

Abstract
We investigate several approaches to coarse grained normal mode analysis on protein residual-level structural fluctuations by choosing different ways of representing the residues and the forces among them. Single-atom representations using the backbone atoms Cα, C, N, and Cβ are considered. Combinations of some of these atoms are also tested. The force constants between the representative atoms are extracted from the Hessian matrix of the energy function and served as the force constants between the corresponding residues. The residue mean-square-fluctuations and their correlations with the experimental B-factors are calculated for a large set of proteins. The results are compared with all-atom normal mode analysis and the residue-level Gaussian Network Model. The coarse-grained methods perform more efficiently than all-atom normal mode analysis, while their B-factor correlations are also higher. Their B-factor correlations are comparable with those estimated by the Gaussian Network Model and in many cases better. The extracted force constants are surveyed for different pairs of residues with different numbers of separation residues in sequence. The statistical averages are used to build a refined Gaussian Network Model, which is able to predict residue-level structural fluctuations significantly better than the conventional Gaussian Network Model in many test cases.
Keywords: Protein structural fluctuation, Normal mode analysis, Gaussian Network Model, Atomic B-factors, Atomic mean-square-fluctuations, Residue mean-square-fluctuations
1 Introduction
Proteins are biomolecules, each formed by a chain of small molecules called amino acids. There are total 20 different amino acids. They can make many different chains of amino acids, and hence different proteins. There are hundreds of thousands of different proteins, with distinct biological functions supporting diverse biological forms and processes. They are key molecular elements of life and are fundamental research subjects of life sciences (Berg et al. 2006).
The biological function of a protein is determined by the protein’s 3D structure. The 3D structure is determined by the protein’s amino acid sequence. In other words, a given protein, with a given amino acid sequence, always assumes a certain 3D structure, which then helps to perform certain functions. Therefore, the 3D structure carries important information for the understanding of the protein and in particular, its sequence-structure-function relations (Berg et al. 2006).
While a protein folds to an unique structure, it also fluctuates and makes thermodynamic movements. The movements can be different for different atoms or residues or fragments of the protein, and may correspond to certain functional differences. Therefore, the determination of the structural fluctuation is an equally important issue as the determination of the structure itself in protein modeling. In X-ray crystallography, the structural fluctuation is measured by the so-called atomic B-factors, which are proportional to the atomic mean-square-fluctuations (Drenth 2006).
In contrast to structural determination, which is mainly through physical experiments, such as X-ray crystallography and NMR, the structural fluctuation can be analyzed theoretically for a given structure. For example, it can be traced by molecular dynamics simulation in the force field of the protein. It can in particular be determined in an analytical form using the so-called normal mode analysis (NMA) (Brooks et al. 1989; Schlick 2002).
The normal mode analysis usually requires a singular-value-decomposition of the Hessian matrix of the energy function, which is costly for an all-atom model, yet the estimation on the fluctuation may not necessarily always accurate, because of the errors in the force field approximation. A simplified residue-level model called the Gaussian Network Model (GNM) has been proposed in recent years and proved to be more efficient as well as accurate than NMA (Micheletti et al. 2004; Cui and Bahar 2006).
In this paper, we investigate several new approaches to residue-level normal mode analysis. Similar to GNM, we take the residues as the basic units in a protein and use a single backbone atom (or a combination of several backbone atoms) to represent each residue. Different from GNM, the force constants for pairs of representative atoms are not the same and are instead extracted from the Hessian matrix of the energy function. Therefore, the models can be considered as coarse-grained NMA.
Using the new models, we calculate the mean-square-fluctuations of the residues and their correlations with the experimental B-factors (called the B-factor correlations) for a large set of proteins. We compare them with the all-atom normal mode analysis and the residue-level Gaussian Network Model. We show that our models perform more efficiently than the all-atom normal mode analysis, and the B-factor correlations are also higher. The B-factor correlations are comparable with those estimated by the Gaussian Network Model and in some cases better.
Following the development of the coarse-grained NMA, we conduct a statistical survey on the extracted force constants, for different pairs of residues with different numbers of separation residues in sequence. We then base on their statistical averages to build a refined Gaussian Network Model. We show that the force constants for the neighboring residue pairs are always about one-magnitude larger than other pairs in contact. Therefore, in the refined GNM, the entries of the contact matrix can be defined in the same way as the conventional GNM except that the bi-diagonal entries have large magnitudes. We show that such a simply refined GNM can predict residue-level structural fluctuations significantly better than the conventional GNM in many test cases.
The paper is organized as follows. In Section 2, we provide a general background on normal mode analysis. We describe the system of equations of motion and its linear approximation, and derive the solution to the linear system for normal mode analysis. In Section 3, we introduce the Gaussian Network Model, and discuss its relationship with normal mode analysis. In Section 4, we describe the coarse-grained NMA and present the test results. In Section 5, we discuss the statistics of the force constants. We then describe the refined GNM and show the test results. We make concluding remarks in Sect. 6. Sections 2 and 3 are expository sections. They do not contain new insights into NMA or GNM. However, they do provide a more rigorous mathematical description on these topics than most in current literature, and show mathematically the relationship between the two approaches. Sections 4 and 5 describe the methods of coarse-grained NMA and refined GNM and the test results, and are the sections with new contributions.
2 Normal Mode Analysis
A normal mode is a pattern of motion of an oscillating system in which all parts of system move sinusoidally with the same frequency and a fixed phase relation. An oscillating system may have multiple normal modes. The motion of the system is then a collecting result of all its normal mode motions (Morin 2008). For example, a mass-spring system, i.e., a set of objects connected with springs, would vibrate with different modes corresponding to different amplitudes and frequencies, even when the masses of the objects and the force constants of the springs are all the same.
A protein structure fluctuates around its native state. In other words, the atoms in the protein vibrate around their equilibrium positions, as if they all are connected by some sort of springs. Therefore, protein structural fluctuations can be estimated approximately via normal mode analysis as well. Brooks and Karplus (1983) and Go et al. (1983) are among the first who applied the normal mode analysis to study the structural fluctuations of a small protein bovine pancreatic trypsin inhibitor (BPTI). Levitt et al. (1985) later performed a detailed analysis on larger proteins including BPTI, Crambin, Ribonuclease, Lysozyme. Ever since, the method has been used widely for routine structural analysis in protein modeling (Micheletti et al. 2004; Cui and Bahar 2006).
NMA assumes the protein in a stable state, where the potential energy has reached an energy minimum. Therefore, around this state, the potential energy can be approximated by a quadratic function. The system of equations of motion for the protein can then be solved in an analytical form, and the normal modes of the motion can be extracted from the solution for analysis of atomic vibrations as well as overall structural fluctuations. We provide a brief description on the system of equations of motion used in classical molecular dynamics simulation, the closed form solution to the approximated system, and the formulas for computing the atomic vibrations.
Lemma 2.1
Suppose that a biological molecule has n atoms. Let r(t) be the collection of the coordinates of the atoms in the given molecule at time t such that r(t) = {ri(t) : i = 1,…, 3n}, where r3j−2(t), r3j−1(t), r3j(t) are the x, y, z coordinates of atom j at time t, j = 1,…, n. Let Ep(r) be the potential energy function. Then the molecular motion can be described as a collection of movements of the atoms in the molecule, as given in the following system of equations of motion:
| (1) |
where M is a diagonal matrix with the diagonal element mii being the mass of atom i. Proof Let the Lagrangian of the physical system for the molecule be defined as the following:
Then
and
Based on the Euler–Lagrange equation,
we then have
The potential energy function Ep(r) is usually given in a complicated nonlinear form. Therefore, the system of (1) can only be solved numerically. While many methods have been developed for the solution of the system, they all are computationally costly even for the calculation of very short period trajectories (say in nanoseconds), because they have to use very small time steps (in femtoseconds) to match the fast atomic motions so they can keep the accuracy of the calculations (Brooks et al. 1989; Schlick 2002).
For normal mode analysis, the potential energy function is approximated by a quadratic function at its energy minimum. Let r0 be the minimum energy state, and assume without loss of generality that E(r0) = 0. Then
Let Δr = r − r0. The function can also be written as
| (2) |
By using this approximation, the system of (1) becomes
That is,
| (3) |
The above equation can be solved analytically via the singular-value-decomposition of the Hessian matrix ▿2Ep(r0). Suppose that the molecule has n atoms. Then the dimension of the Hessian matrix is 3n by 3n and has 3n singular-value and singular-vector pairs. Among them, the first 6 values corresponding to 3 translational and 3 rotational motions are equal to zeros, while the remaining 3n − 6 defines 3n − 6 normal modes. The detailed formulas for obtaining the normal modes are given in the following lemma.
Lemma 2.2
Suppose that the singular-value-decomposition of the mass-weighted Hessian matrix, H = M−1▿2Ep(r0), is given by UʌUT . Then the solution of the system of (3) is given by the following functions:
| (4) |
where αj and βj can be determined by the given conditions of the system. Proof Let H = M−1▿2Ep(r0). Then
and
Let . Then
and
proving that
is a solution to the system of (3), where αj is called the amplitude, ωj the angular frequency, and βj the phase of the jth normal-mode of motion.
Now let qj = αj cos(ωjt + βj). Then
Also,
Then the time-averaged potential energy for each mode is
Since at the thermal equilibrium, the averaged potential energy should be equal to the kinetic energy kBT/2 for each mode, we then have
| (5) |
Once the solution f’:r(t) for the system of (3) is obtained, the average fluctuation of r(t) from its equilibrium position r0(t) can be calculated using a simple formula, as given in the following theorem.
Theorem 2.3
Let the solution to the system of (3) be given by . Let UʌUT be the singular-value-decomposition of the mass-weighted Hessian matrix H = M−1▿2Ep(r0). Then the mean-square-fluctuation of the ith mode motion of the molecule is
| (6) |
Proof
If we use νj(t) to represent the position vector of atom j at time t, then νj(t) = (r3j−2(t), r3j−1(t), r3j(t))T, and the mean-square-fluctuation of atom j can be calculated as
| (7) |
Note that if the singular value ʌjj is small, then the frequency ωj is small and, therefore the corresponding mode is slow. Thus, the smaller the singular value, the slower the corresponding mode; and the slower a mode is, the larger mean-square-fluctuation it makes. Also, the atomic mean-square-fluctuation is usually related to the experimentally detected average atomic fluctuation, which, in X-ray crystallography, is represented by the so-called temperature factor or B-factor. Let the B-factor for atomj be denoted by Bj. Then
| (8) |
From equations (6), (7), and (8) above, we see that the B-factors are proportional to the atomic mean-square-fluctuations, and thus inversely proportional to all the singular values ʌjj (or square frequency ωj2). Therefore, the largest contributions to the atomic displacements come from the lowest frequency normal modes (small ʌjj or ωj2). In addition, the singular vectors corresponding to the lowest frequency normal modes represent the most globally distributed or collective motions. For these reasons, studies on NMA usually focus on only a few low frequency normal modes, and in real applications, only several terms in (6) corresponding to the smallest singular values are used for the evaluation of the atomic mean-square-fluctuations.
3 Gaussian Network Model
NMA requires the availability of the potential energy function and the computation of the Hessian matrix of the function. Tirion (1996) demonstrated that an approximation to the Hessian can be obtained by using a single parameter for all the atomic pairs in a close distance. The analysis with such an approximation can be as accurate as using the exact Hessian. Along this line, Bahar et al. (1997, 1998), Haliloglu et al. (1997) later made further simplification on conventional normal mode analysis using the Gaussian Network Model (GNM). In this model, a protein structure is described at a coarse level with the amino acid residues as the basic units and is considered as an elastic network with the residues as nodes and the contact connections among them as springs. Each residue can be represented by one of its backbone atoms, say Cα, or a selected point in the residue, say the geometric center of the side-chain. Two residues are said to be in contact if they are close in distance (usually, in a 7 Å cutoff distance). All the springs are assumed to be the same, i.e., have the same force constant. Figure 1 shows an example elastic network model for a protein structure 1HEL.
Fig. 1.
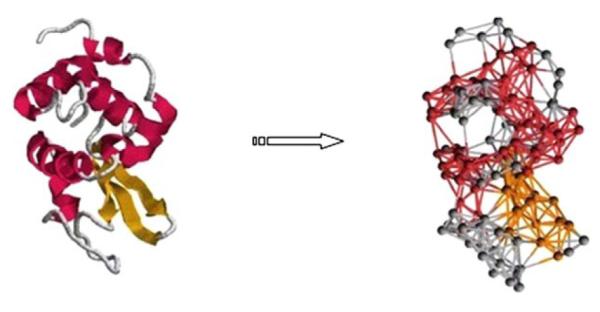
The picture on the left shows the Cα trace of 1HEL. The one on the right shows all connections between Cα nodes for 1HEL to indicate the nature of the elastic network analyzed by GNM (Cui and Bahar 2006).
Once an elastic network is constructed, the energy function for the protein can be defined without referring to the protein’s atomic level potentials: Assume that the protein has m residues. Let r be the collection of the coordinates of the residues in the protein such that r = {ri : i = 1,…, 3m}, where r3i−2, r3i−1, r3i are the x, y, z coordinates of residue i. Correspondingly, let ν be the collection of the coordinate vectors of the residues such that ν = {νi = (νi1, νi2, νi3)T = (r3i−2, r3i−1, r3i)T : i = 1,…, m}. Let Δνi = νi - ν0 be the displacement of residue i from its equilibrium position ν0 and kij be the force constant for the spring between residues i and j. Then the total force on residue i would be
and the potential energy contributed from residue i would be
we then have the total potential energy of the protein to be
| (9) |
Since in GNM, the spring forces are kept constant and are zeros if the distances between any pairs of residues are larger than 7 Å, we can define kij = kΓij for some constant k and
| (10) |
Here, Γ = {Γij : i,j = 1,…, m} form a matrix called the contact matrix. Let Δνj = {Δνij : i = 1,…, m}, j = 1, 2, 3. Then the energy function in (9) can be written as
| (11) |
An example contact matrix can be found in Fig. 2.
Fig. 2.

This is an example of a contact matrix.
Based on the theory of statistical physics, at a given temperature, the probability of a physical state r to occur is subject to the Gibbs–Boltzmann distribution
| (12) |
where kB is the Boltzmann constant, T the temperature, and Z the partition function.
Given the energy function in (11) and the fact that ∫R3m p(r)dr = ∫R3m p(r)dΔr = 1, it follows that
By changing all the variables Δνl to Δu, we then have
Let Γ = UʌUT be the singular-value-decomposition of Γ. Then
| (13) |
where Δω = UT Δu. Then , with
| (14) |
Note that given the probability distribution of a physical state, p(r), we can evaluate the expectation value of any property related to this physical state, f(r), using the integral ∫R3m f(r)p(r)dr It follows that if we consider the fluctuation of residue , as a function f(r) of state r, we can calculate the mean-square-fluctuation using the integral . We give a formula for the calculation of this integral in the following theorem.
Theorem 3.1
Let the potential energy function Ep(r) for a given protein be defined as (11). Let the singular-value-decomposition of Γ be given by UʌUT . Then the mean-square-fluctuation (Δνi, Δνi) can be calculated by the formula:
| (15) |
where k is a force constant, kB the Boltzmann constant, and T the absolute temperature. Proof Note that for any i = 1,…, m,
Since and Z = Z’3 by (13),
By changing all the variables Δνl to Δu, we then have
Let Δu = UΔω. Then
Note that if j ≠ k for ΔωjΔωk,
But if j = k for ΔωjΔωk,
It follows that for any i = 1,…, m
Note that the formula for calculating the mean-square-fluctuations of the residues in (15) is very similar to that for calculating the mean-square-fluctuations of the atoms in (6). It is not a coincident because GNM can indeed be considered as residue level NMA with the energy function approximated by using the contact matrix as the Hessian matrix. In other words, if we consider the function Ep(r) in (11) to be a harmonic approximation to the potential energy function for the protein at the residue level, we can perform NMA for the protein at its residue level in the same way as that at the atomic level. We provide some further justifications in the following.
Note that the energy function Ep(r) in (11) is symmetric with respect to Δνl, l = 1, 2, 3. Therefore, the system of equations of motion for the residues can be reduced to three independent and identical subsystems:
| (16) |
Theorem 3.2
Consider Ep(r) in (11) to be a harmonic approximation to the potential energy function of the protein at an equilibrium state r0. Let M be the mass matrix of the residues and H = M−1Γ. Then, the solution to the system of equations of motion for the residues in (16) is given by
| (17) |
where αj and βj can be determined by the given conditions of the system.
Proof Since the system of equations of motion can be reduced to three independent and identical subsystems, we only need to solve any of them, which can be put into the following general form:
Let H = M−1Γ. Let UʌUT be the singular value decomposition of H. Then, by using a similar argument for Lemma 2.2, we can have
meaning that
Based on the argument that at the thermal equilibrium, the averaged potential energy should be equal to the kinetic energy kBT/2 for each mode, we then have
| (18) |
Therefore, we can have the following theorem for the calculation of the residue mean-square-fluctuations.
Theorem 3.3
Let the solution to the system of (16) be given by . Let UʌUT be the singular-value-decomposition of the matrix H = M−1Γ. Then the mean-square-fluctuations of the residues can be calculated by the formula,
| (19) |
Proof Based on the discussion in Theorem 3.2,
with . Then
proving that
Note that if M is an identity matrix, then H = Γ and the formula in (19) for calculating the mean-square-fluctuations of the residues using NMA will be the same as that in (15) using GNM. In other words, GNM for calculating the residue mean-square-fluctuations is equivalent to NMA with the contact matrix used as the Hessian approximation and with all the residues assumed to have a unit mass.
4 Coarse-Grained Normal Mode Analysis
As described in Sect. 2, in classical NMA, the system of equations of motion is solved with a quadratic approximation to the potential energy function. This approximation is accurate enough only in a small neighborhood of the energy minimum state (or structure). It also depends on the original potential energy function, which itself is an approximation obtained semi-empirically (Brooks et al. 1989; Schlick 2002). In addition, the classical NMA is usually conducted at the atomic level, which is subject to all the atomic level errors induced in energy calculations. As a result, the structural fluctuations predicted by NMA are often even worse than those by GNM, which is in fact a coarser model than NMA (Micheletti et al. 2004; Cui and Bahar 2006).
As analyzed in Sect. 3, the GNM method can be considered as a type of residue level NMA. In GNM, a residue contact matrix is defined using the distances between the residues (the distances between the representative atoms Cα, N, C, or Cβ). If the distance between two different residues is less or greater than the given cutoff value, say 7 Å, then a constant, −1 or 0 (or more accurately, −k or 0), is allocated for the entry in the matrix, respectively. In other words, constants are assigned to the entries throughout the matrix according to the contact distances. The advantage of using GNM is that the model is simple and easy to construct, the dimension of the model is much smaller (proportional to the number of residues) than atomic level NMA, and the computation is efficient. In addition, GNM does not require the exact potential energy and the Hessian, reducing not only computational cost but also possible errors in atomic energy calculations.
Physically speaking, in GNM, the protein is viewed as an elastic network with the residues as nodes and the contact distances as the links. The links are approximated by springs and assigned with some spring constants. In other words, the forces between the residues in contact are approximated by some harmonic forces. Mathematically speaking, this is equivalent to saying that a quadratic approximation is used to represent the potential energy for the residue interactions of the protein. As a first approximation, in GNM, a single spring constant is assigned to all the residue pairs in contact. However, different pairs of residues may interact differently with different force constants. A more accurate model may be built if these differences can be considered.
Here, we investigate an alternative approach to coarse-grained normal mode analysis. Instead of using a homogeneous force constant for all the interactions among the residues in contact, we assign different force constants for different types of interactions in terms of related residue types and distances. In a certain sense, it can also be considered as a refined, nonhomogeneous Gaussian Network Model. There have been several efforts made in the past to refine GNM: For example, Taner and Jernigan (2006) defined the force constants in terms of the residue contact distances, and Kondrashov et al. (2006) assigned different force constants for residue pairs of different sequence distances. We derive the force constants from the atomic level Hessian matrix of the potential energy function. Therefore, our approach can be considered to be more physics-based.
Much work has been done in the past on coarse-grained normal mode analysis at different coarse levels and with different calculation schemes. Early work includes using graph theoretical methods to estimate protein shapes and structural flexibility by Mitchell et al. (2001) and by Jacobs et al. (2001). Tama et al. (2000), and Li and Cui (2002) proposed an approach to use residue fragments as building blocks for computing low frequency normal modes. Approaches have been studied using rigid components of proteins as basic units for normal mode analysis (Schuyler and Chirikjian 2004; Ahmed and Gohlke 2006; Demerdash and Mitchell 2012). Work has also been done in Lu and Ma (2008, 2011) with the Hessian elements derived from all-atom normal mode analysis and some universal force constants. Our method differs from these approaches in using the Hessian matrix of the energy function to extract the force constants for residue interactions and further determine them based on their probability distributions in a large set of know protein structures.
For a given structure, we first perform sufficient steps of energy minimization in an atomic-level force field. Once an energy minimum is reached, we save the Hessian matrix of the energy function and use it to derive the force constants for residue interactions. Let Ri and Rj be two residues represented by two atoms Ai and Aj, where Ai and Aj must have two position vectors (νi1, νi2, νi3)T and (νj1, νj2, νj3)T and correspond to a 3 by 3 submatrix S(ij) of the Hessian H. We then define the force constant for residues Ri and Rj to be the average value of the entries in S(ij). In other words, if kij is the force constant for residues Ri and Rj, then kij = − ∥ S(ij) ∥ F, where ∥ · ∥ F is the matrix Frobenius norm. More precisely,
where m is the number of residues in the protein. Then, the matrix Γ = {kij : i,j = 1,…, m} can serve as a reduced Hessian matrix for coarse-grained NMA.
We have applied this new model to a set of protein structures downloaded from the PDB (Berman et al. 2010). The resolutions of the structures are 1.5 Å or higher and the sequence similarity is 30 % or lower. The sizes of the structures are from small to large, with the numbers of atoms ranging from 35 to 2387. We have employed the CHARMM version 27b2 (Brooks et al. 2009) for energy minimization and implemented a Matlab NMA code for residue normal mode calculations. For each structure, we have used different representative atoms Cα, N, C, or Cβ for residues, to obtain the reduced Hessian. We have computed the residue mean-square-fluctuations and their correlations with the B-factors of the corresponding representative atoms of the residues (B-factor correlations). We call our model the coarse-grained NMA or cgNMA for short. A cgNMA with Cα, N, C, or Cβ as the representative atoms for the residues is denoted as cgNMA(Cα), cgNMA(N), cgNMA(C), cgNMA(Cβ), respectively.
For comparison, we have also computed the residue mean-square-fluctuations and their B-factor correlations for each structure, using GNM with Cα, N, C, Cβ as the representative atoms for the residues. A GNM with Cα, N, C, or Cβ as the representative atoms for the residues is denoted as GNM(Cα), GNM(N), GNM(C), GNM(Cβ), respectively. An atomic level NMA has also been performed for each structure with the CHARMM NMA routine. The mean-square-fluctuations of Cα atoms are recorded and compared with their experimental B-factors as well.
A more sophisticated cgNMA is to use a set of atoms to represent each residue. Let two residues Ri and Rj be represented by the same types of N atoms, Ai1,…, AiN and Aj1 …, AjN, respectively. Let S(ij) be the 3N by 3N submatrix of the Hessian matrix H corresponding to the representative atoms of Ri and Rj. Wereduce S(ij) to a 3 by 3 matrix T(ij), with
We then define the force constant kij for residues Ri and Rj to be the average value of the entries in T(ij), i.e.,
where m is the number of residues in the protein. We denote this model by cgNMA(M), meaning the coarse-grained NMA with multiple representative atoms. In particular, we have used Cα, N, C for cgNMA(M) in our test.
We have divided the test structures into three groups of relatively small-,medium-, and large-sized structures. We then summarize the test results for each of them separately. Tables 1, 2, and 3 show the test results on small, medium, and large structures, respectively. In these tables, we have listed the names of the proteins (ID), the numbers of the atoms and residues in the proteins (TA and TR), and the B-factor correlations of the computed residue mean-square-fluctuations for the structures using different methods. The methods include GNM with different representative atoms, GNM(Cα), GNM(N), GNM(C), GNM(Cβ), the coarse-grained NMA with different representative atoms, cgNMA(Cα), cgNMA(N), cgNMA(C), cgNMA(Cβ), cgNMA(M), and classical NMA using CHARMM. The B-factor correlations are calculated against the B-factors of the representative atoms except for NMA for which the B-factors of Cα atoms are compared with. For cgNMA(M), the averages of fluctuations of representative atoms of residues are compared with the averages of B-factors of representative atoms of residues.
Table 1.
B-factor correlations for small-sized structuresa
| ID | TA | TR | GNM(Cα) | GNM(N) | GNM(C) | GNM(Cβ) | cgNMA(Cα) | cgNMA(N) | cgNMA(C) | cgNMA(Cβ) | cgNMA(M) | NMA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2OLX | 35 | 4 | 0.885 | 0.857 | 0.410 | 0.925 | 0.776 | 0.867 | 0.664 | 0.859 | 0.738 | −0.933 |
| 2OL9 | 51 | 6 | 0.689 | 0.970 | 0.831 | 0.741 | 0.886 | 0.989 | 0.898 | 0.722 | 0.965 | 0.881 |
| 1YJO | 55 | 6 | 0.434 | 0.568 | 0.543 | −0.054 | 0.445 | 0.238 | 0.524 | 0.549 | 0.423 | −0.149 |
| 1XY2 | 69 | 8 | 0.562 | 0.474 | 0.354 | 0.596 | 0.458 | −0.104 | 0.722 | 0.507 | 0.771 | 0.081 |
| 1NOT | 96 | 13 | 0.523 | 0.508 | 0.305 | −0.005 | 0.567 | 0.971 | 0.405 | 0.010 | 0.814 | 0.056 |
| 1PEN | 109 | 16 | 0.270 | 0.406 | −0.099 | 0.149 | 0.056 | 0.707 | −0.164 | −0.033 | 0.193 | 0.699 |
| 1OB4 | 110 | 5 | −0.744 | −0.721 | −0.402 | NaN | 0.930 | 0.956 | −0.091 | 0.845 | 0.543 | 0.463 |
| 1OB7 | 111 | 5 | −0.463 | −0.371 | −0.711 | NaN | 0.952 | 0.843 | 0.947 | 0.381 | 0.926 | −0.001 |
| 1AKG | 112 | 16 | 0.185 | 0.483 | 0.225 | 0.593 | −0.229 | 0.621 | 0.025 | 0.141 | 0.071 | −0.027 |
| 1KYC | 138 | 15 | 0.754 | 0.863 | 0.625 | 0.586 | 0.784 | 0.538 | 0.725 | 0.664 | 0.706 | 0.600 |
| 1ETN | 147 | 12 | −0.274 | −0.107 | −0.377 | 0.288 | −0.537 | 0.019 | −0.298 | −0.358 | −0.490 | 0.463 |
| 1ETL | 147 | 12 | 0.628 | 0.658 | 0.214 | 0.655 | 0.355 | 0.540 | 0.583 | −0.258 | 0.606 | 0.847 |
| 1PEF | 153 | 18 | 0.808 | 0.810 | 0.828 | 0.710 | 0.888 | 0.897 | 0.701 | 0.761 | 0.790 | 0.609 |
| 1ETM | 160 | 12 | 0.432 | 0.365 | −0.135 | 0.287 | 0.027 | 0.496 | 0.400 | 0.081 | 0.462 | 0.896 |
| 1O06 | 172 | 20 | 0.844 | 0.881 | 0.782 | 0.812 | 0.900 | 0.779 | 0.873 | 0.488 | 0.923 | 0.750 |
| 1HJE | 214 | 13 | 0.616 | 0.526 | 0.701 | 0.057 | 0.562 | 0.741 | 0.686 | 0.652 | 0.838 | 0.209 |
| 1P9I | 225 | 29 | 0.625 | 0.678 | 0.606 | 0.524 | 0.603 | 0.805 | 0.885 | 0.092 | 0.813 | 0.405 |
| 2JKU | 255 | 35 | 0.656 | 0.689 | 0.487 | 0.515 | 0.850 | 0.480 | 0.647 | 0.396 | 0.654 | 0.698 |
| 1RJU | 257 | 36 | 0.431 | 0.425 | 0.367 | 0.285 | 0.235 | −0.070 | 0.358 | 0.168 | 0.274 | −0.041 |
| 2NLS | 273 | 36 | 0.530 | 0.658 | 0.507 | 0.384 | 0.088 | 0.653 | 0.618 | 0.233 | 0.701 | 0.430 |
| 1VRZ | 277 | 13 | 0.327 | 0.420 | −0.193 | 0.313 | −0.203 | 0.511 | 0.398 | −0.157 | 0.271 | 0.807 |
| 1AIE | 295 | 31 | 0.155 | 0.250 | 0.178 | 0.365 | 0.712 | 0.447 | 0.566 | 0.322 | 0.584 | 0.670 |
| 1GK7 | 335 | 39 | 0.821 | 0.790 | 0.872 | 0.748 | 0.822 | 0.763 | 0.806 | 0.670 | 0.808 | 0.690 |
| 1Q9B | 345 | 43 | 0.656 | 0.699 | 0.864 | 0.509 | 0.646 | 0.614 | 0.830 | 0.291 | 0.789 | 0.570 |
| 1YZM | 361 | 46 | 0.901 | 0.853 | 0.900 | 0.789 | 0.939 | 0.938 | 0.849 | 0.549 | 0.925 | 0.472 |
| 6RXN | 367 | 45 | 0.594 | 0.497 | 0.593 | 0.487 | 0.304 | 0.369 | 0.332 | 0.216 | 0.415 | 0.254 |
| 1USE | 382 | 40 | −0.142 | 0.006 | −0.174 | −0.230 | −0.399 | −0.003 | −0.132 | 0.075 | −0.236 | 0.384 |
| 1BX7 | 387 | 51 | 0.706 | 0.639 | 0.717 | 0.478 | 0.868 | 0.417 | 0.653 | 0.221 | 0.637 | 0.507 |
| 2DSX | 399 | 52 | 0.127 | −0.010 | −0.046 | 0.272 | 0.433 | 0.407 | 0.536 | 0.107 | 0.487 | −0.079 |
| 1UOY | 452 | 64 | 0.671 | 0.674 | 0.630 | 0.053 | 0.628 | 0.654 | 0.719 | 0.760 | 0.746 | 0.609 |
| 1U06 | 470 | 55 | 0.434 | 0.422 | 0.544 | 0.492 | 0.377 | 0.285 | 0.374 | 0.111 | 0.322 | 0.553 |
| 1GVD | 491 | 52 | 0.591 | 0.316 | 0.555 | 0.772 | 0.570 | 0.658 | 0.808 | 0.579 | 0.747 | 0.421 |
| 1FF4 | 503 | 65 | 0.674 | 0.651 | 0.624 | 0.578 | 0.555 | 0.314 | 0.509 | 0.700 | 0.512 | 0.564 |
ID-protein ID, TA-total number of atoms, TR-total number of residues, GNM(Cα, N, C, Cβ)—residue B-factor correlations using GNM with different choices of representative atoms, cgNMA(Cα, N, C, Cβ)—residue B-factor correlations using cgNMA with different choices of representative atoms, cgNMA(M)—residue B-factor correlations using cgNMA with a combination of Cα, N, C atoms as the representative atom, NMA—correlations using NMA
Table 2.
Factor correlations for medium-sized structuresa
| ID | TA | TR | GNM(Cα) | GNM(N) | GNM(C) | GNM(Cβ) | cgNMA(Cα) | cgNMA(N) | cgNMA(C) | cgNMA(Cβ) | cgNMA(M) | NMA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1LR7 | 522 | 73 | 0.620 | 0.667 | 0.751 | 0.624 | 0.795 | 0.704 | 0.628 | 0.449 | 0.712 | 0.629 |
| 1ZVA | 551 | 75 | 0.690 | −0.078 | 0.510 | 0.775 | 0.579 | 0.736 | 0.845 | 0.312 | 0.812 | 0.714 |
| 2BF9 | 562 | 35 | 0.419 | 0.249 | −0.021 | 0.147 | 0.521 | 0.556 | 0.840 | 0.197 | 0.762 | 0.367 |
| 2EHS | 598 | 75 | 0.747 | 0.269 | 0.302 | 0.570 | 0.565 | 0.717 | 0.720 | 0.386 | 0.761 | 0.330 |
| 1UHA | 623 | 82 | 0.583 | 0.491 | 0.463 | 0.540 | 0.308 | 0.568 | 0.387 | 0.216 | 0.498 | 0.749 |
| 1FK5 | 626 | 93 | 0.485 | 0.414 | 0.378 | 0.441 | 0.362 | 0.554 | 0.516 | 0.489 | 0.539 | 0.351 |
| 1OPD | 642 | 85 | 0.398 | 0.559 | 0.382 | 0.317 | 0.796 | 0.298 | 0.255 | 0.072 | 0.355 | 0.372 |
| 1X3O | 642 | 80 | 0.654 | 0.606 | 0.694 | 0.550 | 0.453 | 0.409 | 0.514 | 0.339 | 0.542 | 0.177 |
| 2E3H | 651 | 81 | 0.605 | 0.775 | 0.652 | 0.540 | 0.632 | 0.654 | 0.665 | 0.488 | 0.741 | 0.680 |
| 1USM | 661 | 77 | 0.798 | 0.669 | 0.738 | 0.719 | 0.780 | 0.761 | 0.840 | 0.350 | 0.865 | 0.613 |
| 1ULR | 677 | 87 | 0.495 | 0.387 | 0.354 | 0.548 | 0.223 | 0.421 | 0.648 | 0.482 | 0.695 | 0.561 |
| 1I71 | 683 | 83 | 0.549 | 0.608 | 0.466 | 0.334 | 0.380 | 0.355 | 0.325 | 0.283 | 0.334 | 0.353 |
| 2PKT | 688 | 91 | −0.286 | −0.209 | −0.288 | −0.042 | −0.165 | −0.220 | −0.279 | −0.257 | −0.247 | −0.245 |
| 1N7E | 700 | 95 | 0.497 | 0.511 | 0.454 | 0.431 | 0.385 | 0.583 | 0.409 | 0.074 | 0.534 | 0.301 |
| 2IP6 | 702 | 87 | 0.572 | 0.404 | 0.549 | 0.493 | 0.826 | 0.644 | 0.772 | 0.473 | 0.733 | 0.322 |
| 2EAQ | 705 | 89 | 0.695 | 0.631 | 0.659 | 0.483 | 0.688 | 0.622 | 0.743 | 0.336 | 0.775 | 0.707 |
| 2FQ3 | 721 | 85 | 0.348 | 0.270 | 0.190 | 0.549 | 0.508 | 0.659 | 0.697 | 0.290 | 0.679 | 0.609 |
| 2RB8 | 723 | 93 | 0.517 | 0.432 | 0.530 | 0.653 | 0.485 | 0.314 | 0.530 | 0.269 | 0.515 | 0.292 |
| 1GXU | 726 | 88 | 0.421 | 0.388 | 0.288 | 0.557 | 0.581 | 0.825 | 0.643 | 0.530 | 0.762 | 0.756 |
| 2PLT | 727 | 98 | 0.481 | 0.360 | 0.410 | 0.161 | 0.187 | 0.450 | 0.493 | 0.058 | 0.543 | 0.248 |
| 1ABA | 728 | 87 | 0.613 | 0.556 | 0.547 | 0.554 | 0.057 | 0.602 | 0.636 | 0.438 | 0.649 | 0.648 |
| 1R7J | 729 | 90 | 0.368 | 0.377 | 0.405 | 0.489 | 0.078 | 0.277 | 0.344 | 0.325 | 0.244 | 0.4 |
| 1V05 | 731 | 96 | 0.632 | 0.670 | 0.662 | 0.511 | 0.389 | 0.382 | 0.506 | 0.166 | 0.482 | 0.5 |
| 1CYO | 731 | 88 | 0.741 | 0.800 | 0.807 | 0.682 | 0.774 | 0.683 | 0.803 | 0.618 | 0.763 | 0.694 |
| 3BZQ | 742 | 99 | 0.466 | 0.344 | 0.356 | 0.562 | 0.351 | 0.176 | 0.110 | 0.057 | 0.167 | 0.492 |
| 1W2L | 746 | 97 | 0.397 | 0.421 | 0.284 | 0.432 | 0.432 | 0.611 | 0.582 | 0.117 | 0.647 | 0.130 |
| 2MCM | 769 | 112 | 0.820 | 0.770 | 0.787 | 0.778 | 0.643 | 0.673 | 0.757 | 0.509 | 0.821 | 0.789 |
| 1Z21 | 771 | 96 | 0.433 | 0.214 | 0.253 | 0.626 | 0.289 | 0.403 | 0.484 | 0.352 | 0.490 | 0.540 |
| 2BRF | 775 | 100 | 0.710 | 0.764 | 0.770 | 0.693 | 0.535 | 0.561 | 0.711 | 0.400 | 0.742 | 0.612 |
| 1NOA | 778 | 113 | 0.615 | 0.578 | 0.651 | 0.406 | 0.485 | 0.485 | 0.589 | 0.213 | 0.615 | 0.531 |
| 1NNX | 780 | 93 | 0.631 | 0.711 | 0.661 | 0.314 | 0.517 | 0.584 | 0.550 | 0.515 | 0.581 | 0.665 |
| 5CYT | 800 | 103 | 0.331 | 0.333 | 0.266 | 0.435 | 0.102 | 0.402 | 0.458 | 0.327 | 0.431 | 0.248 |
| 1QAU | 812 | 112 | 0.620 | 0.580 | 0.617 | 0.563 | 0.533 | 0.526 | 0.546 | 0.286 | 0.587 | 0.758 |
| 2QJL | 816 | 99 | 0.594 | 0.407 | 0.546 | 0.327 | 0.497 | 0.403 | 0.620 | 0.048 | 0.610 | 0.297 |
| 2NUH | 818 | 104 | 0.771 | 0.756 | 0.696 | 0.738 | 0.685 | 0.834 | 0.871 | 0.508 | 0.878 | 0.734 |
| 2CE0 | 828 | 99 | 0.529 | 0.710 | 0.629 | 0.326 | 0.628 | 0.626 | 0.651 | 0.479 | 0.726 | 0.307 |
See descriptions in Table 1
Table 3.
B-factor correlations for large-sized structuresa
| ID | TA | TR | GNM(Cα) | GNM(N) | GNM(C) | GNM(Cβ) | cgNMA(Cα) | cgNMA(N) | cgNMA(C) | cgNMA(Cβ) | cgNMA(M) | NMA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1V70 | 832 | 105 | 0.162 | 0.166 | 0.190 | 0.424 | 0.285 | 0.279 | 0.281 | 0.395 | 0.282 | 0.265 |
| 1RRO | 846 | 108 | 0.418 | 0.616 | 0.257 | 0.277 | 0.546 | 0.305 | 0.216 | 0.272 | 0.200 | 0.115 |
| 3VUB | 855 | 101 | 0.607 | 0.512 | 0.618 | 0.273 | 0.365 | 0.278 | 0.338 | 0.295 | 0.324 | 0.300 |
| 1CCR | 855 | 111 | 0.351 | 0.322 | 0.331 | 0.323 | 0.530 | 0.288 | 0.541 | 0.343 | 0.509 | 0.379 |
| 1EW4 | 863 | 106 | 0.547 | 0.314 | 0.357 | 0.625 | 0.447 | 0.645 | 0.671 | 0.200 | 0.693 | 0.542 |
| 2VIM | 864 | 104 | 0.212 | 0.094 | 0.160 | 0.347 | 0.221 | 0.312 | 0.282 | 0.104 | 0.357 | 0.243 |
| 2I24 | 872 | 113 | 0.494 | 0.443 | 0.515 | 0.288 | 0.441 | 0.334 | 0.436 | 0.119 | 0.417 | 0.296 |
| 1UKU | 873 | 102 | 0.742 | 0.721 | 0.773 | 0.710 | 0.720 | 0.335 | 0.486 | 0.255 | 0.462 | 0.519 |
| 1IFR | 878 | 113 | 0.637 | 0.689 | 0.590 | 0.652 | 0.330 | 0.413 | 0.524 | 0.405 | 0.534 | 0.462 |
| 2CG7 | 879 | 90 | 0.379 | 0.455 | 0.436 | 0.286 | 0.308 | 0.349 | 0.315 | 0.256 | 0.364 | 0.355 |
| 1PZ4 | 890 | 113 | 0.843 | 0.838 | 0.809 | 0.766 | 0.844 | 0.801 | 0.861 | 0.731 | 0.873 | 0.702 |
| 1WPA | 906 | 107 | 0.417 | 0.254 | 0.214 | 0.385 | 0.380 | 0.476 | 0.638 | 0.261 | 0.568 | 0.459 |
| 1AHO | 925 | 64 | 0.562 | 0.393 | 0.413 | 0.504 | 0.339 | 0.349 | 0.474 | 0.217 | 0.496 | 0.552 |
| 1QTO | 934 | 122 | 0.334 | 0.374 | 0.293 | 0.422 | 0.725 | 0.599 | 0.622 | 0.462 | 0.670 | 0.672 |
| 1WHI | 937 | 122 | 0.270 | 0.307 | 0.224 | 0.382 | 0.414 | 0.348 | 0.222 | 0.068 | 0.290 | 0.492 |
| 1PMY | 937 | 123 | 0.685 | 0.575 | 0.660 | 0.478 | 0.702 | 0.530 | 0.699 | 0.282 | 0.715 | 0.262 |
| 2PPN | 963 | 107 | 0.668 | 0.590 | 0.579 | 0.476 | 0.468 | 0.281 | 0.535 | 0.287 | 0.492 | 0.542 |
| 1NKO | 991 | 122 | 0.368 | 0.409 | 0.437 | 0.411 | 0.322 | 0.525 | 0.476 | 0.383 | 0.539 | 0.583 |
| 1E5K | 1423 | 188 | 0.859 | 0.662 | 0.727 | 0.716 | 0.620 | 0.620 | 0.625 | 0.380 | 0.684 | 0.741 |
| 2R16 | 1476 | 175 | 0.616 | 0.625 | 0.580 | 0.355 | 0.411 | 0.545 | 0.444 | 0.136 | 0.487 | 0.579 |
| 2VYO | 1609 | 206 | 0.761 | 0.768 | 0.773 | 0.621 | 0.739 | 0.642 | 0.732 | 0.308 | 0.760 | 0.645 |
| 2IMF | 1700 | 203 | 0.514 | 0.503 | 0.513 | 0.565 | 0.401 | 0.514 | 0.552 | 0.020 | 0.541 | 0.491 |
| 1O08 | 1722 | 221 | 0.309 | 0.308 | 0.410 | 0.452 | 0.616 | 0.408 | 0.562 | 0.296 | 0.521 | 0.281 |
| 2C71 | 1736 | 205 | 0.560 | 0.506 | 0.600 | 0.614 | 0.584 | 0.547 | 0.769 | 0.351 | 0.742 | 0.491 |
| 1ATG | 1749 | 231 | 0.497 | 0.553 | 0.543 | 0.409 | 0.154 | 0.288 | 0.233 | 0.188 | 0.323 | 0.465 |
| 2VPA | 1750 | 204 | 0.576 | 0.609 | 0.443 | 0.613 | 0.594 | 0.593 | 0.644 | 0.440 | 0.669 | 0.591 |
| 1WBE | 1774 | 204 | 0.549 | 0.559 | 0.539 | 0.495 | 0.574 | 0.522 | 0.485 | 0.411 | 0.538 | 0.575 |
| 2HQK | 1810 | 213 | 0.365 | 0.553 | 0.272 | 0.677 | 0.743 | 0.733 | 0.588 | 0.333 | 0.716 | 0.715 |
| 2CWS | 1847 | 227 | 0.696 | 0.690 | 0.640 | 0.566 | 0.524 | 0.644 | 0.596 | 0.346 | 0.642 | 0.618 |
| 1BYI | 1848 | 224 | 0.552 | 0.365 | 0.411 | 0.489 | 0.133 | 0.290 | 0.369 | 0.184 | 0.372 | 0.223 |
| 1NLS | 1872 | 237 | 0.515 | 0.580 | 0.486 | 0.502 | 0.385 | 0.568 | 0.554 | 0.484 | 0.628 | 0.813 |
| 2AGK | 1885 | 233 | 0.512 | 0.524 | 0.564 | 0.587 | 0.514 | 0.727 | 0.781 | 0.392 | 0.820 | 0.687 |
| 2HYK | 1893 | 237 | 0.515 | 0.550 | 0.483 | 0.556 | 0.593 | 0.396 | 0.491 | 0.351 | 0.523 | 0.392 |
| 3SEB | 1948 | 238 | 0.826 | 0.844 | 0.859 | 0.709 | 0.720 | 0.666 | 0.699 | 0.538 | 0.745 | 0.709 |
| 2V9V | 2387 | 135 | 0.528 | 0.578 | 0.424 | 0.514 | 0.594 | 0.663 | 0.471 | 0.216 | 0.572 | 0.663 |
See description in Table 1
In Tables 1, 2, and 3, we see consistently that the results from cgNMA methods are comparable with GNM except when Cβ atoms are used as the representative atoms. Both types of methods performed much better than NMA. Among all these methods, it seems that cgNMA(M) performed the best for all three categories of structures. Tables 4, 5, and 6 compare the test results for different pairs of methods for small, medium, and large structures, respectively. In these tables, we compare the B-factor correlations of the residue mean-square-fluctuations of the structures produced by each pair of methods, method A vs. method B, denoted as A-B. We list the numbers of B-factor correlations of method A that are significantly higher (or lower (−) than that of method B. If the difference of two B-factor correlations is within 1.0e–02, we consider them to be comparable (=). From these tables, we see more clearly how different methods performed and compared to each other. It seems that GNM using Cα as the representative atom performed the best among all GNM methods; cgNMA using N or C as the representative atom performed better than using Cα or Cβ; cgNMA(M) performed better than GNM(Cα) and NMA consistently.
Table 4.
Comparison of B-factor correlations for small structuresa
| cgNMA(Cα)-GNM(Cα) | cgNMA(N)-GNM(N) | cgNMA(C)-GNM(C) | cgNMA(M)-GNM(Cα) | |
|---|---|---|---|---|
| + | 13 | 17 | 17 | 16 |
| − | 17 | 13 | 12 | 14 |
| = | 3 | 3 | 4 | 3 |
|
| ||||
| cgNMA(Cα)-NMA | cgNMA(N)-NMA | cgNMA(C)-NMA | cgNMA(M)-NMA | |
|
| ||||
| + | 22 | 19 | 22 | 23 |
| − | 9 | 12 | 11 | 10 |
| = | 2 | 2 | 0 | 0 |
+: number of structures whose B-factor correlations for the two different models are positive; −: number of structures whose B-factor correlations for the two different models are negative; =: number of structures whose B-factor correlations for the two different models are comparable
Table 5.
Comparison of B-factor correlations for medium-sized structuresa
| cgNMA(Cα)-GNM(Cα) | cgNMA(N)-GNM(N) | cgNMA(C)-GNM(C) | cgNMA(M)-GNM(Cα) | |
|---|---|---|---|---|
| + | 10 | 19 | 19 | 23 |
| − | 24 | 16 | 13 | 9 |
| = | 2 | 1 | 4 | 4 |
|
| ||||
| cgNMA(Cα)-NMA | cgNMA(N)-NMA | cgNMA(C)-NMA | cgNMA(M)-NMA | |
|
| ||||
| + | 15 | 18 | 21 | 25 |
| − | 20 | 16 | 13 | 9 |
| = | 1 | 2 | 2 | 2 |
See descriptions in Table 4
Table 6.
Comparison of B-factor correlations for large-sized structuresa
| cgNMA(Cα) - GNM(Cα) |
cgNMA(N) - GNM(N) |
cgNMA(C) - GNM(C) |
cgNMA(M) - GNM(Cα) |
|
|---|---|---|---|---|
| + | 13 | 13 | 19 | 18 |
| − | 19 | 22 | 15 | 15 |
| = | 3 | 0 | 1 | 2 |
|
| ||||
| cgNMA(Cα)-NMA | cgNMA(N)-NMA | cgNMA(C)-NMA | cgNMA(M)-NMA | |
|
| ||||
| + | 15 | 15 | 19 | 22 |
| − | 18 | 15 | 16 | 11 |
| = | 2 | 5 | 0 | 2 |
See descriptions in Table 4
Figures 3, 4, and 5 demonstrate the test results on three example structures, 1HJE, 2BF9, and 2HQK, each representing a small, medium, and large structure. For each structure, the residue mean-square-fluctuations calculated by GNM(Cα), cgNMA(M), and NMA are shown in red curves. The experimental B-factors of the representative atoms are shown in black curves. In Fig. 3, the B-factor correlations of the residue mean-square-fluctuations of the structure calculated by GNM(Cα), cgNMA(M), and NMA are 0.616, 0.838, and 0.209, respectively. In Fig. 4, the B- factor correlations of the residue mean-square-fluctuations of the structure calculated by GNM(Cα), cgNMA(M), and NMA are 0.419, 0.762, and 0.367, respectively. In Fig. 5, the B-factor correlations of the residue mean-square-fluctuations of the structure calculated by GNM(Cα), cgNMA(M), and NMA are 0.365, 0.716, and 0.715, respectively.
Fig. 3.

(a) The residue mean-square-fluctuations calculated by GNM(Cα) for 1HJE are compared with the experimental B-factors of Cα. (b) The residue mean-square-fluctuations calculated by cgNMA(M) for 1HJE are compared with the experimental B-factors of Cα. (c) The residue mean-square-fluctuations calculated by NMA for 1HJE are compared with the experimental B-factors of Cα
Fig. 4.

(a) The residue mean-square-fluctuations calculated by GNM(Cα) for 2BF9 are compared with the experimental B-factors of Cα. (b) The residue mean-square-fluctuations calculated by cgNMA(M) for 2BF9 are compared with the experimental B-factors of Cα. (c) The residue mean-square-fluctuations calculated by NMA for 2BF9 are compared with the experimental B-factors of Cα
Fig. 5.

(a) The residue mean-square-fluctuations calculated by GNM(Cα) for 2HQK are compared with the experimental B-factors of Cα. (b) The residue mean-square-fluctuations calculated by cgNMA(M) for 2HQK are compared with the experimental B-factors of Cα. (c) The residue mean-square-fluctuations calculated by NMA for 2HQK are compared with the experimental B-factors of Cα
5 Refined Gaussian Network Model
In this section, we present a new Gaussian Network Model based on our study on coarse-grained NMA as described in Sect. 4. We consider the requirement on an energy function as a key distinction between GNM and NMA. Indeed, the coarse-grained NMA in Sect. 4 still requires the availability of an energy function to obtain the Hessian matrix, although it conducts the same analysis as GNM at the residue level. However, we do see that with nonhomogeneous force constants extracted from the Hessian matrix, the coarse-grained NMA such as cgNMA(M) performs better than GNM. The question then is whether or not GNM can be improved by introducing some nonhomogeneous force constants, yet without requiring the Hessian matrix from an energy function.
In order to answer this question, we have examined the Hessian matrices of our downloaded structures. We collected all the force constants (the entries in the Hessian matrices) defined in cgNMA(M). For each structure, we have a matrix of force constants {kij : i,j = 1,…, m} where m is the number of residues in the structures. We then grouped the constants for all the residue pairs in contact but separated by s residues in sequence, where s = 0, 1, 2, 3, etc. For a fixed s, different residue pairs do not have significantly different force constants. However, for different s, the force constants are different, especially between the directly connected pairs (s = 0) and other types of pairs (s > 0). Figure 6 shows the distributions of the force constants for residue pairs in contact but separated by 0, 1, 2, 3 residues in sequence. Clearly, the constants are around 12 units when s = 0, and are all around 1 unit when s > 0. Based on the above survey, we have a reason to suggest that the forces between the residue pairs in contact should not be treated as the same. At the very least, the forces between directly connected residue pairs should be stronger than others in a magnitude. In other words, the force constants for directly connected residue pairs should be at least one magnitude larger than others. These constants correspond exactly to the bidiagonal elements of the contact matrices. Therefore, a simple way to build a new Gaussian Network Model that incorporates this nonhomogeneity of the force constants is to define all the entries of the contact matrix in the same way as the conventional GNM except for the bidiagonal entries set to a number of larger mag nitude, say −10. Such a model may reflect more accurately the real residue interactions in proteins. We call it a GNM model with nonhomogeneous force constants or nhGNM for short. An example nhGNM contact matrix can be found in Fig. 7. We have tested the nhGNM model on a large set of protein structures, each time with the force constants for directly connected residue pairs set to −8, −9, −10, −11, −12, −13, −14, or −15, to see if the results vary with varying these constants. Again, we divide the structures into three groups corresponding to relatively small-, medium-, and large-sized structures. The test results on each group are shown in Tables 7, 8, and 9 separately. In each table, we show the B-factor correlations of the residue mean-square-fluctuations of the proteins calculated by nhGNM. For comparison, we have also listed the B-factor correlations obtained using GNM. For both types of models, Cα atoms were used as the representative atoms for the residues. We use “+” to mean that nhGNM with one of the selected force constants has a higher B-factor correlation than GNM,“−” to mean the opposite. We use “=” to mean that the difference in the B-factor correlations computed by GNM and nhGNM is small within 1.0e–02.
Fig. 6.

The distributions of the force constants for pairs of residues separated by 0,1,2,3 residues in sequence.
Fig. 7.
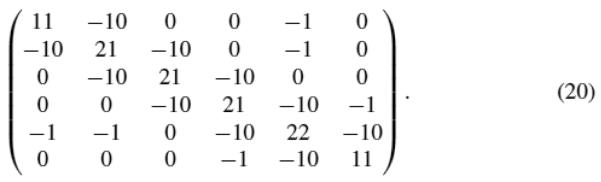
An example of an nhGNM contact matrix.
Table 7.
B-factor correlations of nhGNM for small-sized structuresa
| ID | TA | TR | GNM | nhGNM-8 | nhGNM-9 | nhGNM-10 | nhGNM-11 | nhGNM-12 | nhGNM-13 | nhGNM-14 | nhGNM-15 | nhGNM-GNM |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2OLX | 35 | 4 | 0.885 | 0.885 | 0.885 | 0.885 | 0.885 | 0.885 | 0.885 | 0.885 | 0.885 | = |
| 2OL9 | 51 | 6 | 0.689 | 0.891 | 0.894 | 0.897 | 0.898 | 0.9 | 0.901 | 0.902 | 0.903 | + |
| 1YJO | 55 | 6 | 0.434 | 0.43 | 0.43 | 0.43 | 0.43 | 0.43 | 0.43 | 0.43 | 0.43 | = |
| 1XY2 | 69 | 8 | 0.562 | 0.557 | 0.548 | 0.54 | 0.532 | 0.525 | 0.518 | 0.512 | 0.506 | = |
| 1NOT | 96 | 13 | 0.523 | 0.72 | 0.724 | 0.726 | 0.728 | 0.729 | 0.729 | 0.728 | 0.727 | + |
| 1PEN | 109 | 16 | 0.270 | 0.236 | 0.228 | 0.22 | 0.213 | 0.207 | 0.201 | 0.196 | 0.191 | − |
| 1OB4 | 110 | 5 | −0.744 | −0.744 | −0.744 | −0.744 | −0.744 | −0.744 | −0.744 | −0.744 | −0.744 | = |
| 1OB7 | 111 | 5 | −0.463 | −0.463 | −0.463 | −0.463 | −0.463 | −0.463 | −0.463 | −0.463 | −0.463 | = |
| 1AKG | 112 | 16 | 0.185 | −0.038 | −0.054 | −0.068 | −0.079 | −0.09 | −0.099 | −0.107 | −0.115 | − |
| 1KYC | 138 | 15 | 0.754 | 0.744 | 0.745 | 0.746 | 0.747 | 0.748 | 0.748 | 0.749 | 0.75 | = |
| 1ETN | 147 | 12 | −0.274 | −0.503 | −0.51 | −0.515 | −0.518 | −0.521 | −0.523 | −0.524 | −0.526 | = |
| 1ETL | 147 | 12 | 0.628 | 0.5 | 0.483 | 0.468 | 0.455 | 0.444 | 0.435 | 0.426 | 0.419 | − |
| 1PEF | 153 | 18 | 0.808 | 0.853 | 0.856 | 0.859 | 0.862 | 0.864 | 0.866 | 0.868 | 0.869 | + |
| 1ETM | 160 | 12 | 0.432 | 0.159 | 0.138 | 0.121 | 0.106 | 0.094 | 0.084 | 0.075 | 0.067 | − |
| 1O06 | 172 | 20 | 0.844 | 0.907 | 0.91 | 0.912 | 0.914 | 0.916 | 0.917 | 0.918 | 0.919 | + |
| 1HJE | 214 | 13 | 0.616 | 0.856 | 0.86 | 0.862 | 0.864 | 0.864 | 0.863 | 0.862 | 0.86 | + |
| 1P9I | 225 | 29 | 0.625 | 0.61 | 0.608 | 0.607 | 0.605 | 0.604 | 0.603 | 0.602 | 0.601 | − |
| 2JKU | 255 | 35 | 0.656 | 0.621 | 0.621 | 0.621 | 0.622 | 0.622 | 0.623 | 0.624 | 0.624 | − |
| 1RJU | 257 | 36 | 0.431 | 0.42 | 0.416 | 0.413 | 0.409 | 0.406 | 0.402 | 0.398 | 0.395 | = |
| 2NLS | 273 | 36 | 0.530 | 0.55 | 0.545 | 0.539 | 0.532 | 0.525 | 0.517 | 0.508 | 0.499 | + |
| 1VRZ | 277 | 13 | 0.327 | 0.134 | 0.105 | 0.079 | 0.055 | 0.032 | 0.012 | −0.006 | −0.023 | − |
| 1AIE | 295 | 31 | 0.155 | 0.287 | 0.3 | 0.312 | 0.323 | 0.333 | 0.343 | 0.351 | 0.36 | + |
| 1GK7 | 335 | 39 | 0.821 | 0.821 | 0.82 | 0.82 | 0.82 | 0.82 | 0.82 | 0.82 | 0.819 | = |
| 1Q9B | 345 | 43 | 0.656 | 0.814 | 0.818 | 0.821 | 0.823 | 0.824 | 0.824 | 0.824 | 0.823 | + |
| 1YZM | 361 | 46 | 0.901 | 0.94 | 0.941 | 0.943 | 0.944 | 0.944 | 0.945 | 0.946 | 0.946 | + |
| 6RXN | 367 | 45 | 0.594 | 0.534 | 0.529 | 0.524 | 0.52 | 0.516 | 0.512 | 0.509 | 0.505 | − |
| 1USE | 382 | 40 | −0.142 | −0.303 | −0.311 | −0.318 | −0.323 | −0.328 | −0.333 | −0.337 | −0.34 | = |
| 1BX7 | 387 | 51 | 0.706 | 0.801 | 0.806 | 0.811 | 0.815 | 0.818 | 0.821 | 0.823 | 0.825 | + |
| 2DSX | 399 | 52 | 0.127 | 0.057 | 0.059 | 0.061 | 0.064 | 0.067 | 0.071 | 0.075 | 0.079 | − |
| 1UOY | 452 | 64 | 0.671 | 0.691 | 0.691 | 0.691 | 0.691 | 0.691 | 0.691 | 0.69 | 0.689 | + |
| 1U06 | 470 | 55 | 0.434 | 0.43 | 0.428 | 0.427 | 0.426 | 0.425 | 0.424 | 0.424 | 0.423 | = |
| 1GVD | 491 | 52 | 0.591 | 0.361 | 0.35 | 0.341 | 0.334 | 0.328 | 0.322 | 0.318 | 0.314 | − |
| 1FF4 | 503 | 65 | 0.674 | 0.755 | 0.76 | 0.765 | 0.769 | 0.773 | 0.777 | 0.78 | 0.783 | + |
ID-protein ID, TA-total number of atoms, TR-total number of residues, GNM—residue B-factor correlations using GNM, nhGNM-k—residue B-factor correlations using nhGNMwith -k as the value for the bidiagonal elements in the contact matrix, nhGNM-GNM—comparison between nhGNM and GNM: “+”—nhGNM has a higher B-factor correlation than GNM, “−”—nhGNM has a lower B-factor correlation than GNM, “=”—nhGNM has a comparable B-factor correlation with GNM
Table 8.
B-factor correlations of nhGNM for medium-sized structuresa
| ID | TA | TR | GNM | nhGNM -8 |
nhGNM-9 | nhGNM- 10 |
nhGNM- 11 |
nhGNM- 12 |
nhGNM- 13 |
nhGNM- 14 |
nhGNM- 15 |
nhGNM- GNM |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1LR7 | 522 | 73 | 0.620 | 0.693 | 0.7 | 0.706 | 0.712 | 0.717 | 0.722 | 0.727 | 0.731 | + |
| 1ZVA | 551 | 75 | 0.690 | 0.626 | 0.616 | 0.606 | 0.597 | 0.588 | 0.579 | 0.571 | 0.563 | − |
| 2BF9 | 562 | 35 | 0.419 | 0.596 | 0.601 | 0.605 | 0.607 | 0.608 | 0.609 | 0.609 | 0.608 | + |
| 2EHS | 598 | 75 | 0.747 | 0.648 | 0.637 | 0.627 | 0.618 | 0.609 | 0.601 | 0.593 | 0.586 | − |
| 1UHA | 623 | 82 | 0.583 | 0.578 | 0.573 | 0.568 | 0.563 | 0.558 | 0.554 | 0.549 | 0.544 | = |
| 1FK5 | 626 | 93 | 0.485 | 0.44 | 0.434 | 0.427 | 0.421 | 0.415 | 0.409 | 0.403 | 0.398 | − |
| 1OPD | 642 | 85 | 0.398 | 0.583 | 0.598 | 0.611 | 0.623 | 0.634 | 0.644 | 0.653 | 0.661 | + |
| 1X3O | 642 | 80 | 0.654 | 0.589 | 0.58 | 0.572 | 0.564 | 0.557 | 0.55 | 0.543 | 0.537 | − |
| 2E3H | 651 | 81 | 0.605 | 0.663 | 0.665 | 0.666 | 0.666 | 0.665 | 0.665 | 0.663 | 0.661 | + |
| 1USM | 661 | 77 | 0.798 | 0.727 | 0.719 | 0.712 | 0.705 | 0.699 | 0.693 | 0.688 | 0.683 | − |
| 1ULR | 677 | 87 | 0.495 | 0.433 | 0.42 | 0.407 | 0.395 | 0.383 | 0.371 | 0.359 | 0.349 | − |
| 1I71 | 683 | 83 | 0.549 | 0.508 | 0.51 | 0.512 | 0.515 | 0.517 | 0.519 | 0.522 | 0.524 | − |
| 2PKT | 688 | 91 | −0.286 | −0.246 | −0.242 | −0.237 | −0.234 | −0.23 | −0.226 | −0.223 | −0.22 | = |
| 1N7E | 700 | 95 | 0.497 | 0.473 | 0.474 | 0.474 | 0.474 | 0.475 | 0.475 | 0.475 | 0.476 | − |
| 2IP6 | 702 | 87 | 0.572 | 0.654 | 0.661 | 0.667 | 0.673 | 0.679 | 0.684 | 0.689 | 0.694 | − |
| 2EAQ | 705 | 89 | 0.695 | 0.797 | 0.797 | 0.796 | 0.794 | 0.792 | 0.79 | 0.788 | 0.786 | + |
| 2FQ3 | 721 | 85 | 0.348 | 0.259 | 0.252 | 0.246 | 0.24 | 0.235 | 0.23 | 0.225 | 0.221 | − |
| 2RB8 | 723 | 93 | 0.517 | 0.564 | 0.564 | 0.565 | 0.564 | 0.564 | 0.564 | 0.563 | 0.562 | + |
| 1GXU | 726 | 88 | 0.421 | 0.352 | 0.347 | 0.341 | 0.337 | 0.332 | 0.328 | 0.325 | 0.322 | − |
| 2PLT | 727 | 98 | 0.481 | 0.479 | 0.476 | 0.472 | 0.469 | 0.466 | 0.462 | 0.459 | 0.456 | = |
| 1ABA | 728 | 87 | 0.613 | 0.412 | 0.392 | 0.372 | 0.354 | 0.337 | 0.321 | 0.306 | 0.291 | − |
| 1R7J | 729 | 90 | 0.368 | 0.219 | 0.231 | 0.208 | 0.204 | 0.2 | 0.197 | 0.194 | 0.191 | − |
| 1V05 | 731 | 96 | 0.632 | 0.641 | 0.636 | 0.632 | 0.627 | 0.622 | 0.618 | 0.613 | 0.608 | + |
| 1CYO | 731 | 88 | 0.741 | 0.733 | 0.731 | 0.729 | 0.727 | 0.725 | 0.724 | 0.722 | 0.721 | = |
| 3BZQ | 742 | 99 | 0.466 | 0.495 | 0.497 | 0.499 | 0.501 | 0.502 | 0.503 | 0.504 | 0.505 | + |
| 1W2L | 746 | 97 | 0.397 | 0.388 | 0.384 | 0.38 | 0.377 | 0.374 | 0.372 | 0.369 | 0.367 | = |
| 2MCM | 769 | 112 | 0.820 | 0.875 | 0.876 | 0.877 | 0.878 | 0.878 | 0.878 | 0.878 | 0.878 | + |
| 1Z21 | 771 | 96 | 0.433 | 0.305 | 0.297 | 0.29 | 0.283 | 0.277 | 0.272 | 0.267 | 0.262 | − |
| 2BRF | 775 | 100 | 0.710 | 0.716 | 0.713 | 0.711 | 0.708 | 0.706 | 0.703 | 0.701 | 0.699 | + |
| 1NOA | 778 | 113 | 0.615 | 0.661 | 0.662 | 0.663 | 0.663 | 0.663 | 0.663 | 0.663 | 0.662 | + |
| 1NNX | 780 | 93 | 0.631 | 0.566 | 0.556 | 0.546 | 0.537 | 0.528 | 0.519 | 0.511 | 0.504 | − |
| 5CYT | 800 | 103 | 0.331 | 0.235 | 0.223 | 0.212 | 0.202 | 0.192 | 0.183 | 0.173 | 0.165 | − |
| 1QAU | 812 | 112 | 0.620 | 0.615 | 0.613 | 0.611 | 0.609 | 0.607 | 0.605 | 0.604 | 0.602 | = |
| 2QJL | 816 | 99 | 0.594 | 0.569 | 0.56 | 0.552 | 0.544 | 0.537 | 0.53 | 0.523 | 0.517 | − |
| 2NUH | 818 | 104 | 0.771 | 0.791 | 0.788 | 0.784 | 0.78 | 0.777 | 0.773 | 0.769 | 0.765 | + |
| 2CE0 | 828 | 99 | 0.529 | 0.606 | 0.612 | 0.616 | 0.621 | 0.625 | 0.628 | 0.631 | 0.634 | + |
See descriptions in Table 7.
Table 9.
B-factor correlations of nhGNM for large-sized structuresa
| ID | TA | TR | GNM | nhGNM-8 | nhGNM-9 | nhGNM-10 | nhGNM-11 | nhGNM-12 | nhGNM-13 | nhGNM-14 | nhGNM-15 | nhGNM-GNM |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1V70 | 832 | 105 | 0.162 | 0.171 | 0.17 | 0.17 | 0.17 | 0.17 | 0.17 | 0.169 | 0.169 | = |
| 1RRO | 846 | 108 | 0.418 | 0.626 | 0.641 | 0.654 | 0.665 | 0.676 | 0.685 | 0.693 | 0.701 | + |
| 3VUB | 855 | 101 | 0.607 | 0.622 | 0.614 | 0.605 | 0.597 | 0.589 | 0.581 | 0.573 | 0.566 | + |
| 1CCR | 855 | 111 | 0.351 | 0.398 | 0.402 | 0.407 | 0.411 | 0.414 | 0.418 | 0.421 | 0.425 | + |
| 1EW4 | 863 | 106 | 0.547 | 0.369 | 0.355 | 0.343 | 0.333 | 0.323 | 0.314 | 0.306 | 0.299 | − |
| 2VIM | 864 | 104 | 0.212 | 0.252 | 0.253 | 0.254 | 0.254 | 0.254 | 0.253 | 0.253 | 0.252 | + |
| 2I24 | 872 | 113 | 0.494 | 0.476 | 0.474 | 0.472 | 0.47 | 0.469 | 0.468 | 0.467 | 0.466 | − |
| 1UKU | 873 | 102 | 0.742 | 0.77 | 0.771 | 0.772 | 0.772 | 0.773 | 0.773 | 0.773 | 0.773 | + |
| 1IFR | 878 | 113 | 0.637 | 0.561 | 0.551 | 0.541 | 0.532 | 0.524 | 0.516 | 0.508 | 0.501 | − |
| 2CG7 | 879 | 90 | 0.379 | 0.366 | 0.367 | 0.368 | 0.368 | 0.369 | 0.37 | 0.371 | 0.371 | = |
| 1PZ4 | 890 | 113 | 0.843 | 0.858 | 0.857 | 0.855 | 0.853 | 0.852 | 0.85 | 0.848 | 0.846 | + |
| 1WPA | 906 | 107 | 0.417 | 0.446 | 0.446 | 0.446 | 0.446 | 0.445 | 0.445 | 0.444 | 0.444 | + |
| 1AHO | 925 | 64 | 0.562 | 0.521 | 0.516 | 0.511 | 0.506 | 0.502 | 0.497 | 0.493 | 0.489 | − |
| 1QTO | 934 | 122 | 0.334 | 0.362 | 0.366 | 0.369 | 0.372 | 0.375 | 0.378 | 0.381 | 0.384 | + |
| 1WHI | 937 | 122 | 0.270 | 0.3 | 0.305 | 0.31 | 0.314 | 0.319 | 0.323 | 0.326 | 0.33 | + |
| 1PMY | 937 | 123 | 0.685 | 0.667 | 0.661 | 0.656 | 0.651 | 0.647 | 0.643 | 0.639 | 0.636 | − |
| 2PPN | 963 | 107 | 0.668 | 0.638 | 0.635 | 0.633 | 0.631 | 0.629 | 0.627 | 0.625 | 0.624 | − |
| 1NKO | 991 | 122 | 0.368 | 0.347 | 0.344 | 0.341 | 0.338 | 0.335 | 0.332 | 0.329 | 0.326 | − |
| 1E5K | 1423 | 188 | 0.859 | 0.827 | 0.821 | 0.815 | 0.809 | 0.804 | 0.798 | 0.793 | 0.789 | − |
| 2R16 | 1476 | 175 | 0.616 | 0.608 | 0.601 | 0.594 | 0.588 | 0.582 | 0.576 | 0.57 | 0.564 | − |
| 2VYO | 1609 | 206 | 0.761 | 0.787 | 0.784 | 0.78 | 0.777 | 0.773 | 0.77 | 0.767 | 0.763 | + |
| 2IMF | 1700 | 203 | 0.514 | 0.492 | 0.49 | 0.488 | 0.486 | 0.484 | 0.483 | 0.482 | 0.48 | − |
| 1O08 | 1722 | 221 | 0.309 | 0.362 | 0.368 | 0.373 | 0.378 | 0.383 | 0.388 | 0.392 | 0.397 | + |
| 2C71 | 1736 | 205 | 0.560 | 0.607 | 0.606 | 0.604 | 0.602 | 0.6 | 0.598 | 0.596 | 0.595 | + |
| 1ATG | 1749 | 231 | 0.497 | 0.52 | 0.518 | 0.515 | 0.513 | 0.51 | 0.507 | 0.504 | 0.501 | + |
| 2VPA | 1750 | 204 | 0.576 | 0.593 | 0.594 | 0.594 | 0.594 | 0.593 | 0.592 | 0.591 | 0.59 | + |
| 1WBE | 1774 | 204 | 0.549 | 0.557 | 0.555 | 0.553 | 0.551 | 0.55 | 0.548 | 0.546 | 0.545 | + |
| 2HQK | 1810 | 213 | 0.365 | 0.244 | 0.237 | 0.231 | 0.225 | 0.221 | 0.216 | 0.212 | 0.208 | − |
| 2CWS | 1847 | 227 | 0.696 | 0.699 | 0.697 | 0.696 | 0.694 | 0.692 | 0.69 | 0.688 | 0.686 | = |
| 1BYI | 1848 | 224 | 0.552 | 0.454 | 0.444 | 0.435 | 0.426 | 0.418 | 0.411 | 0.404 | 0.397 | − |
| 1NLS | 1872 | 237 | 0.515 | 0.579 | 0.578 | 0.577 | 0.575 | 0.574 | 0.572 | 0.57 | 0.568 | + |
| 2AGK | 1885 | 233 | 0.512 | 0.579 | 0.573 | 0.567 | 0.561 | 0.554 | 0.549 | 0.543 | 0.538 | + |
| 2HYK | 1893 | 237 | 0.515 | 0.569 | 0.569 | 0.568 | 0.567 | 0.566 | 0.565 | 0.563 | 0.562 | + |
| 3SEB | 1948 | 238 | 0.826 | 0.853 | 0.851 | 0.848 | 0.845 | 0.842 | 0.838 | 0.835 | 0.832 | + |
| 2V9V | 2387 | 135 | 0.528 | 0.555 | 0.556 | 0.556 | 0.557 | 0.558 | 0.559 | 0.56 | 0.561 | + |
See descriptions in Table 7
We see from Tables 7, 8, and 9 that in many test cases, the nhGNM performed significantly better than GNM. Out of total 33 relatively small-sized structures, nhGNM predicted the residue mean-square-fluctuations no worse than GNM for 23 cases (Table 7). nhGNM outperformed GNM significantly for 12 cases. Out of total 36 relatively medium-sized structures, nhGNM predicted the residue mean-square-fluctuations no worse than GNM for 19 cases. nhGNM outperformed significantly GNM for 13 cases. Out of total 35 relatively large-sized structures, nhGNM predicted the residue mean-square-fluctuations no worse than GNM for 23 cases. nhGNM out-performed GNM significantly for 20 cases.
Note that in general, nhGNM performed similarly for the tested force constants for directly connected residue pairs {−8, −9, −10, −11, −12, −13, −14, −15}. We were not able to identify an optimal value, but we would suggest just using −12 for such contact pairs while −1 for all other types. It is also possible to run nhGNM multiple times for this whole set of force constants, remove obvious outliers, and take the average results.
6 Concluding Remarks
In this paper, we have investigated several possible approaches to coarse-grained normal mode analysis. Similar to GNM, we take the residues as the basic units in a protein and use a single backbone atom (or a combination of several backbone atoms) to represent each residue. Different from GNM, the force constants for pairs of representative atoms are not the same and are instead extracted from the Hessian matrix of the energy function.
Using the new models, we have calculated the mean-square-fluctuations of the residues and their correlations with the experimental B-factors (called the B-factor correlations) for a large set of proteins. We have compared them with the all-atom normal mode analysis and the residue-level Gaussian Network Model. We have shown that our models performed more efficiently than the all-atom normal mode analysis, and the B-factor correlations are also higher. The B-factor correlations are comparable with those estimated by the Gaussian Network Model and in some cases better.
Following the development of the coarse-grained NMA, we have conducted a statistical survey on the extracted force constants, for different pairs of residues with different numbers of separation residues in sequence. We then based on their statistical averages to build a refined Gaussian Network Model. We have shown that the force constants for the neighboring residue pairs are always about one-magnitude larger than other pairs in contact. Therefore, in the refined GNM, the entries of the contact matrix could be defined in the same way as the conventional GNM except that the bidiagonal entries have large magnitudes. We have shown that such a simply refined GNM could predict residue-level structural fluctuations significantly better than the conventional GNM in many test cases.
The coarse-grained NMA and refined GNM do not always outperform conventional methods such GNM. In our future efforts, we would like to examine each of our test cases more carefully and find the causes for the disagreements between the predicted structural fluctuations and the experimental B-factors, and to further improve our predictions. We would also like to apply our methods to a few biologically interesting proteins such as the HIV-1 protease and the human telomerase and analyze the results in great details. The structures of these proteins have just been modeled in recent studies (Yang et al. 2008; Steczkiewicz et al. 2011). Accurate predictions on their structural fluctuations may provide great insights into how the dynamics of the structures relate to their functions.
Another direction to explore is to develop a relatively accurate residue-level energy function so that the residue-level NMA can be carried out exactly as the atomic-level NMA. This could be difficult but challenging because of the lack of complete physical understanding of the residue interactions. Much work has been done for developing residue-level potentials using statistical approaches such as residue-residue contact potentials (Miyazawa and Jernigan 1985; Sippl 1990), residue distance or angle potentials (Kuszewski et al. 1996; Wu et al. 2007a, 2007b; Huang et al. 2011), and residue Cα only potentials (Wu et al. 2007a, 2007b). These residue-level potentials may provide a basis for defining a complete residue-level energy function, with which a residue-level NMA can be performed.
Note that the B-factor is a consequence of the dynamic disorder in the crystal caused by the temperature-dependent vibration of the atoms in the structure (Drenth 2006). This is the reason that we can compare the computed atomic or residue mean-square fluctuations with the B-factors detected by X-ray crystallography. However, protein crystals have also static disorder: molecules, or parts of molecules, in different unit cells do not occupy exactly the same position or have exactly the same orientation (Drenth 2006). With this static disorder also included, the B-factor may not always reflect the atomic fluctuations correctly. In future work, we will consider to compare the computed atomic or residue mean-square fluctuations with the atomic or residue fluctuations in NMR structural ensembles, for which a better correlation between theoretically computed and experimentally estimated structural fluctuations may be obtained.
Finally, we would like to point out that the phrase “coarse-grained NMA” can be confusing. In general, by “coarse-grained NMA” we mean the normal mode analysis beyond the atomic level. More specifically, we mean the residue-level normal mode analysis requiring the Hessian or partial Hessian of the energy function, which is different from the analysis via other nonenergy-based methods such as GNM. In this sense, the residue-level normal mode analysis we have conducted using the force constants extracted from the Hessian of the energy function is still called the coarse-grained NMA. So is the residue-level NMA to be explored using a residue-level energy function.
Acknowledgements
The authors would like to acknowledge the support by the NIH/ /NIGMS grant R01GM081680 and by the NSF/ /DMS grant DMS0914354.
References
- Ahmed A, Gohlke H. Multiscale modeling of macromolecular conformational changes combining concepts from rigidity and elastic network theory. Proteins. 2006;63:1038–1051. doi: 10.1002/prot.20907. [DOI] [PubMed] [Google Scholar]
- Bahar I, Atilgan AR, Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential, folding. Design. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- Bahar I, Atilgan AR, Demirel M, Erman B. Vibrational dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 1998;80:2733–2736. [Google Scholar]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. Freeman; New York: 2006. [Google Scholar]
- Berman H, et al. Pdb data bank annual report. 2010 http://www.rcsb.orgpdb.
- Brooks BR, Karplus M. Harmonic dynamics of proteins: normal mode and fluctuations in bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA. 1983;80:6571–6575. doi: 10.1073/pnas.80.21.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, III, Karplus M, Pettitt BM. Proteins: a theoretical perspective of dynamics, structure, and thermodynamics. Wiley; New York: 1989. [Google Scholar]
- Brooks BR, et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Bahar I. Normal mode analysis: theory and application to biological and chemical systems. Chapman & Hall/CRC Press; London/Boca Raton: 2006. [Google Scholar]
- Demerdash ONA, Mitchell JC. Density-cluster NMA: a new protein decomposition technique for coarse-grained normal mode analysis. Proteins. 2012;80:1766–1779. doi: 10.1002/prot.24072. [DOI] [PubMed] [Google Scholar]
- Drenth J. Principles of protein X-ray crystallography. Springer; Berlin: 2006. [Google Scholar]
- Go N, Noguti T, Nishikawa T. Dynamics of a small globular protein in terms of low-frequency vibrational modes. Proc. Natl. Acad. Sci. USA. 1983;80:3696–3700. doi: 10.1073/pnas.80.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haliloglu T, Bahar I, Erman B. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 1997;79:3090–3092. [Google Scholar]
- Huang Y, Bonett S, Kloczkowski A, Jernigan R, Wu Z. Statistical measures on protein residue-level structural properties. J. Struct. Funct. Genomics. 2011;12:119–136. doi: 10.1007/s10969-011-9104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs DJ, Rader AJ, Kuhn LA, Thorpe MF. Protein flexibility predictions using graph theory. Proteins. 2001;44:150–165. doi: 10.1002/prot.1081. [DOI] [PubMed] [Google Scholar]
- Kondrashov D, Cui Q, Phillips G., Jr. Optimization and evaluation of a coarse-grained model of protein motion using X-ray crystal data. Biophys. J. 2006;91:2760–2767. doi: 10.1529/biophysj.106.085894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszewski J, Gronenborn AM, Clore GM. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci. 1996;5:1067–1080. doi: 10.1002/pro.5560050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M, Sander C, Stern PS. Protein normal-mode dynamics: trypsin inhibitor, crambin, ribonuclease and lysozyme. J. Mol. Biol. 1985;181:423–447. doi: 10.1016/0022-2836(85)90230-x. [DOI] [PubMed] [Google Scholar]
- Li G, Cui Q. A coarse-grained normal mode approach for macromolecules: an efficient implementation and application to Ca(21)-ATPase. Biophys. J. 2002;83:2457–2474. doi: 10.1016/S0006-3495(02)75257-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Ma J. A minimalist network model for coarse-grained normal mode analysis and its application to biomolecular x-ray crystallography. Proc. Natl. Acad. Sci. USA. 2008;105:15358–15363. doi: 10.1073/pnas.0806072105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Ma J. Normal mode analysis with molecular geometry restraints: bridging molecular mechanics and elastic models. Arch. Biochem. Biophys. 2011;508:64–71. doi: 10.1016/j.abb.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheletti C, Carloni P, Maritan A. Accurate and efficient description of protein vibrational dynamics: comparing molecular dynamics and Gaussian models. Proteins. 2004;55:635–645. doi: 10.1002/prot.20049. [DOI] [PubMed] [Google Scholar]
- Mitchell JC, Kerr R, Ten Eyck LF. Rapid atomic density methods for molecular shape characterization. J. Mol. Graph. Model. 2001;19:325–330. doi: 10.1016/s1093-3263(00)00079-6. [DOI] [PubMed] [Google Scholar]
- Miyazawa S, Jernigan RL. Estimation of effective inter-residue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules. 1985;18:534–552. [Google Scholar]
- Morin D. Introduction to classical mechanics. Cambridge University Press; Cambridge: 2008. [Google Scholar]
- Schlick T. Molecular modeling and simulation—an interdisciplinary guide. Springer; Berlin: 2002. [Google Scholar]
- Schuyler AD, Chirikjian GS. Normal mode analysis of proteins: a comparison of rigid cluster modes with Ca coarse graining. J. Mol. Graph. Model. 2004;22:183–193. doi: 10.1016/S1093-3263(03)00158-X. [DOI] [PubMed] [Google Scholar]
- Sippl MJ. Calculation of conformational ensembles from potentials of mean force. J. Mol. Biol. 1990;213:859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- Steczkiewicz K, et al. Human telomerase model shows the role of the TEN domain in advancing the double helix for the next polymerization step. Proc. Natl. Acad. Sci. USA. 2011;108:9443–9448. doi: 10.1073/pnas.1015399108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tama F, Gadea FX, Marques O, Sanejouand YH. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins. 2000;41:1–7. doi: 10.1002/1097-0134(20001001)41:1<1::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Taner ZS, Jernigan RL. Optimizing the parameters of the Gaussian network model for ATP-binding proteins. In: Cui Q, Bahar I, editors. Normal mode analysis: theory and applications to biological and chemical systems. 2006. pp. 171–186. [Google Scholar]
- Tirion M. Large amplitude elastic motions in proteins from a single-parameter atomic analysis. Phys. Rev. Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- Wu D, Jernigan R, Wu Z. Refinement of NMR-determined protein structures with database derived mean force potentials. Proteins. 2007a;68:232–242. doi: 10.1002/prot.21358. [DOI] [PubMed] [Google Scholar]
- Wu Y, Lu M, Chen M, Li J, Ma J. OPUS-Ca: a knowledge-based potential function requiring only ca positions. Protein Sci. 2007b;16:1449–1463. doi: 10.1110/ps.072796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Song G, Carriquiry A, Jernigan RL. Close correspondence between the motions from principal component analysis of multiple HIV-1 protease structures and elastic network modes. Structure. 2008;16:321–330. doi: 10.1016/j.str.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]