Several types of interactions between dimerized GPCRs have been proposed, and two main dimerization models have subsequently been described: a contact dimer model, and a domain-swapped dimer [9]. The domain-swapped model proposes that TM6 and TM7 are exchanged between monomers to from a dimer [16]. The contact dimer model proposes that dimerization occurs through direct contact between the different interfaces of helixes of GPCRs: TM5/TM6, TM3/TM4, and TM1/TM2 interfaces have been postulated and observed [13,14,17,18]. Both hypotheses have been supported by mutation and computational studies, but due to observations of GPCR crystal structure, the contact dimer may represent a more realistic model.
An important aspect of GPCR dimerization is its effect on receptor function and signaling. As alluded to earlier, a possible outcome of dimerization is positive and negative cooperativity (Fig. 2) [7,8]. Positive cooperativity occurs when binding of a ligand to one receptor leads to partial, full, or enhanced activation of the second receptor [7,8]. It may also occur when two ligands bind both receptors, and an enhanced action is seen. Negative cooperativity can occur when one bound ligand leads to either inhibition of a second ligand binding to the dimer, or inhibition of signaling from a second bound ligand [7,8].
Fig. (2).

Positive and negative cooperativity in GPCR dimerization. a) Agonist A binding to one GPCR (in green) results in partial activation of another GPCR (in blue). b) When two agonists, A and B, bind to the GPCRs there will be enhanced activation, synergism. c) In negative cooperativity binding of A to one GPCR (in green) leads to inhibition of the binding of B to another GPCR (in blue), leading to suppression of ligand B related signaling. d) Binding of A leads to inhibition of signaling from the GPCR (in blue) even with B bound to it.
Functionally, heterodimers may allow for different mechanisms of signal regulation for GPCRs [19]. For example, within the CCR2-CCR5 heterodimer, dimerization led to the receptors being able to couple with Gαq/11, which, as individual receptors, they normally do not couple with [20]. A similar effect was seen for the MOR-delta opioid receptor (DOR) heterodimer; when the receptors were expressed alone, pertussis toxin inhibited agonist stimulated Gα-dependent signaling from both receptors, but when expressed together, pertussis toxin did not inhibit their Gα-dependent signaling [21]. These results suggested that the heterodimer could couple to different G proteins than the monomers by themselves. Dimerization of GPCRs may also affect receptor desensitization and internalization [7,8,21]. There are several comprehensive reviews on chemokine receptor homodimerization and heterodimerization, which reveal the extent of their dimerization and the functional consequences [20,22–29]. Similarly, chemokine receptor dimers led to unique pharmacological profiles which can add upon the already intricate receptor-ligand interactions. Targeting these chemokine dimer interactions may lead to unique therapies with marked potential. Currently, the most direct way to monitor these interactions is to target them with chemical probes such as bivalent compounds [5]. A bivalent compound is defined as a compound that contains two distinct pharmacophores which can interact simultaneously with two receptors at once [6].
2. Chemokine Receptor Bivalent Ligands
Bivalents compounds are indispensable for studying the relationship between GPCRs in both homodimers and heterodimers pairs [5]. By targeting dimers of GPCRs, new pharmacological profiles are obtainable because of their unique properties [30]. Using bivalent ligands may lead to ligands that have higher affinity, higher selectivity, and improved or altered physiological responses. The possible synergistic effects are due to the cooperativity between the receptors and an overall drop in the entropy of interaction by targeting two receptors at once [6]. As such, it is imperative and advantageous to target homodimers or heterodimers of chemokine receptors with bivalent compounds [20,23,25,29,31].
Generally, bivalent compounds can either be classified as homo-bivalent or hetero-bivalent, that is, they either have two of the same pharmacophores, or two different ones. These two pharmacophores are attached to each other with a linker that will not interfere with receptor binding and is the appropriate length to allow the two pharmacophores to interact with both receptors. The average distance between GPCR dimers is thought to be between 27 Å and 32 Å [5]; therefore, the linker length should ideally be in that range. Several different linker types have been reported, and range from aliphatic chains to polyethers [30]. The pharmacophores of choice usually have high affinity and selectivity for the targeted receptor(s) dimer and can tolerate added substitutions onto their structure to facilitate the addition of the linker. Currently, two chemokine receptor dimers have been targeted with bivalent compounds: the CXCR4-CXCR4 homodimer and the CCR5-MOR heterodimer. However, more examples are expected in the future with the expanding field of GPCR dimerization and the examples below are very typical within bivalent ligand studies.
2.1. Bivalent Ligands Targeting the CXCR4-CXCR4 Homodimer
Chemokine receptor CXCR4 is one of the two main co-receptors for the human immunodeficiency virus type 1 (HIV-1) for viral invasion into human cells [32–35]. Homodimerization of CXCR4 has been shown to contribute to “warts, hypogammaglobulinemia, infections and myelokathexis” (WHIM) syndrome and may be involved in other physiological functions [36,37]. Bivalent ligands have been specifically designed to interact with the CXCR4 homodimer in order to study its biological significance [5,30].
The first report of a chemokine receptor CXCR4 bivalent ligand was by Tanaka et al. in 2010 [38]. Using an analogue of the cyclic peptide CXCR4 antagonist FC131 [cyclo(-D-Tyr1-Arg2-Arg3-Nal4-Gly5-)] as the principle pharmacophore, different length of poly(L-proline) linkers were used to estimate the distances between binding pockets of CXCR4 homodimers. The rigid poly(L-proline) linkers are known to maintain a predetermined, constant distance between two ligands and therefore, could act as a molecular ruler. Fig. 3 shows the two bivalent ligand scaffolds with either a poly(L-proline) linker (1-8) or a polyethylene glycolated (PEG) poly(L-proline) linker (9-14) and the two monovalent controls (15, 16). The first difficulty in designing these bivalent ligands was finding high affinity ligands with linker attachment site(s) that did not affect their affinity. The CXCR4 antagonist FC131 was chosen for the bivalent compound due to its high binding affinity (Ki = 31.5 nM) [38–40] and an accessible attachment site. Previous structure-activity relationship (SAR) studies of FC131 have shown that the carbonyl oxygen of Gly5, but not the side chain’s hydrogen atoms, plays a role in binding [41–44]. Therefore, different amino acids can take the place of Gly5 without drastically altering the binding affinity. Replacing Gly5 with a D-Cys only had a 2-fold decrease in its binding affinity to CXCR4 and maintained a nanomolar Ki value of 53.4 nM [38].
Fig. (3).

Bivalent ligands for the CXCR4 homodimer with various linker lengths [38].
Once the pharmacophore and the linker attachment site were chosen the linker was then designed based upon proline oligomers. Proline oligomers can adopt a constant helical structure that maintains a length of 0.9 nm per turn by which three prolines may give a rise of 3.1 Å per residue [38,45]. This structure allowed for two pharmacophores to be separated by specific distances. Previous bivalent ligand studies have shown that the length of the linker between pharmacophores was essential for activity [5,30,46–49]. The poly(L-proline) linker in compounds 1-8 and 9-14 varied in length from 2 to 8 nm. Since the poly(L-proline) linker is rigid, more flexible alkyl chains (1-8) or PEG groups (9-14) attached the linker to the FC131 units making the bivalent ligand less rigid and allowing them to adopt more confirmations.
Fig. 4 shows the effects that linker length had on binding affinity of the two sets of compounds. For both series the optimum linker length was approximately 6 nm with compounds 6 and 12 having a Ki value of 9.9 nM and 13.9 nM respectively [38]. Increasing or decreasing the linker length from 6 nm led to loss of binding affinity, resulting in a maximum 6-fold lose in activity. The 2 to 3-fold increase in binding affinity for the bivalent compounds compared to FC131 may due to the synergistic effect of binding two receptors at once [5,30]. Additionally, the two control compounds, 15 and 16, had Ki values of 294 nM and 72 nM, which was less than FC131, and thus, showed that the synergistic effects were not due to the poly(L-proline) linkers.
Fig. (4).
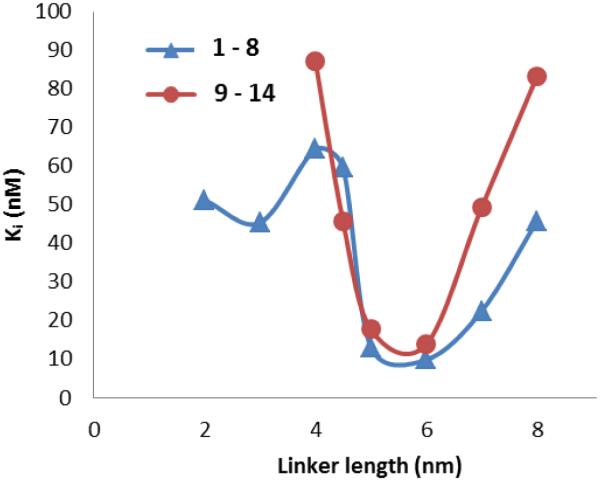
Relationship of CXCR4 binding affinity (Ki) versus the linker length of compounds 1 through 14 [38].
Compounds 1 through 14 revealed several important aspects of designing chemokine receptor dimer bivalent compounds. First, linker composition is pivotal; when comparing the two series of compounds, there was a general trend that the non-PEGylated linker (1-8) was up to two times more potent than the PEGylated linker (9-14). One explanation of the loss of potency was that the PEGylated linker added more rotatable bonds making the linker less rigid, which had been shown to decrease binding affinity [30,50]. Second, linker length is essential for the binding affinity of CXCR4 bivalent ligands. As shown in figure 4, there was a global minimum for both sets of compounds at a length of 6 nm. Overall, this study demonstrated the importance of linker length and composition in designing chemokine receptor bivalent ligands.
While linker length and composition are important in bivalent ligand development, Choi et al. showed that synthetic linkers may not be needed at all for chemokine receptor bivalent ligands [37]. Utilizing a small, all D-amino acid peptide (Fig. 5, DV1, 17) based upon the N-terminus of viral macrophage protein II (vMIP-II) they were able to synthesize a bivalent ligand which lacked the classical linker seen in traditional bivalent compounds. The DV1 dimer, 18, consists of two DV1 peptides linked together through a disulfide bond between cysteines already present in the peptides. Competition binding assays utilizing a CXCR4 specific antibody indicated that bivalent peptide (18) had 14 times greater affinity for CXCR4 than the monomer (17), with IC50 values of 3 nM and 43 nM respectively [37]. A similar trend was seen in antiviral activity against the HIV-1 IIIB strain; compound 18 had an IC50 value of 4.4 µM, whereas 17 had an IC50 value of 12.1 µM [37]. The synergistic increase in binding affinity and functional activity may be from the bivalent peptide by simultaneously interacting with both binding pockets within a CXCR4 homodimer. Guided by site directed mutagenesis, molecular modeling verified that 18 could interact with both binding pockets of a CXCR4 homodimer [37].
Fig. (5).
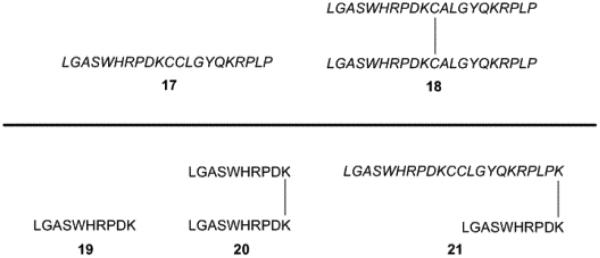
Peptide based bivalent ligands for the CXCR4 homodimer [37,51].
Further studies utilizing peptides for the basis of bivalent CXCR4 homodimer ligands led to simplifying the DV1 (17) peptide into a shortened version named DV3 (19) [51]. Using competition binding assays utilizing a CXCR4 specific antibody, DV1 (17) and DV3 (19) showed IC50 values of 236 nM and 440 nM respectively [51]. The dimer version of the DV3 peptide (20) had an IC50 value of 133 nM, which was 3 times higher than the monomer 19, and was consistent with the increase in binding affinity seen for bivalent compounds [51]. The binding data for the DV3 peptide dimer (20) indicated that the residues after the cysteine reside of dimeric DV1 (18) were essential for CXCR4 binding. An additional bivalent ligand consisting of DV1 and DV3 linked through C-terminus lysine residues, DV1-K-DV3 (21), had an IC50 of 4 nM, which was 33 times higher than the DV3 dimer (20) [51]. These results suggested that 21 was more capable of interacting with both binding pockets within the CXCR4 homodimer. Reasonable explanations for the increase in interaction could be the increase in length of the bivalent compound or a different tertiary structure that was only adopted in 21.
Both examples of peptide-based bivalent ligands for CXCR4 homodimers revealed that bivalent compounds can be synthesized without the traditional linker connecting two pharmacophores. Compounds 18, 20, and 21 illustrate a logical progression which suggested that optimization of chemokine receptor bivalent ligands may rely heavily on pharmacophore choice and attachment site.
2.2 Bivalent Ligands Targeting the CCR5-MOR Heterodimer
Due to modern antiretroviral therapies, HIV-1 infected patients have longer lifespans and a better quality of life [52]. However, several neurological complications are now being seen due to HIV-1 associated injury of neurons by infected microglia and astrocytes (neuroAIDS) [53–55]. Furthermore, these effects are further exacerbated with opiate use and abuse [54,56–59]. A possible mechanism for the potentiation effects of opiates is the interaction of the mu opioid receptor (MOR) with chemokine receptor CCR5 (CCR5), a known HIV-1 co-receptor in the CNS [35,54,60–64]. The progression of neuroAIDS has been linked to opiate abuse that may arise from the synergistic interactions between CCR5 and MOR [54,57–59,65,66]. A key example of this was that MOR agonists can up-regulate the expression of CCR5 and promote HIV-1 infection, which can be blocked by MOR antagonists [67]. Opiates can also exacerbate the amount of indirect neuronal injury in neurons and glia through HIV-1 induced CNS inflammation.[68,69]. The specific opioid dependent neuronal injury may be primarily induced by MOR expressing glia in the CNS [70].
Importantly, MOR and CCR5 have been shown to heterodimerize with each other and undergo crosstalk [71,72]. The interaction has been shown to affect immune cell function and may produce the synergistic effects seen in neuroAIDS progression.[67,73]. In order to explore the pharmacological profile of the CCR5-MOR heterodimerization and its relation with neuroAIDS, Yuan et al. designed a bivalent ligand (22) containing both MOR and CCR5 antagonist pharmacophores [49].
The premise of the bivalent compound 22 was to use both a MOR and a CCR5 antagonist to try to inhibit both receptors at the same time (Fig. 7). Naltrexone (28) and maraviroc (29) were chosen for their high binding affinities and well known pharmacological profiles [52,74]. However, both molecules had to be functionalized with an amine group in order to allow for attachment of the linker. 6β-Naltrexamine has been synthesized before, but 4-aminophenyl-maraviroc had never been reported, so a new synthetic route was devised [49]. The linker connecting the two pharmacophores was chosen based on the work of Daniels et al. with MOR-DOR bivalent compounds [48]. They found that a 21-atom spacer made of an aliphatic diamine flanked by two diglycolic groups was optimal for opioid receptor heterodimers [48].
Fig. (7).

Bivalent compound strategy for targeting the CCR5-MOR heterodimer [49].
In order to test if the bivalent ligand still recognized both receptors, binding assays were first conducted. Table 1 shows the results of both CCR5 and MOR radiobinding assays for selected compounds. Within the MOR binding assay, all of the compounds showed higher Ki values than naltrexone (28) [75]. The bivalent compound 22 had a 70-fold loss in binding affinity to the MOR, whereas the monovalent compound 24 had a 13-fold loss in affinity. The data for the CCR5 binding assay indicated that any substitution on maraviroc’s phenyl ring is detrimental; there was a clear trend of decreasing affinity with increasing size of the group at the 4-position [75]. Overall, there was about a 1000-fold loss of affinity seen for 22 compared to maraviroc [75]. However, it did still bind CCR5 at a nanomolar level, meaning that its affinity wasn’t completely abolished.
Table 1.
CCR5 and MOR radioligand binding assay results.
| Compound | MOR Ki (nM)a | CCR5 Ki (nM)b |
|---|---|---|
| 28 | 0.7 ± 0.1 | - |
| 29 | - | 0.24 ± 0.06 |
| 22 | 51.8 ± 7.9 | 239 ± 56 |
| 23 | - | 151 ± 44 |
| 24 | 9.2 ± 3.4 | - |
| 27 | - | 15.3 ± 4.8 |
[3H]naloxone was used in hMOR-CHO membranes.
[125I]MIP-1α was used in CCR5 rhesus macaque membranes. All values are means ± S.E.M. of three independent experiments [75].
Calcium mobilization assays were used to determine the functional activity of the compounds to both the MOR and the CCR5 (Table 2). Compounds containing a morphinan group (22, 24, 28) were tested for their MOR antagonism [75]. Overall, substitution on naltrexone (28) was much more tolerated than for maraviroc as seen in Table 1 [75]. All of the compounds had similar IC50 values which meant that the difference in maraviroc attachment sites and lack of maraviroc did not affect MOR antagonism.
Table 2.
Antagonism of DAMGO and RANTES stimulated calcium mobilization in hMOR-CHO and MOLT-4 cells respectively.
| Compound | MOR IC50 (nM)a | CCR5 IC50 (nM)b |
|---|---|---|
| 28 | 8.9 ± 0.9 | - |
| 29 | - | 2.2 ± 0.3 |
| 22 | 40.0 ± 4.8 | 126 ± 28 |
| 23 | - | 622 ± 36 |
| 24 | 37.8 ± 4.4 | - |
| 25 | - | 7.91 ± 0.76 |
| 26 | - | 1.57 ± 0.18 |
| 27 | - | 14.2 ± 1.9 |
hMOR-CHO cells were stimulated with DAMGO,
MOLT-4 cells were stimulated with RANTES, (-) denotes that the compound was not tested [75].
The CCR5 antagonism results from the calcium mobilization assays indicated that modification of maraviroc (29) with phenyl substituents was not well tolerated. When an amino group was added to the 4-position (27), there was about 7-fold loss in CCR5 inhibition [75]. An even more drastic effect was seen for the bulkier substituents in 25 and 26, with losses in activity of 3600-fold and 700-fold respectively [75]. Therefore, smaller substituents on the phenyl ring of maraviroc might be better tolerated compared than more sterically bulky groups. However, this observation was not seen to the same extent for the bivalent and monovalent compound. The monovalent compound 23 had a larger substituent than 25 and 26, but they showed only a 200-fold and 60-fold decrease in activity compared to 29 [75]. These results suggest that the longer monovalent compounds may adopt a different binding mode than 25 and 26 and retain some of their CCR5 antagonism.
A HIV-1 infection assay was conducted using primary human astrocytes; primary human astrocytes were chosen because they are one of the primary sites of infection in NeuroAIDS. They are localized on the blood brain barrier and are the sites where opioids can synergistically potentiate the pathophysiological effects of HIV-1 infection [54]. Upon infection with R5 HIVSF162 (with and without morphine), there was a significant increase in Tat (transactivator of transcription) expression in astrocytes that coincided with virus invasion (Fig. 8) [76]. When maraviroc was added, virus invasion was decreased, as expected. However, when morphine was added along with maraviroc (29), its antiviral effects were completely abolished, which was indicated by a significant 4-fold increase in HIV Tat expression in the astrocytes. Treatment with naltrexone (28), or a combination of naltrexone (28) and maraviroc (29), had no effect on virus invasion with and without morphine’s presence. On the other hand, addition of the bivalent compound 22 (“bivalent”) had a significant effect compared to maraviroc and maraviroc with morphine stimulation. Overall, there was a 3.3-fold decrease in virus entry compared to maraviroc alone and a 7-fold decrease when compared to maraviroc with morphine [76]. Importantly, morphine stimulation had no effect on the bivalent compound’s viral entry inhibition activity. Cytotoxicity assays indicated neither maraviroc nor 22 had any toxicity in the astrocytes [76]. The results showed that in a native system, the bivalent compound could act as a potent virus invasion inhibitor without deleterious effects caused by morphine stimulation.
Fig. (8).

HIV-1 infection assay. HIV-1SF162 infectivity in human glial was determined based on the relative amount of Tat protein expressed by the virus using a luciferase based assay. (HA) human astrocytes, (R5) HIV-1SF162, (M) morphine at 500 nM, (MVC) maraviroc at 100 nM, (bivalent) compound 49 at 100 nM, and (NTX) naltrexone at 1500 nM. Values are absorbance ± SEM of 3 independent experiments at 18 h post-infection (*p < 0.005 vs. un-infected cells; $p < 0.05 vs. R5 HIV-1; # p <0.05 vs. opioid; ¶p < 0.05 vs. maraviroc (MVC); §p < 0.05 vs. morphine + MVC; ^ p <0.05 vs. MVC + NTX; ^^ p <0.05 vs. morphine + MVC + NTX; Ω p <0.05 vs. bivalent) [76].
While astrocytes harbor HIV in a more latent state, microglia and macrophages are the primary location for viral production in the brain [76,77]. Accordingly, bivalent compound 22 was also tested in microglia to elucidate cell-specific interactions. As previously observed in astrocytes, the antiviral effect of maraviroc (29) was significantly decreased by morphine administration. However, unlike in astrocytes, the bivalent compound 22 was unable to prevent HIV-1 infection in microglia [76]. Several explanations exist for the differential effects seen in astrocytes and microglia for 22. One possibility for the difference is that the expression levels of CCR5 and MOR differ greatly in the two cell types. The ratio of CCR5:MOR in astrocytes is roughly 2:1, while in microglia it is roughly 4:1 [76]. This difference may lead to more CCR5-MOR heterodimers forming in astrocytes than microglia. Bivalent compound 22 was designed to selectively target those heterodimers; therefore, it should have a greater affect in astrocytes where there are potentially more CCR5-MOR heterodimers present [49,75,76].
Another plausible reason for the difference in compound 22’s anti viral potency between the cell types are the different MOR splice variants present in them. The MOR undergoes extensive alternative splicing and these splice variants may lead to cell-specific effects and unique pharmacological profiles [78–80]. Increasing evidence has shown that MOR splice variants could directly affect HIV infection, susceptibility, and progression [78,79]. When comparing the expression rates of three different MOR splice variants (MOR-1, MOR-1A, and MOR-1X), Dever et al. found that they were differentially expressed in the individual CNS cell types [79]. MOR-1 is the canonical variant while MOR-1A is the shortest C-terminal splice variant and MOR-1X is the longest C-terminal variant. Importantly, astrocytes were found to express all three subtypes, while microglia only express MOR-1A [79]. The MOR-1 splice variant is known to dimerize with CCR5, while the other splice variants have not been fully studied for CCR5-MOR heterodimerization [72]. Therefore, the MOR-1A variant might not be able to heterodimerize with CCR5 in microglia, which would lead to the lack of antiviral activity for bivalent compound 22 seen in microglia. Both the differences in expression ratios of CCR5 to MOR and differences in splice variants between astrocytes and microglia helped explain the cell specific effects for 22.
3. Current Chemokine Receptor Crystal Structures
In 2010, the first chemokine receptor (CXCR4) was crystallized by the Stevens lab at the Scripps Research Institute [14]. The CXCR4 crystal structure was the first peptide GPCR to be solved and represented a major breakthrough in this field. It is important to note that several structural changes were used in order to stabilize the receptor for crystallography [13,14,81–91]. Using the T4 Lysozime (T4L) strategy, intracellular 3 (IL3) of CXCR4 was replaced with T4L along with truncating its C-terminal and using point mutations for stabilizing [14]. While these techniques have been used successfully to crystallize GPCRs, they may introduce or induce unnatural receptor conformations [18,92]. However, GPCR crystal structures have provided a wealth of knowledge concerning ligand binding that have confirmed or disproved modeling and mutagenesis data [18,93]. More importantly, the chemokine receptor CXCR4 was crystallized in two dimeric forms with resolutions of 2.5 and 3.2 Å and these two dimers had either a TM5 and TM6 interface or a TM3 and TM4 interface respectively [14].
The antagonists utilized in the CXCR4 crystallization process were a small molecule antagonist IT1t and a cyclic peptide antagonist CVX15 [14]. In the presence of either ligand the binding pocket was shown to be much larger than comparable aminergic receptors, which was most likely due to its much larger endogenous peptide agonists [14,93]. The increase in the size of the binding pocket presumably led to both antagonists binding shallowly near extracellular loop 2 (ECL2), which is important in ligand recognition and receptor activation [14,93–95]. While the large binding pocket may make computational modeling and docking difficult, the CXCR4 crystal structure has led to a magnitude of studies using structural based drug design in order to make new CXCR4 specific ligands [51,93,96–108].
In 2013 chemokine receptor CCR5 (CCR5) was crystallized by Tan et al. and revealed both similarities and differences within the chemokine receptor family [91]. Like CXCR4, the binding pocket of CCR5 was large due to its endogenous peptide agonists, but the crystallized antagonist, maraviroc, occupied a deeper domain of the pocket compared to CXCR4. Due to the depth of the maraviroc binding pocket, ECL2 did not play a role in binding, which was in contrast to the CXCR4 structure [91]. The variances could reflect the differences in the mechanisms of antagonists used in the crystallization processes or general structural differences between the receptors. In all, both chemokine receptor crystal structures will allow for the rational design of new ligands and for the homology modeling of other chemokine receptors.
In addition to modeling ligand binding, the CXCR4 crystal structure allows for a unique approach to model chemokine receptor dimers. Previously, the only methods to model GPCR dimers were either sequence-based or docking-based, which are largely pragmatic and easily biased [18]. However, crystallized GPCR homodimers permit a new method of modeling other GPCR dimers. For example, using the crystal structure of the bovine rhodopsin homodimer, Gorinski et al. were able to model the 5-HT1A homodimer by superimposing monomer units over the dimer based upon sequence similarities [109]. When combined with site directed mutagenesis, the work supported a TM4/TM5 interface for 5-HT1A homodimers [109].
Presumably different receptor types may lead to different dimer interfaces; therefore, it is critical to choose a dimer template that is similar to the target dimer being modeled [18]. The CXCR4 homodimer crystal structure thus allows for other chemokine receptor dimers to be more confidently modeled. For example, the CCR5-MOR heterodimer was recently modeled utilizing the CXCR4 homodimer crystal structure as the template (Fig. 9a) [76]. The heterodimer was based on the TM5-TM6 dimer interface that was seen in the IT1t bound CXCR4 crystal structure [14]. Utilizing this method the CCR5-MOR heterodimer model showed favorable electrostatic and hydrophobic interactions between receptors and could represent a possible conformation of the heterodimer. Furthermore, such models can be used to map the interactions between bivalent compounds and their respective receptor dimer. Fig. 9b and 9c show the CCR5-MOR selective ligand (22) bound to both receptors by spanning across the TM5-TM6 interface [110]. By further understanding the interactions between bivalent compounds and dimers, insight can be gained both in receptor function and dimer interaction. For example, the observed synergism in the HIV-1 inhibitory effects of ligand 22 was explained through dynamic simulations of 22 bound to the CCR5-MOR dimerization model [110].
Fig. (9).

CCR5-MOR heterodimer model based on CXCR4 dimer crystal structure (PDB code: 3ODU) with bivalent compound 22 bound. The CCR5 is colored in blue whereas the MOR in green. Compound 22 is colored in yellow [110].
4. Conclusion
Bivalent ligands represent a very promising technique for the study of GPCR dimerization. As indicated in this review, the groundwork for discovering new bivalent ligands has been accomplished within the field of chemokine receptor dimerization. First these studies indicated, a rational process must drive the bivalent ligand design in order to achieve interpretive results. Aspects such as pharmacophore identification, linker attachment site, linker composition, and linker length must be addressed first, as seen in the examples outlined in this review. Secondly, biological techniques such as co-immunoprecipitation, fluorescent lifetime imaging microscopy, FRET, protein fragment complementation assays, and even crystallization studies will be imperative to confirm the receptor(s) of interest may actually dimerize [20,22–29]. Thirdly, in order to fully explore the pharmacological and therapeutic implication of these dimers, bivalent ligands should be exploited in disease relevant cellular and molecular models.
For chemokine receptors, there are currently bivalent ligands targeting the CXCR4-CXCR4 homodimer and the CCR5-MOR heterodimer. For the CXCR4 homodimer three independent studies synthesized bivalent ligands that successfully targeted the homodimer. Linker length and composition proved to be essential when combining CXCR4 antagonist pharmacophores; more rigid linkers having a length of approximately 6 nm were favored [38]. Peptide based bivalent ligands were also capable of interacting with the homodimer. Both the attachment site and peptide lengths were influential in binding affinity [37,51]. For the CCR5-MOR heterodimer a bivalent compound was synthesized containing two antagonists linked together with an aliphatic linker [49]. The CCR5-MOR bivalent ligand proved to be a potent and cell type specific inhibitor for NeuroAIDS where the known treatment, maraviroc, is less efficacious and fails to inhibit virus entry in the presence of morphine [75,76]. These studies show the effectiveness and potential of targeting chemokine receptor dimers with bivalent ligands and have laid the foundation for future studies.
The exponential increase of GPCR crystal structures within the last decade will undoubtedly aid in the design and implementation of bivalent ligands targeting chemokine receptor dimers. This is especially true since two chemokine receptors have been successfully crystallized and one of them was crystallized as a homodimer. When combined, these discoveries will allow for more reliable homology models of chemokine receptors and the ability to study their dimerized state. By utilizing computational modeling to support bivalent ligand design, a better understanding of receptor(s)-ligand(s) interaction can be gained. Furthermore, dynamics simulations of dimer-bivalent ligand complex models can improve our understanding on the dimerization mechanisms and its relationship to the synergism seen in bivalent ligands. This strategy has become even more advantageous due to the availability of chemokine receptor crystal structures.
Fig. (6).

Library of compounds for the study of the CCR5-MOR heterodimer. The library consists of the bivalent compound (22), the monovalent controls (23, and 24) and the 4-substituted maraviroc compounds (25, 26, and 27) [49,75].
Acknowledgements
The work was partially supported by PHS grant DA024022 (Y. Z.). The content in this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse or the National Institutes of Health.
Footnotes
Conflict of Interest
The author declares no conflicts of interest.
References
- [1].Bjarnadottir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schioth HB. Comprehensive Repertoire and Phylogenetic Analysis of the G Protein-Coupled Receptors in Human and Mouse. Genomics. 2006;88:263–273. doi: 10.1016/j.ygeno.2006.04.001. [DOI] [PubMed] [Google Scholar]
- [2].Borish LC, Steinke JW. 2. Cytokines and Chemokines. J. Allergy Clin. Immunol. 2003;111:S460–S475. doi: 10.1067/mai.2003.108. [DOI] [PubMed] [Google Scholar]
- [3].Zlotnik A, Yoshie O. Chemokines: A New Classification System and Their Role in Immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- [4].Horuk R. Chemokine Receptors. Cytokine Growth Factor Rev. 2001;12:315–335. doi: 10.1016/s1359-6101(01)00014-4. [DOI] [PubMed] [Google Scholar]
- [5].Portoghese PS. From Models to Molecules: Opioid Receptor Dimers, Bivalent Ligands, and Selective Opioid Receptor Probes. J. Med. Chem. 2001;44:2259–2269. doi: 10.1021/jm010158+. [DOI] [PubMed] [Google Scholar]
- [6].Portoghese PS. Bivalent Ligands and the Message-Address Concept in the Design of Selective Opioid Antagonists. Trends Pharmacol. Sci. 1989;10:230–235. doi: 10.1016/0165-6147(89)90267-8. [DOI] [PubMed] [Google Scholar]
- [7].Bai M. Dimerization of G-Protein-Coupled Receptors: Roles in Signal Transduction. Cell. Signal. 2004;16:175–186. doi: 10.1016/s0898-6568(03)00128-1. [DOI] [PubMed] [Google Scholar]
- [8].Fuxe K, Borroto-Escuela DO, Marcellino D, Romero-Fernandez W, Frankowska M, Guidolin D, Filip M, Ferraro L, Woods AS, Tarakanov A, Ciruela F, Agnati LF, Tanganelli S. GPCR Heteromers and Their Allosteric Receptor-Receptor Interactions. Curr. Med. Chem. 2012;19:356–363. doi: 10.2174/092986712803414259. [DOI] [PubMed] [Google Scholar]
- [9].George SR, O’Dowd BF, Lee SP. G-Protein-Coupled Receptor Oligomerization and Its Potential for Drug Discovery. Nat. Rev. Drug Disc. 2002;1:808–820. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- [10].Herbert TE, Moffett S, Morello JP, Losiel TP, Bichet DG, Barret C, Bouvier M. A Peptide Derived from a beta(2)-Adrenergic Receptor Transmembrane Domain Inhibits Both Receptor Dimerization and Activation. J. Biol. Chem. 1996;271:16384–16392. doi: 10.1074/jbc.271.27.16384. [DOI] [PubMed] [Google Scholar]
- [11].Pfleger KDG, Eidne KA. Monitoring the Formation of Dynamic G-Protein-Coupled Receptor-Protein Complexes in Living Cells. Biochem. J. 2005;385:625–637. doi: 10.1042/BJ20041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Angers S, Salahpour A, Joly E, Hilairet S, Chelsky D, Dennis M, Bouvier M. Detection of Beta 2-Adrenergic Receptor Dimerization in Living Cells Using Bioluminescence Resonance Energy Transfer (BRET) Proc. Natl. Acad. Sci. USA. 2000;97:3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal Structure of the Μ-Opioid Receptor Bound to a Morphinan Antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science (80-. ) 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. Atomic-Force Microscopy: Rhodopsin Dimers in Native Disc Membranes. Nature. 2003;421:127–128. doi: 10.1038/421127a. [DOI] [PubMed] [Google Scholar]
- [16].Gouldson PR, Higgs C, Smith RE, Dean MK, Gkoutos GV, Reynolds CA. Dimerization and Domain Swapping in G-Protein-Coupled Receptors: A Computational Study. Neuropsychopharmacology. 2000;23:S60–S77. doi: 10.1016/S0893-133X(00)00153-6. [DOI] [PubMed] [Google Scholar]
- [17].Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal Structure of the Ligand-Free G-Protein-Coupled Receptor Opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- [18].Fanelli F, Benedetti PGD. Update 1 of: Computational Modeling Approaches to Structure – Function Analysis of G Protein-Coupled Receptors. Chem. Rev. 2011;111:PR438–PR535. doi: 10.1021/cr100437t. [DOI] [PubMed] [Google Scholar]
- [19].Jordan BA, Devi AL. G-Protein-Coupled Receptor Heterodimerization Modulates Receptor Function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mellado M, Rodriguez-Frade JM, Manes S, Martinez AC. Chemokine Signaling and Functional Responses: The Role or Receptor Dimerization and TK Pathway Activation. Annu. Rev. Immunol. 2001;19:397–421. doi: 10.1146/annurev.immunol.19.1.397. [DOI] [PubMed] [Google Scholar]
- [21].George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, O’Dowd BF. Oligomerization of Mu- and Delta-Opioid Receptors. Generation of Novel Functional Properties. J. Biol. Chem. 2000;275:26128–26135. doi: 10.1074/jbc.M000345200. [DOI] [PubMed] [Google Scholar]
- [22].Kramp BK, Sarabi A, Koenen RR, Weber C. Heterophilic Chemokine Receptor Interactions in Chemokine Signaling and Biology. Exp. Cell Res. 2011;317:655–663. doi: 10.1016/j.yexcr.2010.11.014. [DOI] [PubMed] [Google Scholar]
- [23].Wang J, Norcross M. Dimerization of Chemokine Receptors in Living Cells: Key to Receptor Function and Novel Targets for Therapy. Drug Discov. Today. 2008;13:625–632. doi: 10.1016/j.drudis.2008.04.004. [DOI] [PubMed] [Google Scholar]
- [24].Munoz LM, Holgado BL, Martinez-A C, Rodriguez-Frade JM, Mellado M. Chemokine Receptor Oligomerization: A Further Step toward Chemokine Function. Immunol. Lett. 2012;145:23–29. doi: 10.1016/j.imlet.2012.04.012. [DOI] [PubMed] [Google Scholar]
- [25].Salanga CL, O’Hayre M, Handel TM. Modulation of Chemokine Receptor Activity through Dimerization and Crosstalk. Cell Mol. Life Sci. 2010;66:1370–1386. doi: 10.1007/s00018-008-8666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Springael J-Y, Urizar E, Parmentier M. Dimerization of Chemokine Receptors and Its Functional Consequences. Cytokine Growth Factor Rev. 2005;16:611–623. doi: 10.1016/j.cytogfr.2005.05.005. [DOI] [PubMed] [Google Scholar]
- [27].Thelen M, Munoz LM, Rodriguez-Frade JM, Mellado M. Chemokine Receptor Oligomerization: Function Considerations. Curr. Opin. Pharmacol. 2010;8:38–43. doi: 10.1016/j.coph.2009.09.004. [DOI] [PubMed] [Google Scholar]
- [28].Mellado M, Martinez-A C, Miguel J, Rodriguez-Frade JM. Analysis of G-Protein-Coupled Receptor Dimerization Following Chemokine Signaling. Methods. 2002;27:349–357. doi: 10.1016/s1046-2023(02)00093-2. [DOI] [PubMed] [Google Scholar]
- [29].Rodriguez-Frade JM, Mellado M, Martinez-A C. Chemokine Receptor Dimerization: Two Are Better than One. Trends Immunol. 2001;22:612–617. doi: 10.1016/s1471-4906(01)02036-1. [DOI] [PubMed] [Google Scholar]
- [30].Shonberg J, Scammells PJ, Capuano B. Design Stratagies for Bivalent Ligands Targeting GPCRs. ChemMedChem. 2011;6:963–974. doi: 10.1002/cmdc.201100101. [DOI] [PubMed] [Google Scholar]
- [31].Munoz LM, Holgado BL, Martinez-A C. Chemokine Receptor Oligomerization: A Further Step toward Chemokine Function. Immunol. Lett. 2012;145:23–29. doi: 10.1016/j.imlet.2012.04.012. Rod. [DOI] [PubMed] [Google Scholar]
- [32].Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 Entry Cofactor: Functional cDNA Cloning of a Seven Transmembrane, G Protein-Coupled Receptor. Science (80-. ) 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- [33].Chinen J, Shearer WT. Molecular Virology and Immunology of HIV Infection. Allergy Clin. Immunol. 2002;110:189–198. doi: 10.1067/mai.2002.126226. [DOI] [PubMed] [Google Scholar]
- [34].Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous Defect in HIV-1 Coreceptor Accounts for Resistance of Some Mutiply-Exposed Individuals to HIV-1 Infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- [35].Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Shall TJ, Littman DR, Landau NR. Identification of a Major Co-Receptor for Primary Isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- [36].Hernandez PA, Grolin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, Klotman ME, Diaz GA. Mutations in the Chemokine Receptor Gene CXCR4 Are Associated with WHIM Syndrome, a Combined Immunodeficiency Disease. Nat. Genet. 2003;34:70–74. doi: 10.1038/ng1149. [DOI] [PubMed] [Google Scholar]
- [37].Choi H-J, Kumar S, Madani N, Han X, Tian S, Dong C-Z, Liu D, Duggineni S, Yuan J, Sodroski JG, Huang Z, An J. A Novel Synthetic Bivalent Ligand to Probe Chemokine Receptor CXCR4 Dimerization and Inhibit HIV-1 Entry. Biochemistry. 2012;51:7078–7086. doi: 10.1021/bi2016712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tanaka T, Nomura W, Narumi T, Masuda A, Tamamura H. Bivalent Ligands of CXCR4 with Rigid Linkers for Elucidation of Dimerization State in Cells. J. Am. Chem. Soc. 2010;132:15899–15901. doi: 10.1021/ja107447w. [DOI] [PubMed] [Google Scholar]
- [39].Kobayashi K, Oishi S, Hayashi R, Tomita K, Kubo T, Tanahara N, Ohno H, Yoshikawa Y, Furuya T, Hoshino M, Fujii N. Structure-Activity Relationship Study of a CXC Chemokine Receptor Type 4 Antagonist, FC131, Using a Series of Alkene Dipeptide Isosteres. J. Med. Chem. 2012;55:2746–2757. doi: 10.1021/jm2016914. [DOI] [PubMed] [Google Scholar]
- [40].Tamamura H, Hiramatsu K, Ueda S, Wang Z, Kusano S, Terakubo S, Trent JO, Peiper SC, Yamamoto N, Nakashima H, Otaka A, Fujii N. Stereoselective Synthesis of [L-Arg-L/D-3-(2-Naphthyl)alanine]-Type (E)-Alkene Dipeptide Isosteres and Its Application to the Synthesis and Biological Evaluation of Pseudopeptide Analogues of the CXCR4 Antagonist FC131. J. Med. Chem. 2005;48:380–391. doi: 10.1021/jm049429h. [DOI] [PubMed] [Google Scholar]
- [41].Demmer O, Dijkgraaf I, Schumacher U, Marinelli L, Cosconati S, Gourni E, Wester HJ, Kessler H. Design, Synthesis, and Functionalization of Dimeric Peptides Targeting Chemokine Receptor CXCR4. J. Med. Chem. 2011;54:7648–7662. doi: 10.1021/jm2009716. [DOI] [PubMed] [Google Scholar]
- [42].Yoshikawa Y, Kobayashi K, Oishi S, Fujii N, Furuya T. Molecular Modeling Study of Cyclic Pentapeptide CXCR4 Antagonists: New Insight into CXCR4-FC131 Interactions. Bioorg. Med. Chem. Lett. 2012;22:2146–2150. doi: 10.1016/j.bmcl.2012.01.134. [DOI] [PubMed] [Google Scholar]
- [43].Vabeno J, Nikiforovich GV, Marshall GR. Insight into the Binding Mode for Cyclopentapeptide Antagonists of the CXCR4 Receptor. Chem. Biol. Drug. Des. 2006;67:346–354. doi: 10.1111/j.1747-0285.2006.00387.x. [DOI] [PubMed] [Google Scholar]
- [44].Vabeno J, Nikiforovich GV, Marshall GR. A Minimalistic 3D Pharmacophore Model for Cyclopentapeptide CXCR4 Antagonists. Biopolymers. 2006;84:459–471. doi: 10.1002/bip.20508. [DOI] [PubMed] [Google Scholar]
- [45].Bonger KM, Kapoerchan VV, Grotenberg GM, Van Koppen CJ, Timmers CM, Van der Marel GA, Overkleeft HS. Oligoproline Helices as Structurally Defined Scaffolds for Oligomeric G Protein-Coupled Receptor Ligands. Org. Biomol. Chem. 2010;8:1881–1884. doi: 10.1039/b923556f. [DOI] [PubMed] [Google Scholar]
- [46].Zheng Y, Akgun E, Harikumar KG, Hopson J, Powers MD, Lunzer MM, Miller LJ, Portoghese PS. Induced Association of Mu Opioid (MOP) and Type 2 Cholecystokinin (CCK2) Receptors by Novel Bivalent Ligands. J. Med. Chem. 2009;52:247–258. doi: 10.1021/jm800174p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang S, Yekkirala A, Tang Y, Portoghese PS. A Bivalent Ligand (KMN-21) Antagonist for Mu/kappa Heterodimeric Opioid Receptors. Bioorg. Med. Chem. Lett. 2009;19:6978–6980. doi: 10.1016/j.bmcl.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Daniels DJ, Lenard NR, Etienne CL, Law P-Y, Roerig SC, Portoghese PS. Opioid-Induced Tolerance and Dependence in Mice Is Modulated by the Distance between Pharmacophores in a Bivalent Ligand Series. Proc. Natl. Acad. Sci. U. S. A. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yuan Y, Arnatt CK, Li G, Haney KM, Ding D, Jacob JC, Selley DE, Zhang Y. Design and Synthesis of a Bivalent Ligand to Explore the Putative Heterodimerization of the Mu Opioid Receptor and the Chemokine Receptor CCR5. Org. Biomol. Chem. 2012;10:2633–2646. doi: 10.1039/c2ob06801j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bobronik SA. The Influence of Rigid or Flexible Linkage between Two Ligands on the Effective Affinity and Avidity for Reversible Interactions with Bivalent Receptors. J. Mol. Recognit. 2007;20:253–262. doi: 10.1002/jmr.836. [DOI] [PubMed] [Google Scholar]
- [51].Xu Y, Duggineni S, Espitia S, Richman DD, An J, Huang Z. A Synthetic Bivalent Ligand of CXCR4 Inhibits HIV Infection. Biochem. Biophys. Res. Commun. 2013;435:646–650. doi: 10.1016/j.bbrc.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori G, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. Maraviroc (UK-427,857), a Potent, Orally Bioavailable, and Selective Small-Molecule Inhibitor of Chemokine Receptor CCR5 with Broad-Spectrum Anti-Human Immunodeficiency Virus Type 1 Activity. Antimicrob. Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Minagar A, Commins D, Alexander JS, Hoque R, Chiappelli F, Signer EJ, Nikbin B, Shapshak P. NeuroAIDS: Characteristics and Diagnosis of the Neurological Complications of AIDS. Mol. Diagn. Ther. 2008;12:25–43. doi: 10.1007/BF03256266. [DOI] [PubMed] [Google Scholar]
- [54].Hauser KF, Fitting S, Dever SM, Podhaizer EM, Knapp PE. Opiate Drug Use and the Pathophysiology of neuroAIDS. Curr. HIV Res. 2012;10:435–452. doi: 10.2174/157016212802138779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lindl KA, Marks DR, Kolson DL, Jordan-Sciutto KL. HIV-Associated Neurocognitive Disorder: Pathogenesis and Therapeutic Opportunities. J. Neuroimmune. Pharmacol. 2010;5:294–309. doi: 10.1007/s11481-010-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, Cass W, Turchan JT. Molecular Basis for Interactions of HIV and Drugs of Abuse. JAIDS, J. Acquir. Immune Defic. Syndr. 2002;31:S62–S69. doi: 10.1097/00126334-200210012-00006. [DOI] [PubMed] [Google Scholar]
- [57].Hauser KF, El-Hage N, Buch S, Berger JR, Tyor WR, Nath A, Bruce-Keller AJ, Knapp PE. Molecular Targets of Opiate Drug Abuse in neuroAIDS. Neurotoxic. Res. 2005;8:63–80. doi: 10.1007/BF03033820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Norman KF, Basso M, Kurmar A, Malow R. Neuropsychological Consequences of HIV and Substance Abuse: A Literature Review and Implications for Treatment and Future Research. Curr. Drug Abus. Rev. 2009;2:143–156. doi: 10.2174/1874473710902020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Anthony IC, Arango JC, Stephens B, Simmonds P, Bell JE. The Effects of Illicit Drugs on the HIV Infected Brain. Front. Biosci. 2008;13:1294–1307. doi: 10.2741/2762. [DOI] [PubMed] [Google Scholar]
- [60].Alkhatib G, Comadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: A RANTES, MIP-1alpha, MIP-1beta Receptor as a Fusion Cofactor for Macrophage-Tropic HIV-1. Science (80-. ) 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- [61].Gabuzda D, Wang J. Chemokine Receptors and Virus Entry in the Central Nervous System. J. Neurovirol. 1999;5:643–658. doi: 10.3109/13550289909021293. [DOI] [PubMed] [Google Scholar]
- [62].Luster AD. Chemokines - Chemotactic Cytokines That Mediate Inflammation. N. Engl. J. Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- [63].Schmidhammer H, Burkard WP, Eggstein-Aeppli L, Smith CF. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 2. (−)-N-(cyclopropylmethyl)-4,14-Dimethoxymorphinan-6-One, a Selective Mu Opioid Receptor Antagonist. J. Med. Chem. 1989;32:418–421. doi: 10.1021/jm00122a021. [DOI] [PubMed] [Google Scholar]
- [64].Noel RJJ, Rivera-Amill V, Buch S, Kurmar A. Opiates, Immune System, Acquired Immunodeficiency Syndrome, and Nonhuman Primate Model. J. Neurovirol. 2008;14:279–285. doi: 10.1080/13550280802078209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hauser KF, El-Hage N, Steine-Martin A, Maragos WF, Nath A, Peridsky Y, Volsky DJ, Knapp PE. HIV-1 Neuropathogenesis: Glial Mechanisms Revealed through Substance Abuse. J. Neurochem. 2007;100:567–586. doi: 10.1111/j.1471-4159.2006.04227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF. HIV-1 Tat and Opiate-Induced Changes in Astrocytes Promote Chemotaxis of Microglia through the Expression of MCP-1 and Alternative Chemokines. Glia. 2006;53:132–146. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Suzuki S, Chuang LF, Yau P, Doi RH, Chuang RY. Interactions of Opioid and Chemokine Receptors: Oligomerization of Mu, Kappa, and Delta with CCR5 on Immune Cells. Exp. Cell Res. 2002;280:192–200. doi: 10.1006/excr.2002.5638. [DOI] [PubMed] [Google Scholar]
- [68].Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, Hauser KF. Synergistic Neurotoxicity of Opioids and Human Immunodeficiency Virus-1 Tat Protein in Striatal Neurons in Vitro. Neuroscience. 2001;102:555–563. doi: 10.1016/s0306-4522(00)00461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Turchan-Cholewo J, Liu Y, Gartner S, Reid R, Jie C, Peng X, Chen KC, Chauhan A, Haughey N, Cutler R, Mattson MP, Pardo C, Conant K, Sacktor N, McArthur JC, Hauser KF, Gairola C, Nath A. Increased Vulnerability of ApoE4 Neurons to HIV Proteins and Opiates: Protection by Diosgenin and L-Deprenyl. Neurobiol. Dis. 2006;23:109–119. doi: 10.1016/j.nbd.2006.02.005. [DOI] [PubMed] [Google Scholar]
- [70].Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, Knapp PE. Morphine Potentiates Neurodegenerative Effects of HIV-1 Tat through Actions at M-Opioid Receptor-Expressing Glia. Brain. 2011;134:3613–3628. doi: 10.1093/brain/awr281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mahajan SD, Schwartz SA, Shanahan TC, Chawda RP, Nair MPN. Morphine Regulates Gene Expression of Α- and Β-Chemokines and Their Receptors on Astroglial Cells via the Opioid Receptor. J. Immunol. 2002;169:3589–3599. doi: 10.4049/jimmunol.169.7.3589. [DOI] [PubMed] [Google Scholar]
- [72].Chen C, Li J, Bot G, Szabo I, Rogers TJ, Liu-Chen L-Y. Heterodimerization and Cross-Desensitization between the Mu-Opioid Receptor and the Chemokine CCR5 Receptor. Eur. J. Pharmacol. 2004;483:175–186. doi: 10.1016/j.ejphar.2003.10.033. [DOI] [PubMed] [Google Scholar]
- [73].Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, Rogers TJ. Heterologous Desensitization of Opioid Receptors by Chemokines Inhibits Chemotaxis and Enhances the Perception of Pain. Proc. Natl. Acad. Sci. U. S. A. 2002;99:10276–10281. doi: 10.1073/pnas.102327699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sayre LM, Portoghese PS. Stereospecific Synthesis of 6 Alpha and 6 Beta-Amino Derivatives of Naltrexone and Oxymorphone. J. Org. Chem. 1980;45:3366–3368. [Google Scholar]
- [75].Yuan Y, Arnatt CK, El-Hage N, Dever SM, Jacob JC, Selley DE, Hauser KF, Zhang Y. A Bivalent Ligand Targeting the Putative Mu Opioid Receptor and Chemokine Receptor CCR5 Heterodimer: Binding Affinity versus Functional Activities. Med. Chem. Comm. 2013;4:847–851. doi: 10.1039/C3MD00080J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].El-Hage N, Dever SM, Podhaizer EM, Arnatt CK, Zhang Y, Hauser KF. A Novel Bivalent HIV-1 Entry Inhibitor Reveals Fundamental Differences in CCR5 - [mu]- Opioid Receptor Interactions in Human Glia. AIDS. 2013;27:2181–2190. doi: 10.1097/QAD.0b013e3283639804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Brack-Werner R. Astrocytes: HIV Cellular Reservoirs and Important Participants in Neuropathogenesis. AIDS. 1999;13:1–22. doi: 10.1097/00002030-199901140-00003. [DOI] [PubMed] [Google Scholar]
- [78].Dever SM, Costin BN, Xu R, El-Hage N, Balinang J, Samoshkin A, O’Brien MA, McRae MP, Diatchenko L, Knapp PE, Hauser KF. Differential Expression of the Alternatively Spliced OPRM1 Isoform M-Opioid Receptor-1K in HIV-Infected Individuals. AIDS. 2014;28:19–30. doi: 10.1097/QAD.0000000000000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Dever SM, Xu R, Fitting S, Knapp PE, Hauser KF. Differential Expression and HIV-1 Regulation of M-Opioid Receptor Splice Variants across Human Central Nervous System Cell Types. J. Neurovirol. 2012;18:181–190. doi: 10.1007/s13365-012-0096-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Pasternak GW, Pan YX. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013;65:1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Rasmussen SGF, Choi H-J, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, Schertler GFX, Weis WI, Kobilka BK. Crystal Structure of the Human β2 Adrenergic G-Protein-Coupled Receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- [82].Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-Resolution Crystal Structure of an Engineered Human β¬2-Adrenergic G Protein-Coupled Receptor. Science (80-. ) 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-Adrenergic G-Protein-Coupled Receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 Angstrom Crystal Structure of a A2A Adenosine Receptor Bound to an Antagonist. Science (80-. ) 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the Human Dopamine d3 Receptor in Complex with d2/d3 Selective Antagonist. Science (80-. ) 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S. Structure of the Human Histamine H1 Receptor Complex with Doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the Δ-Opioid Receptor Bound to Naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal Structure of the β2 Adrenergic Receptor-Gs Protein Complex. Nature. 2011;477:549–557. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang X-H, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the Human Κ-Opioid Receptor in Complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang X-P, Trapella C, Guerrini R, Calo G, Roth BL, Cherezov V, Stevens RC. Structure of the Nociception/orphanin FQ Receptor in Complex with a Peptide Mimetic. Nature. 2012;485:395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, Li T, Ma L, Fenalti G, Zhang W, Xie X, Yang H, Jiang H, Cherezov V, Liu H, Stevens RC, Zhao Q, Wu B. Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science (80-. ) 2013;341:1387–1390. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Arnatt CK, Zhang Y. A Nuclear G Protein-Coupled Estrogen Receptor, GPER. Homology Modeling Studies toward Its Ligand-Binding Mode Characterization. In: Cozzini P, Kellogg GE, editors. Computational approaches to nuclear receptors. The Royal Society of Chemistry; Cambridge, UK: 2013. pp. 117–137. [Google Scholar]
- [93].Congreve M, Langmead CJ, Mason JS, Marshall FH. Progress in Structure Based Drug Design for G Protein-Coupled Receptors. J. Med. Chem. 2011;54:4283–4311. doi: 10.1021/jm200371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Clark-Lewis L, Kim KS, Rajarathnam K, Gong JH, Dewald B, Moser B, Baggiolini M, Sykes BD. Structure-Activity Relationships of Chemokines. J. Leukoc. Biol. 1995;57:703–711. doi: 10.1002/jlb.57.5.703. [DOI] [PubMed] [Google Scholar]
- [95].Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC, 3rd, Basnet H, Sakmar TP, Volkman BF. Structural Basis of CXCR4 Sulfotyrosine Recognition by the Chemokine SDF-1/CXCL12. Sci. Signal. 2008;37:ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Choi W-T, Duggineni S, Xu Y, Huang Z, An J. Drug Discovery Research Targeting the CXC Chemokine Receptor 4 (CXCR4) J. Med. Chem. 2012;55:977–994. doi: 10.1021/jm200568c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Planesas JM, Perez-Nueno VI, Borrell JI, Teixido J. Impact of the CXCR4 Structure on Docking-Based Virtual Screening of HIV Entry Inhibitors. J. Mol. Graph. Model. 2012;38:123–136. doi: 10.1016/j.jmgm.2012.06.010. [DOI] [PubMed] [Google Scholar]
- [98].Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, Heveker N, Shoichet BK, Volkman BF. Structure-Based Ligand Discovery for the Protein–protein Interface of Chemokine Receptor CXCR4. Proc. Natl. Acad. Sci. U. S. A. 2012;109:5517–5522. doi: 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Small Molecule Inhibitors of CXCR4. Theranostics. 2013;3:47–75. doi: 10.7150/thno.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Peled A, Wald O, Burger J. Development of Novel CXCR4-Based Therapeutics. Expert Opin. Invest. Drugs. 2012;21:341–353. doi: 10.1517/13543784.2012.656197. [DOI] [PubMed] [Google Scholar]
- [101].Aboye TL, Ha H, Majumder S, Christ F, Debyser Z, Shekhtman A, Neamati N, Camarero JA. Design of a Novel Cyclotide-Based CXCR4 Antagonist with Anti-Human Immunodeficiency Virus (HIV)-1 Activity. J. Med. Chem. 2012;55:10729–10734. doi: 10.1021/jm301468k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Vinader V, Ahmet DS, Ahmed MS, Patterson LH, Afarinkia K. Discovery and Computer Aided Potency Optimization of a Novel Class of Small Molecule CXCR4 Antagonists. PLoS One. 2013;8:1–18. doi: 10.1371/journal.pone.0078744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Wilkinson RA, Pincus SH, Song K, Shepard JB, Weaver AJJ, Labib ME, Teintze M. Improved Guanide Compounds Which Bind the CXCR4 Co-Receptor and Inhibit HIV-1 Infection. Bioorg. Med. Chem. Lett. 2014;23:2197–2201. doi: 10.1016/j.bmcl.2013.01.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Vitale RM, Gatti M, Carbone M, Barbieri F, Felicita V, Gavagnin M, Florio T, Amodeo P. Minimalist Hybrid Ligand/receptor-Based Pharmacophore Model for CXCR4 Applied to a Small-Library of Marine Natural Products Led to the Identification of Phidianidine A as a New CXCR4 Ligand Exhibiting Antagonist Activity. ACS Chem. Biol. 2013;8:2762–2770. doi: 10.1021/cb400521b. [DOI] [PubMed] [Google Scholar]
- [105].Truax VM, Zhao H, Katzman BM, Prosser AR, Alcaraz AA, Saindane MT, Howard RB, Culver D, Arrendale RF, Gruddanti PR, Evers TJ, Natchus MG, Snyder JP, Liotta DC, Wilson LJ. Discovery of Tetrahydroisoquinoline-Based CXCR4 Antagonists. ACS Med. Chem. Lett. 2013;4:1025–1030. doi: 10.1021/ml400183q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Zhang C, Hou T, Feng Z, Li Y. Structure-Based Development of Antagonists for Chemokine Receptor CXCR4. Curr. Comput. Aided Drug Des. 2013;9:60–75. [PubMed] [Google Scholar]
- [107].Zhu L, Zhao Q, Wu B. Structure-Based Studies of Chemokine Receptors. Curr. Opin. Struct. Biol. 2013;23:539–546. doi: 10.1016/j.sbi.2013.05.003. [DOI] [PubMed] [Google Scholar]
- [108].Roumen L, Scholten DJ, de Kruijf P, de Esch IJP, Leurs R, de Graaf C. C(X)CR in Silico: Computer-Aided Prediction of Chemokine Receptor–ligand Interactions. Drug Discov. Today Technol. 2012;9:e281–e291. doi: 10.1016/j.ddtec.2012.05.002. [DOI] [PubMed] [Google Scholar]
- [109].Gorinski N, Kowalsman N, Renner U, Wirth A, Reinartz MT, Seifert R, Zeug A, Ponimaskin E, Niv MY. Computational and Experimental Analysis of the Transmembrane Domain 4/5 Dimerization Interface Fo the Serotonin 5-HT1a Receptor. Mol. Pharmacol. 2012;82:448–463. doi: 10.1124/mol.112.079137. [DOI] [PubMed] [Google Scholar]
- [110].Arnatt CK. Development of Antagonists Targeting Chemokine Receptor CCR5 and the Chemokine Receptor CCR5-Mu Opioid Receptor Heterodimer. Virginia Commonwealth University; 2013. pp. 92–104. [Google Scholar]