

Abstract
Background
Thrombelastography (TEG) allows for rapid global assessment of coagulation function. Our previous work demonstrated that a hypercoagulable state identified by TEG’s G value was associated with thromboembolic events in a cohort of critically ill surgical patients despite routine chemoprophylaxis. We hypothesized that a hypercoagulable state could be differentiated into enzymatic or platelet etiology through the use of thrombus velocity curves; specifically the time to maximum rate of thrombus generation (TMRTG) and the novel TEG parameter, delta. (Δ)
Methods
We retrospectively studied 10 critically ill surgical patients receiving thromboprophylaxis for at least 72 h by TEG, using kaolin activated citrated samples. Thrombus velocity curves were plotted for each patient, and delta was calculated as the difference between the TEG parameters R and SP, corresponding to the time to maximum rate of thrombus generation (TMRTG), which reflects the enzymatic contribution to clot formation. The TEG parameter G, (G = 5000 × A/100-A) also was determined for each patient. As G is derived from amplitude (A), it reflects overall net clot strength. A hypercoagulable state was defined as delta < 0.6 min and/or G >11 dynes/cm2.
Results
A hypercoagulable state was identified via delta in 6 patients (60%); all of whom remained hypercoagulable following heparinase addition, suggesting chemoprophylaxis was ineffective. Of six patients with a hypercoagulable G value, 50% had a normal delta suggesting the presence of platelet hypercoagulability. Delta closely correlated with TMRTG (r = 0.94). However, the varying contribution of platelets to hypercoagulability, was shown by a nonlinear, weak correlation of delta and TMRTG with G (r = 0.11 and r = 0.14, respectively).
Conclusion
Delta reflects changes in thrombin generation as measured by TMRTG, allowing for differentiation of enzymatic from platelet hypercoagulability. Future studies will be required to validate these findings.
Keywords: thrombelastography, delta, trauma, thrombin generation, hypercoagulability, thromboprophylaxis, deep vein thrombosis, thromboembolism
INTRODUCTION
Multiple factors in the critically ill surgical patient may disrupt regulatory mechanisms of hemostasis, including endothelial injury, stasis, platelet activation, decreased levels of endogenous anticoagulants, and impaired fibrinolysis. These factors may result in a hypercoagulable state, leaving patients at risk for thromboemobolic (TE) events. Objectively, confirmed deep vein thrombosis (DVT) rates were found to be in the range of 10% to 80% for patients admitted to ICUs or following trauma, neurosurgery, or spinal cord injury in a systematic review [1]. Further, pulmonary embolism (PE) has been observed in 2% to 22% of patients with trauma, and fatal PE is the third most common cause of death in patients who survive the first 24 h [2]. Current guidelines recommend chemoprophylaxis either using low dose unfractionated heparin (UH) or low molecular weigh heparin (LMWH) for prevention of these TE complications in high risk patient populations [3]. Despite adoption of these guidelines, several studies [4–6] suggest that the frequency of TE events has not changed.
Thrombelastography (TEG) allows for a rapid global assessment of haemostatic function from initial thrombin activation to fibrinolysis [7]. Although TEG is recognized for monitoring hypocoagulable states in the operating room, recent studies suggest this technique may identify hypercoagulable states in a variety of clinical settings [8–10]. Furthermore, our previous work has demonstrated that a hypercoagulable state identified by the TEG total clot strength parameter, G, is associated with TE events in a cohort of critically ill surgical patients despite standard chemoprophylaxis [11].
Since Virchow initially identified the triad of factors associated with thrombosis in 1856 [12], hypercoagulability has been implicated in the pathogenesis of TE events, and current studies confirm this concept [13]. Although endothelial injury and platelet activation are known contributors towards arterial thromboemboli [13], the sequence of events leading to venous thrombosis is not well known. Specifically, the platelet’s underlying contributions to thrombosis may be underestimated. Thrombin, the primary driving force of the coagulation system, has a significant role in the processes of initiation and propagation of a thrombus [14]. Thrombus velocity curves (V-curve) derived from standard TEG parameters have been described [15], and recent reports suggest that such measurements may represent surrogate markers of thrombin generation [16, 17].
The purpose of this pilot investigation was to determine if a hypercoagulable state could be differentiated into enzymatic or platelet etiology through the use of thrombus velocity curves; specifically the time to maximum rate of thrombus generation (TMRTG) and the novel TEG parameter delta (Δ), which represent thrombin generation.
METHODS
We studied critically ill surgical patients from the surgical ICU of our level 1 academic trauma center during a 1-wk period. Patients on chemoprophylaxis for TE events for at least 72 h were included in this study, while patients who received therapeutic anticoagulation for established TE events or for blunt carotid/vertebral injuries were excluded. Patients receiving aspirin or other platelet inhibitors prior to or during hospitalization where not included in this study. A total of 10 patients meeting these criteria were selected. A computerized thrombelastograph coagulation analyzer (TEG model 5000; Haemoscope Corporation, Niles, IL) was used to assess haemostatic function while on pharmacologic thromboprophylaxis. Quality control checks were completed within 8 h of blood collection per the manufacturer. Blood samples were collected at bedside into standard 3 mL phlebotomy collection tubes containing 0.3 mL of 3.2% (109 mM) buffered sodium citrate. Once samples were allowed to sit for 15 min per manufacturer recommendations, 1 mL of blood was drawn from the citrated tubes and placed into manufacturer-supplied kaolin vials (containing kaolin, phospholipids, and buffered stabilizers), which accelerate the coagulation process. From the resulting specimen 340 μL of sample were drawn and re-calcified with 20 μL of calcium chloride (200 mM) within a plastic cup standardized for TEG use. On the TEG analyzer, sample cup temperature was then adjusted to that of the patient’s and the assay was initiated. Each sample was run simultaneously with and without added heparinase to neutralize the presence of UH/LMWH and determine baseline coagulation profile. The assay was stopped once a complete tracing was generated by the software.
All TEG parameters were recorded from standard tracings: reaction time (R, minutes), alpha angle (α, degrees), coagulation time (K, min), maximum amplitude (MA, mm), clot strength (G, dynes/cm2), and estimated percent lysis (EPL, %). The various components of the TEG tracing are depicted in Fig. 1. The R value, is the time elapsed from the initiation of the test until the point were the onset of clotting provides enough resistance to produce a 2 mm amplitude reading on the TEG tracing, and is most representative of the initiation phase of enzymatic clotting factors; this is the point at which all other platelet-poor plasma clotting assays are stopped (e.g., PT and aPTT). K is a measurement of the time interval from the R time to the point where fibrin cross-linking provides enough clot resistance to produce a 20 mm amplitude reading. The α is the angle formed by the slope of a tangent line traced from the R to the K time measured in degrees. K time and the α angle denote the rate at which the clot strengthens and is most representative of thrombin’s cleaving of the available fibrinogen into fibrin. The MA indicates the point at which clot strength reaches its maximum measure in millimeters on the TEG tracing, and reflects the end result of maximal platelet-fibrin interaction via the GPIIb-IIIa receptors [18]. Finally, G is a calculated measure of total clot strength derived from amplitude (A, mm) G = (5000 × A)/(100 × A), due to its exponential relationship with A, G is a more realistic representation of overall clot strength [19]. Since G is calculated from the progressive increase of amplitude (clot strength) as the TEG tracing develops, it is inclusive of both platelet and enzymatic contributions to overall clot strength. Delta (Δ) was then calculated from the difference between R time and the time of initial split point (SP, mins) of the TEG tracing (R – SP), representing the time interval of greatest clot growth secondary to peak thrombin generation. Hypercoagulability was defined by a delta ≤0.6 min and/or G ≥ 11 dynes/cm2. For each sample, thrombus velocity curves derived from the above mentioned TEG parameters were plotted using TEG software, as seen in Fig. 2.
FIG. 1.
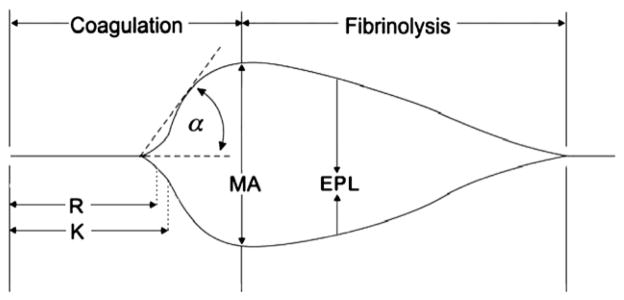
Depiction of thrombelastography (TEG) tracing and parameters measured throughout the clot’s lifespan. R = reaction time; K = coagulation time; α = alpha angle; MA = maximum amplitude; EPL = estimated percent lysis.
FIG. 2.

Sample of a thrombus velocity curve (V-curve, in grey) depicted over a standard thrombelastography (TEG) tracing, showing correlation of delta with thrombus generation parameters. A V-curve is plotted from the first derivative of changes in clot resistance expressed as a change in clot strength per unit of time (dynes/cm2/s), representing the maximum velocity of clot formation. TTG = total thrombus generation; MRTG =maximum rate of thrombus generation; TMRTG = time to maximum rate of thrombus generation; R = reaction time; SP = split point; Δ = delta. (Color version of figure is available online.)
Variables of interest for identifying risk factors of hypercoagulability and subsequent TE events were obtained by electronic chart review; these included sex, age, injury pattern, injury severity score (ISS), hospital days, ICU days, ventilator days, transfusions within the first 24 h, thromboprophylaxis received, and clinical and/or radiologic documentation of TE events.
RESULTS
Ten patients from the surgical ICU were receiving TE chemoprophylaxis; six were receiving a standard thromboprophylactic dose (5000 IU sc qd) of LMWH, and four received low-dose UH (5000 IU sc bid). All but one patient (due to bilateral lower extremity fractures) had lower extremity sequential compression devices (SCDs) in place continuously.
The general demographic characteristics and TE risk factors of these two groups are outlined in Table 1. Despite thromboprophylaxis, six (60%) patients were hypercoagulable by delta (<0.6 min) and four (40%) had a normal delta (0.6–1.2 min). Injury patterns associated with hypercoagulability such as intracranial, spinal, pelvic, and long bone injury, were more common in patients hypercoagulable by delta. Hypercoagulable patients by delta, compared with patients with a normal delta also had an increased mean hospital stay, [ 50.3 (±8.3) versus 22 (±3.2) d ], ICU stay [ 32.8 (±7.7) versus 11 (±3.7) d ], and ventilator days [ 25 (±6.5) versus 6.5 (±3.4) d ]. There were no symptomatic TE events noted in this study, but we do not routinely screen patients for events in our center. Furthermore, no clinical bleeding events were noted. The mean transfusion requirement during the study period was 1.8 (±1.0) packed red blood cell units. No difference in transfusion requirements were noted between normal and hypercoagulable patients by delta (Table 1).
TABLE 1.
Patient Demographics and Venous Thrombosis Risk Factors in Patients with Normal Delta Versus Hypercoagulable Delta
| Variable | Normal delta (0.6–1.2 min.) n = 4 | Hypercoagulable delta (<0.6 min.) n = 6 |
|---|---|---|
| Sex (% male) | 75% | 83% |
| Age (mean, ±SEM) | 50 (±11) | 47 (±5) |
| Trauma service | 50% | 83% |
| Intracranial injury | 25% | 50% |
| Spinal injury | 25% | 33% |
| Pelvic injury | 25% | 50% |
| Long bone injury | 0 | 33% |
| Hospital days (mean, ±SEM) | 22.0 (±3.2) | 50.3 (±8.3) |
| ICU days (mean, ±SEM) | 11 (±3.7) | 32.8 (±7.7) |
| Ventilator days (mean, ±SEM) | 6.5 (±3.4) | 25 (±6.5) |
| Thromboprophylaxis | 100% | 100% |
| LMWH | 50% | 67% |
| UH | 50% | 33% |
| SCDs | 100% | 67% |
| ISS (mean, ±SEM) * | 21 (±3.2) | 30 (±5.4) |
| Blood products within first 24 h * | 25% | 50% |
| Packed red blood cells (mean, ±SEM) | 1.7 (±0.9) | 2.0 (±1.1) |
SEM = standard error of the mean; LMWH low-molecular weight heparin; UH = unfractionated heparin; SCDs = sequential compression devices; ISS = injury severity score.
Applies to trauma population
Table 2 demonstrates that when standard TEG parameters were compared between groups, G values were higher in the hypercoagulable delta group than in the normal group; 16.9 (±2.4) versus 13.0 (±1.8) dynes/cm2. But of six patients with an elevated G, half had a normal delta, suggesting the etiology was predominantly platelet hypercoagulability. Of note, PT and aPTT in both groups were similar. Minimal benefit was realized from anticoagulation, as reflected by virtually no change in standard TEG parameters between active and neutralized UH/LMWH samples by the addition of heparinase to the TEG assay. Furthermore, although the administration of anticoagulation resulted in a minimal increase in delta, the G value still reflected a hypercoagulable state. When V-curves were plotted, peak thrombus generation was reached faster in hypercoagulable delta patients than in those with a normal delta, as measured by TMRTG; 6.9 (±1.1) versus 9.3 (±1.4) min. Figure 3 demonstrates how delta highly correlated with TMRTG (Pearson’s coefficient correlation, r = 0.94). Figure 4 illustrates changes in TEG and V-curve parameters when plotted against an increasing G value. As expected, a linear correlation between the parametric values G and TTG (r = 0.90) occurred. Furthermore, the varying contribution of platelets to hypercoagulability, resulted in a nonlinear, weak correlation of delta and TMRTG with G (r = 0.11 and r = 0.14, respectively).
TABLE 2.
Coagulation Parameters in Patients With Normal Delta Versus Hypercoagulable by Delta
| Variable | Normal delta (0.6–1.2 min.) n = 4 | Hypercoagulable delta (<0.6 min.) n = 6 |
|---|---|---|
| R time (min) | 7.8 (±1.1) | 4.8 (±0.3) |
| K time (min) | 1.2 (±0.5) | 1.0 (±0.3) |
| α (degrees) | 70.9 (±9.1) | 72.5 (±8.4) |
| MA (mm) | 69.7 (±3.5) | 76.18 (±3.1) |
| G (Dynes/cm2) | 13.0 (±1.8) | 16.9 (±2.4) |
| EPL (%) | 1.2 (±0.4) | 1.8 (±0.9) |
| MRTG (Dynes/cm2/s) | 19.7 (±2.2) | 22.9 (±2.6) |
| TMRTG (min) | 9.3 (±1.4) | 6.9 (±1.1) |
| TTG (Dynes/cm2) | 897.7 (±31.3) | 981.5 (±40.6) |
| PT (s) | 16.1 (±1.2) | 15.8 (±1.2) |
| aPTT (s) | 39.4 (±1.5) | 40.8 (±1.9) |
R =reaction time; K =coagulation time; α =alpha angle; MA = maximum amplitude; G = total clot strength; TMRTG = time to maximum rate of thrombus generation; SEM = standard error of the mean.
FIG. 3.

Linear relationship shown between delta and time to maximum thrombus generation (TMRTG). (Color version of figure is available online.)
FIG. 4.

Changes in thrombelastography (TEG) and V-curve parameters are plotted against an increasing G (total clot strength). As expected, a linear correlation between G and TTG (total thrombus generated) occurred (r = 0.90), since G represents total clot strength. The varying contribution of platelets to hypercoagulability, results in a non-linear, weak correlation of the proposed enzymatic markers delta and TMRTG (time to maximum rate of thrombus generation) with G (r = 0.11 and r = 0.14, respectively). (Color version of figure is available online.)
DISCUSSION
The results of the present study demonstrate that in a sample population of critically ill surgical patients receiving TE chemoprophylaxis, delta reflects changes in thrombin generation measured by TMRTG. Of note, despite standard chemoprophylaxis, 60% of patients remained hypercoagulable as measured by total clot strength (G).
Our group recently validated that hypercoagulable G values were predictive of TE events in a cohort of critically ill surgical patients receiving standard chemoprophylaxis. In that study, logistic regression analysis demonstrated that for every 1 point increase in G there was a 25% increased risk of TE complications, when controlling for the presence of chemoprophylaxis [11]. Despite these findings, clear differentiation between the contributions of enzymatic and platelet components to hypercoagulability could not be accomplished based solely on G values, since G represents overall clot strength, reflective of both the platelet and enzymatic contributions. Although the routinely reported TEG parameter MA is often suggested to reflect clot formation, it measures the maximum amplitude of the clot formed, as measured in millimeters, while G is calculated in dynes/cm2, and as a unit of force, we believe this is a more accurate reflection of maximal platelet-fibrin interaction via GPIIb-IIIa receptors [18]. Delta’s linear correlation with TMRTG (Fig. 3) may allow for better identification of thrombin’s contribution to the clot. Further, recent studies have shown that TMRTG correlates with thrombin/anti-thrombin (TAT) levels [15]. Using delta as a measure of enzymatic activity may permit identification of the contributions of platelet reactivity to hypercoagulability in patients who are hypercoagulable by G but with a normal delta.
Current rates of TE events despite DVT chemoprophylaxis, as reported by Geerts et al. [6] and others [4, 5], calls for a broader more inclusive understanding of factors involved in hypercoagulability. While endothelial injury and platelet activation are known triggers for arterial thromboemboli [13], the sequence of events leading to VTE is less clear and little attention has been paid to the role of the platelet. According to the triad of Virchow, three major factors contribute to the development of VTE [12]: stasis of blood flow, hypercoagulability and a damaged endothelium. Several lines of evidence suggest that activation of platelets does indeed contribute to the development and propagation of venous thrombi [13]. In an analysis of 50 venous thrombi obtained from femoral valve pockets, Sevitt et al. showed areas that were characterized by fibrin and erythrocytes, mostly located in proximity of the endothelium, as well as areas dominated by a platelet–fibrin network, located more distally in the growing thrombus [20]. In early thrombus formation, scanning electron microscopy studies show aggregated platelets to be attached to venous endothelium [21]. Moreover, inhibition of P-selectin, a signaling molecule exposed on the surface of an activated platelet, which initiates inflammatory signaling pathways in underlying endothelium and recruits monocytes, results in impaired thrombus formation both in an experimental model of venous thrombosis and in vivo (22). Pharmacologic inhibition of platelet activation and aggregation could reduce the incidence and propagation of thrombus growth and lead to reduction of subsequent DVT and PE. Of note, current TE chemoprophylaxis indications are only based on risk stratification of patient populations [3], and do not include the presence, degree, or differentiation of hypercoagulability.
Current thromboprophylactic regimens using UH or LMWH appear to decrease thrombin levels through antithrombin potentiation and by direct factor Xa inhibition [23]. Given the multitude of factors that activate platelets [14], solely targeting final thrombin production by utilizing these regimens may not provide enough protection against platelet activation; hence, a hypercoagulable state may persist. In a multicenter study involving 12 medical and surgical ICUs where 65% of VTEs occurred in patients receiving standard pharmacologic chemoprophylaxis, Patel et al. concluded that most VTE’s occurred due to prophylaxis failure rather than failure to provide prophylaxis [4]. Although no TE events were reported in our study population, evidence exists that patients with increased platelet reactivity are at increased risk for venous thrombosis [26, 27].
Standard coagulation assays (PT/INR, aPTT) have not been useful as markers of hypercoagulability when compared to TEG [24]. Since they are performed on platelet poor plasma, these tests are sensitive only to the earliest initiation of clot formation. In contrast, more than 95% of thrombin generation takes place after the initial polymerization of fibrinogen [25]. Accordingly, TEG has emerged as a comprehensive rapid global assessment of haemostatic function from initial thrombin activation to fibrinolysis [7]. Further, TEG’s potential capacity to detect a hypercoagulable state via screening and thereby predict TE events has been reported [8, 11]. Sorensen et al. noted that thrombelastographic coagulation kinetics correlated with thrombin generation in vivo. They calculated the first derivative of changes in clot resistance expressed as a change in amplitude per unit of time (mm × 100/s), representing the maximum velocity ofclot formation [16]. Based on these derivatives, clot velocity curves (V-curves) for standard TEG tracings can be plotted utilizing standard TEG software. Rivard et al. demonstrated that the V-curve parameter TMRTG correlated with TAT levels [15], validating TMRTG as potential marker of thrombin generation. Delta is more simply obtained by subtracting SP from R time, and our results suggest a linear correlation between delta and TMRTG (Fig. 3). Thus, delta allows for an assessment of thrombin generation based on TEG parameters reported routinely in a rapid, user-friendly manner, and does not require plotting of V-curves to obtain TMRTG.
This pilot study has certain limitations. First, our study was not powered to detect incidence of TE events, rather we sought proof of concept that critically ill patients had a varying platelet and enzymatic contribution to their hypercoagulable state. Second, our sample was taken from a heterogeneous critically ill surgical population, which included trauma, general surgery, orthopedic surgery and neurosurgery patients. Finally, not all TEG samples were taken at the same time point, and coagulation profiles may vary according to anticoagulation activity at the time samples were obtained.
In summary, our preliminary findings suggest that the derived TEG parameter delta is an indicator of thrombin activity, allowing for differentiation of enzymatic from platelet hypercoagulability. If confirmed by larger studies, such a strategy could result in improved thromboprophylaxis regimens, utilizing anticoagulation, platelet inhibition or both. Accordingly, we have initiated a randomized, prospective trial evaluating the utility of TEG to screen for hypercoagulability and guide thromboprophylactic anticoagulation and platelet inhibition in surgical critically ill patients.
References
- 1.Attia J, Ray JG, Cook DJ, et al. Deep vein thrombosis and its prevention in critically ill patients. Arch Intern Med. 2001;161:1268. doi: 10.1001/archinte.161.10.1268. [DOI] [PubMed] [Google Scholar]
- 2.O’Malley KF, Ross SE. Pulmonary embolism in major trauma patients. J Trauma. 1990;30:748. doi: 10.1097/00005373-199006000-00018. [DOI] [PubMed] [Google Scholar]
- 3.Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: Evidence-based clinical practice guidelines, American College of Chest Physicians. Chest. 2008;133:381. doi: 10.1378/chest.08-0656. [DOI] [PubMed] [Google Scholar]
- 4.Patel R, Cook DJ, Meade MO, et al. Burden of illness in venous thromboembolism in critical care: A multicenter observational study. J Crit Care. 2005;20:341. doi: 10.1016/j.jcrc.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 5.Ibrahim EH, Irequi M, Prentice D, et al. Deep vein thrombosis during prolonged mechanical ventilation despite prophylaxis. Crit Care Med. 2002;30:771. doi: 10.1097/00003246-200204000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Geerts WH, Jay RM, Code KI, et al. A comparison of low-dose heparin with low molecular-weight heparin as prophylaxis against venous thromboembolism after major trauma. N Engl J Med. 1996;335:701. doi: 10.1056/NEJM199609053351003. [DOI] [PubMed] [Google Scholar]
- 7.Whitten CW, Greilich PE. Thromboelastography: Past, present, and future. Anesthesiology. 2000;92:1223. [PubMed] [Google Scholar]
- 8.Mahla E, Lang T, Vicenzi MN, et al. Thromboelastography for monitoring prolonged hypercoagulability after major abdominal surgery. Anesth Analg. 2001;92:572. doi: 10.1097/00000539-200103000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Spiel AO, Mayr FB, Firbas C, et al. Validation of rotation thrombelastography in a model of systemic activation of fibrinolysis and coagulation in humans. J Thromb Haemost. 2006;4:411. doi: 10.1111/j.1538-7836.2006.01715.x. [DOI] [PubMed] [Google Scholar]
- 10.Wilson D, Cooke EA, McNally MA, et al. Changes in coagulability as measured by thromboelastography following surgery for proximal femoral fracture. Injury. 2001;32:7650. doi: 10.1016/s0020-1383(01)00139-5. [DOI] [PubMed] [Google Scholar]
- 11.Kashuk JL, Moore EE, Sabel A, et al. Rapid thrombelastography (r-TEG) identifies hypercoagulability and predicts thromboembolic events in surgical patients. Surgery. 2009;146:764. doi: 10.1016/j.surg.2009.06.054. [DOI] [PubMed] [Google Scholar]
- 12.Virchow RLK, editor. Thrombosis and emboli (1846–1856) Canton, MA: Science History Publications; 1998. Thrombosis and Emboli. [Google Scholar]
- 13.Lopez JA, Kearon C, Lee AY. Deep venous thrombosis. Hematology (Am Soc Hematol Educ Program) 2004;1:439. doi: 10.1182/asheducation-2004.1.439. [DOI] [PubMed] [Google Scholar]
- 14.Furie B, Furie BL. Mechanisms of thrombus formation. N Eng J Med. 2008;359:938. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 15.Rivard GE, Brummel-Ziedins KE, Mann KG, et al. Evaluation of the profile of thrombin generation during the process of whole blood clotting as assessed by thrombelastography. J Thromb Haemost. 2005;3:2039. doi: 10.1111/j.1538-7836.2005.01513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorensen B, Johansen P, Christiansen K, et al. Whole blood coagulation thrombelastographic profiles employing minimal tissue factor activation. J Thromb Haemost. 2003;1:551. doi: 10.1046/j.1538-7836.2003.00075.x. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen VG. Beyond cell based models of coagulation: Analyses of coagulation with clot “lifespan” resistance-time relationships. Thromb Res. 2008;122:145. doi: 10.1016/j.thromres.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Khurana S, Mattson JC, Westley S, et al. Monitoring platelet glycoprotein IIb/IIIa-fibrin interaction with tissue factor-activated thromboelastography. J Lab Clin Med. 1997;130:401. doi: 10.1016/s0022-2143(97)90040-8. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen VG, Geary BT, Band MS. Evaluation of the contribution of platelets to clot strength by thrombelastography in rabbits: The role of tissue factor and cytochalasin D. Anesth Analg. 2000;91:35. doi: 10.1097/00000539-200007000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Sevitt S. Structure and growth of valve-pocket thrombi in femoral veins. J Clin Pathol. 1974;27:517. doi: 10.1136/jcp.27.7.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim Y, Nakase H, Nagata K, et al. Observation of arterial and venous thrombus formation by scanning and transmission electron microscopy. Acta Neurochir (Wien) 2004;146:45. doi: 10.1007/s00701-003-0156-5. [DOI] [PubMed] [Google Scholar]
- 22.Myers DD, Jr, Rectenwald JE, Bedard PW, et al. Decreased venous thrombosis with an oral inhibitor of P selectin. J Vasc Surg. 2005;42:329. doi: 10.1016/j.jvs.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 23.Weitz JI. Low-molecular-weight heparins. N Engl J Med. 1997;337:688. doi: 10.1056/NEJM199709043371007. [DOI] [PubMed] [Google Scholar]
- 24.Park MS, Martini WZ, Dubick MA, et al. Thromboelastography as a better indicator of hypercoagulable state after injury than prothrombin time or activated partial thromboplastin time. J Trauma. 2009;67:266. doi: 10.1097/TA.0b013e3181ae6f1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brummel KE, Paradis SG, Butenas S, et al. Thrombin functions during tissue factor-induced blood coagulation. Blood. 2002;100:148. doi: 10.1182/blood.v100.1.148. [DOI] [PubMed] [Google Scholar]
- 26.Martinelli I, Bucciarelli P, Mannucci PM. Thrombotic risk factors: basic pathophysiology. Crit Care Med. 2010;38(2 Suppl):S3. doi: 10.1097/CCM.0b013e3181c9cbd9. [DOI] [PubMed] [Google Scholar]
- 27.Slotta JE, Braun OO, Menger MD, et al. Capture of platelets to the endothelium of the femoral vein is mediated by CD62P and CD162. Platelets. 2009;20:505. doi: 10.3109/09537100903215417. [DOI] [PubMed] [Google Scholar]