

Abstract
BACKGROUND
Congenital disorders of glycosylation are genetic syndromes that result in impaired glycoprotein production. We evaluated patients who had a novel recessive disorder of glycosylation, with a range of clinical manifestations that included hepatopathy, bifid uvula, malignant hyperthermia, hypogonadotropic hypogonadism, growth retardation, hypoglycemia, myopathy, dilated cardiomyopathy, and cardiac arrest.
METHODS
Homozygosity mapping followed by whole-exome sequencing was used to identify a mutation in the gene for phosphoglucomutase 1 (PGM1) in two siblings. Sequencing identified additional mutations in 15 other families. Phosphoglucomutase 1 enzyme activity was assayed on cell extracts. Analyses of glycosylation efficiency and quantitative studies of sugar metabolites were performed. Galactose supplementation in fibroblast cultures and dietary supplementation in the patients were studied to determine the effect on glycosylation.
RESULTS
Phosphoglucomutase 1 enzyme activity was markedly diminished in all patients. Mass spectrometry of transferrin showed a loss of complete N-glycans and the presence of truncated glycans lacking galactose. Fibroblasts supplemented with galactose showed restoration of protein glycosylation and no evidence of glycogen accumulation. Dietary supplementation with galactose in six patients resulted in changes suggestive of clinical improvement. A new screening test showed good discrimination between patients and controls.
CONCLUSIONS
Phosphoglucomutase 1 deficiency, previously identified as a glycogenosis, is also a congenital disorder of glycosylation. Supplementation with galactose leads to biochemical improvement in indexes of glycosylation in cells and patients, and supplementation with complex carbohydrates stabilizes blood glucose. A new screening test has been developed but has not yet been validated. (Funded by the Netherlands Organization for Scientific Research and others.)
Protein n-glycosylation is a ubiquItous process in all organ systems. During N-glycosylation, glycan precursors are assembled from monosaccharide units and then covalently attached to asparagine residues in the nascent peptide chain of a protein (Fig. 1). The protein-bound glycans undergo further processing to generate mature glycoproteins. Genetic defects in protein N-glycosylation, designated as congenital disorders of glycosylation, lead to multisystem disorders. Mutations of genes involved in N-glycosylation may affect either the biosynthesis of the glycan precursor (congenital disorder of glycosylation type I [CDG-I]) or the processing of the glycan after its attachment to the protein (congenital disorder of glycosylation type II [CDG-II]) (Fig. 1).
Figure 1. Glycoprotein Biosynthesis and Congenital Disorders of Glycosylation.

For the N-glycosylation of proteins, a glycan precursor is assembled from monosaccharide units on an anchoring isoprenoid alcohol, dolichol, in the membrane of the endoplasmic reticulum. Each glycan precursor is then transferred en bloc to a specific asparagine residue in the nascent peptide chain of a protein being synthesized by a ribosome and entering the endoplasmic reticulum. Because transferrin (brown line) is abundant in blood, it is used as an indicator of the efficiency of the glycosylation system. Transferrin has two glycosylation sites. The protein is transported to the Golgi apparatus, and the glycans are modified in multiple steps. Each fully modified glycan is terminated with two negatively charged sialic acid residues. Thus, the transferrin molecule carries a total of four sialic acid residues. If a genetic defect impairs synthesis of the precursor glycan in the endoplasmic reticulum, one or both glycosylation sites of transferrin may remain unoccupied (white arrow). Such defects are classified as congenital disorders of glycosylation type I (CDG-I). In congenital disorders of glycosylation type II (CDG-II), the defect occurs in the modification of the already-transferred glycan. Both glycosylation residues may carry a glycan, but those glycans may be missing terminal sialic acid residues and end with galactose, mannose, or N-acetylglu-cosamine. Isoelectric focusing (IEF), which separates molecules according to differences in their isoelectric point, is used for investigating congenital disorders of glycosylation with the use of an agarose-gel–based system, followed by immunoprecipitation in gel with an antibody to transferrin and final staining of the precipitate with Coomassie blue. Under conditions of normal glycosylation, most transferrin molecules carry four sialic acid residues (tetrasialo-transferrin) and form a single major band on IEF. A few transferrin molecules carry five or six sialic acid residues that appear as minor bands on IEF (gray numbers; not shown in the molecular-structure diagrams). CDG-I defects are easily identified owing to the occurrence of transferrin isoforms with two or no sialic acid residues detected by means of IEF. No signal for monosialotransferrin (transferrin with one sialic acid residue) is seen in such disorders. In CDG-II, the defect can result in transferrin molecules with no, one, two, three, or four sialic acid residues. A patient with a CDG-II defect typically has bands of approximately equal intensity at all positions from 0 through 3 or a smooth gradient of intensities, whereas in most cases, tetrasialotransferrin shows a more intense signal owing to residual activity. In our patients, the signal of transferrin with two sialic acid residues (disialotransferrin) may be more intense than those for either one or three residues (as in the sample from Patient 1.1). The signal with two sialic acid residues presumably arises from a mixture of transferrin molecules that have one unoccupied glycosylation site as well as transferrin molecules that are occupied at both sites by glycans but have only two terminal sialic acid residues. The patients in this study had features of both CDG-I and CDG-II, as shown by IEF, suggesting a novel disorder.
Glucose-1-phosphate is an important intermediate in the pathways leading to protein N-glycosylation and in glucose homeostasis. Phosphoglucomutase 1 catalyzes the interconversion of glucose-6-phosphate and glucose-1-phosphate (Fig. 2).1 In the present study, we evaluated patients with phosphoglucomutase 1 deficiency, which has been identified previously as glycogenosis type XIV (Online Mendelian Inheritance in Man database number, 612934),2 and with glycosylation abnormalities affecting both the attachment and the processing of N-glycans.
Figure 2. Role of Phosphoglucomutase 1 in Sugar Metabolism.

When blood glucose is low, glycogen degradation (yellow box) generates glucose-1-phosphate, which is converted by phosphoglucomutase 1 (PGM1) to glucose-6-phosphate for glucose release from the liver or subsequent catabolism in other tissues. In the reverse reaction, glucose-6-phosphate is converted to glucose-1-phosphate by means of PGM1 and is used as a substrate in the production of uridine diphosphate (UDP)–glucose, which can be used for the synthesis of glycogen or for protein glycosylation. Dietary galactose may be converted to UDP-galactose by galactose-1-phosphate uridyl-transferase (GALT) and used for protein glycosylation. UDP-galactose may also be converted into UDP-glucose by UDP-galactose epimerase (GALE). The GALT reaction is reversible; for clarity, only one direction of the coupled GALT reactions is shown. UGP denotes UDP-glucose pyrophosphorylase.
METHODS
PATIENTS
We initially identified a pair of male siblings (Patients 1.1 and 1.2) from a consanguineous family (the index family; parents were first cousins) with clinical features suggestive of congenital disorders of glycosylation, including myopathy, hepatopathy, short stature, and hypoglycemia. Analysis of serum transferrin by means of isoelectric focusing was performed to screen for congenital disorders of glycosylation.3,4 This evaluation revealed an atypical pattern (Fig. 1), suggesting a novel disorder.
Several additional affected persons were identified subsequently. A total of 19 patients from 16 families (including three sibling pairs) were included in the present study, showing clinical features suggestive of congenital disorders of glycosylation and an atypical pattern on isoelectric focusing (Fig. S1 and Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org; some features of Patients 2, 4, 5.1, 5.2, 6, 8, and 9 have been described previously2,5–8). Approval by a human subjects committee was obtained for all the clinical studies. Written informed consent was obtained from all the participants or, in the case of children, from a parent or legal guardian.
GENETIC STUDIES
Because the parents of the index family were first cousins and were unaffected, we assumed autosomal recessive inheritance of the disease. We therefore performed homozygosity mapping with DNA from the two affected siblings, using Human1M-Duo BeadChips (Illumina; see the Supplementary Appendix). Within the regions of homozygosity thus identified, we used whole-exome sequencing to identify sequence variants that might be associated with the disease. After identification of mutations in the gene for phosphoglucomutase 1 (PGM1) in both siblings (see the Supplementary Appendix), PGM1 in additional patients was analyzed by means of Sanger sequenc ing (see the Supplementary Appendix).
TISSUE SPECIMENS
Blood samples and skin-biopsy and muscle-biopsy specimens were obtained from study participants after informed consent had been provided. Leukocytes were obtained from blood samples. Fibroblast cultures were prepared from skin-biopsy specimens from 15 patients (see the Supplementary Appendix). Biopsy specimens of the vastus lateralis muscle were obtained from 4 patients. Fibroblasts pelleted from cultures, leukocytes, and muscle-biopsy specimens were frozen in liquid nitrogen for use in study assays.
PHOSPHOGLUCOMUTASE 1 EXPRESSION AND ACTIVITY
Total RNA was extracted from fibroblast pellets, and PGM1 messenger RNA (mRNA) was quantified by means of a real-time polymerase chain reaction assay (see the Supplementary Appendix). Western blot analysis was performed on cytosolic proteins extracted from fibroblast pellets with the use of a monoclonal anti-PGM1 antiody (see the Supplementary Appendix). Phos phoglucomutase 1 enzyme activity was assayed spectrophotometrically on extracts from fibroblasts, leukocytes, or skeletal-muscle cells (see the Supplementary Appendix).
GLYCOSYLATION ASSAYS
Analysis of transferrin glycosylation was performed on serum samples with the use of isoelectric focusing,3,4 sodium dodecyl sulfate–polyacrylamide-gel electrophoresis (SDS-PAGE),3 or liquid chromatography–mass spectrometry9 (Fig. 3, and the Supplementary Appendix). The cell-surface glycoprotein intercellular adhesion molecule 1 (ICAM-1), the expression of which is markedly reduced by deficient glycosylation,10 was assayed in fibroblast cultures from patients by means of Western blotting and immunofluorescence analysis with the use of a monoclonal anti–ICAM-1 antibody (see the Supplementary Appendix). Green fluorescent protein engineered for retention in the endoplasmic reticulum was modified to contain an N-glycosylation site that, when glycosylated, causes loss of fluorescence (Glyc-ER-GFP).11 Glyc-ER-GFP was transfected into patient fibroblasts in culture, and fluorescence was measured by means of quantitative microscopy (see the Supplementary Appendix).
Figure 3. Effects of Dietary Galactose on Protein Glycosylation.

The results of mass spectrometry of the intact transferrin protein and corresponding transferrin IEF profiles are shown before (Panel A) and after (Panel B) the intake of supplementary galactose. The glycan structures are shown in detail with the transferrin protein backbone (brown line). Patients with PGM1 deficiency have highly specific transferrin glycosylation profiles. Data from Patient 2 show several peaks indicating the presence of glycoforms completely lacking one or both N-glycans (white arrows), a finding that is also observed in CDG-I. There are also peaks indicating the presence of glycoforms with truncated glycans lacking galactose (ending with N-acetylglucosamine; yellow arrows). Several structures end this way, which is typically seen in CDG-II. Numbers at main peaks are deconvoluted mass (amu). Numbers next to the IEF results indicate individual bands corresponding to numbers of sialic acid residues (black numbers indicate structures shown along the respective mass spectrum, and gray numbers structures not shown). After the patient increased dietary intake of galactose, protein glycosylation was substantially improved (Panel B). Peaks indicating the presence of glycoforms with truncated glycans lacking galactose almost completely disappeared, and peaks indicating the presence of glycoforms with complete loss of one or both N-glycans were considerably reduced. For details on the method and quantification of isoforms by means of liquid chromatography–mass spectrometry, see the Supplementary Appendix.
SUGAR METABOLITE AND GLYCOGEN QUANTIFICATION
The nucleotide sugars uridine diphosphate (UDP)–glucose and UDP-galactose were extracted from cultured fibroblasts and erythrocytes from patients and were quantified by means of reverse-phase high-performance liquid chromatography (see the Supplementary Appendix).12,13 Glucose-1-phosphate was analyzed by means of a photometric method (see the Supplementary Appendix), and galactose-1-phosphate was assayed with the use of 14C-labeled UPD-glucose.14 Glycogen was extracted from fibroblasts and digested with amyloglucosidase as described previously.15 The total amount of glucose was analyzed by means of gas chromatography–mass spectrometry.16 Glycogen content in fibroblasts from patients was also assessed by means of electron microscopy (see the Supplementary Appendix).
GALACTOSE SUPPLEMENTATION IN CULTURE
Galactose (Sigma-Aldrich) was added to fibroblast culture medium to increase the concentration of intracellular UDP-galactose by means of the galac-tose-1-phosphate uridyltransferase (GALT) reaction (Fig. 2). The concentration used was 0.5 mM.
DIETARY SUPPLEMENTATION
Galactose powder was supplied by Falcento. Galactose levels in whole blood were determined in a healthy volunteer after the oral consumption of 250 ml of water in which 0.3 g of galactose per kilogram of body weight had been dissolved. Measurements of galactose levels in blood were made by means of spectrophotometry at intervals of 10 minutes during the first hour and at intervals of 30 minutes for an additional 3 hours. Lactose or galactose supplementation was administered as an aqueous solution at a dose of 0.5 to 1.0 g per kilogram per day, divided into three to six daily doses (on the basis of patient preference).
SCREENING ASSAY
To develop a potential presymptomatic screening test for phosphoglucomutase 1 deficiency, we developed a modified version of the Beutler test, which is used with Guthrie heel-prick test cards (dried blood spots) to screen for galactosemia. The Beutler test is dependent on the generation of glucose-1-phosphate and its metabolites by means of the GALT reaction (Fig. S2 in the Supplementary Appendix). We substituted glucose-1-phosphate for the substrate in the assay. We also developed a similar assay using glucose-6-phosphate, which we anticipated would bypass the enzyme defect in phosphoglucomutase 1 deficiency.
RESULTS
CLINICAL PHENOTYPE
A total of 19 affected family members, 3 to 43 years of age, were identified in 16 families (Table S1 in the Supplementary Appendix). At birth, a bifid uvula with or without cleft palate was the only clinical manifestation.
Subsequent clinical manifestations varied among the patients (Fig. 4). Signs of hepatopathy with moderately elevated serum aminotransferase levels developed in all patients. Dilated cardiomyopathy, cardiac arrest, or both occurred in six patients; three patients were listed for heart transplantation. The majority of the patients had muscle symptoms, including exercise intolerance, muscle weakness, and rhabdomyolysis. Malignant hyperthermia with severe rhabdomyolysis occurred in two patients after the administration of general anesthesia. Growth retardation was reported in all but four patients. Two girls had hypogonadotropic hypogonadism with delayed puberty. Hypoglycemia was common, especially in childhood, requiring treatment with frequent meals, complex carbohydrates, or overnight tube feeding.
Figure 4. Overview of Clinical Features of PGM1 Deficiency.
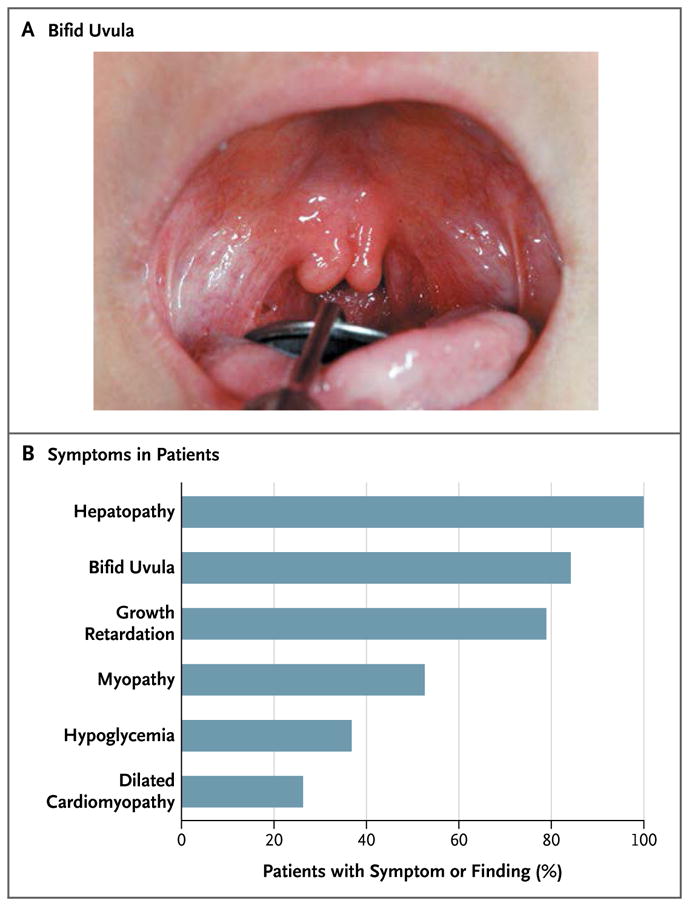
Most patients had a bifid uvula (Panel A). Symptoms or findings related to PGM1 deficiency are listed according to frequency (Panel B). Hepatopathy was defined as elevated aminotransferase levels, steatosis, fibrosis, or a combination of these features. Myopathy was defined as a maximal creatine kinase level of more than 300 U per liter. Growth retardation was defined as a height at or below the 5th percentile. Hypoglycemia was defined as a fasting glucose level of less than 2.2 mmol per liter (40 mg per deciliter).
TRANSFERRIN GLYCOSYLATION
Transferrin analysis by means of isoelectric focusing, supplemented by SDS-PAGE data, revealed that the affected family members in the other families had a distinctive pattern of glycosylation, as did the two siblings in the index family (Fig. S1 in the Supplementary Appendix). Mass spectrometry revealed the presence of a variety of transferrin glycoforms, including forms lacking one or both glycans as well as forms with truncated glycans (Fig. 3A). There was considerable variation in the transferrin-glycoform profile among the patients (Fig. S4 in the Supplementary Appendix).
GENETIC IDENTIFICATION OF PHOSPHOGLUCOMUTASE 1 DEFICIENCY
Homozygosity mapping of the two affected siblings in the index family identified autosomal regions of homozygosity totaling 87.5 mega-bases (Fig. S5 in the Supplementary Appendix). Whole-exome sequencing identified 1516 sequence variants within the regions of homozygosity. These variants were filtered to identify 10 genes carrying homozygous nonsynonymous mutations that had not already been identified as polymorphisms. Two of these genes were considered to be of potential functional relevance, but only the mutation in PGM1 cosegregated with disease. Independently, functional filtering of whole-exome sequencing data for potential CDG-I candidate genes in another patient (Patient 6 in Table S1 in the Supplementary Appendix) led to the identification of a homozygous PGM1 mutation.
A total of 21 different PGM1 mutations were identified in 16 families. Nine patients were compound heterozygotes; all others were homozygotes. The locations of the individual mutations with respect to the structural domains of the phosphoglucomutase 1 protein are shown in Fig. S6 in the Supplementary Appendix.
PHOSPHOGLUCOMUTASE 1 EXPRESSION AND ACTIVITY
Quantification of PGM1 mRNA was performed for 11 patients and showed considerable variation in expression (Fig. S7 in the Supplementary Appendix). Only two homozygous premature stop variants with predicted nonsense-mediated decay showed mRNA levels that were less than 10% of those in healthy controls. Likewise, Western blotting of phosphoglucomutase 1, performed for 13 patients, showed various amounts of protein (Fig. S8 in the Supplementary Appendix). Assays for phosphoglucomutase 1 enzyme activity, however, showed markedly decreased activity in all 17 patients who were tested, amounting to at most 12% of the activity seen in controls (Table S1 in the Supplementary Appendix).
SUGAR-METABOLITE AND GLYCOGEN QUANTIFICATION
Analyses of sugar metabolites in fibroblasts from patients revealed increased concentrations of galactose-1-phosphate and glucose-1 phosphate, as compared with fibroblasts from controls. The ratio of UDP-galactose to UDP-glucose was decreased, suggesting that UDP-galactose is limiting for N-glycan biosynthesis (see the Supplementary Appendix). Glycogen content, as assessed by means of either electron microscopy or gas chromatography–mass spectrometry after amyloglucosidase digestion, was similar to that seen in fibroblasts from controls (see the Supplementary Appendix).
EFFECT OF GALACTOSE SUPPLEMENTATION IN CULTURE
With the addition of galactose to the culture medium of fibroblasts from patients, glycosylation was substantially enhanced, as assessed by means of glycosylation of ICAM-1 (Fig. S9 in the Supplementary Appendix) and Glyc-ER-GFP (Fig. S10 in the Supplementary Appendix). The effect on Glyc-ER-GFP was further enhanced by the addition of uridine to the culture medium, which resulted in essentially normal glycosylation of this test protein (Fig. S10 in the Supplementary Appendix). Glycogen content was not affected by either galactose or glucose supplementation (Table S3 in the Supplementary Appendix).
DIETARY SUPPLEMENTATION
Oral consumption of galactose by a healthy volunteer confirmed the bioavailability of this sugar. The plasma concentration of galactose reached a maximum of 0.7 mM at 50 minutes and decreased to baseline levels after 2 hours (data not shown).
Six study participants (Patients 1.1, 1.2, 2, 5.2, 6, and 11 in Table S1 in the Supplementary Appendix) received dietary supplementation with lactose or galactose. Within 2 weeks, transferrin glycosylation had improved substantially as shown by mass spectrometry (Fig. 3B, and Fig. S11 in the Supplementary Appendix). In addition, there was substantial improvement in glycosylation of the total serum N-glycome (the set of all N-glycosylated proteins) with galactose supplementation (Fig. S12 in the Supplementary Appendix).
In Patients 2 and 6, no further episodes of rhabdomyolysis occurred after the initiation of galactose treatment, and cardiac function remained stable. In these same girls, who were 15 and 17 years of age, respectively, hypogonadotropic hypogonadism resolved within a few weeks after the initiation of galactose supplementation. Levels of luteinizing hormone, which were initially at the 2.5th percentile, increased in these two patients by a factor of 5 and a factor of 10, respectively; in addition, levels of follicle-stimulating hormone (FSH) normalized, and clinical signs of puberty appeared. Ultrasonography of the liver, which was performed every 3 months in Patients 2 and 6 during galactose supplementation, showed no increase in echogenicity and no signs of cholestasis.
SELECTIVE SCREENING
We used the standard Beutler test for galactosemia as well as two modified versions to determine whether such a test could reliably identify patients with phosphoglucomutase 1 deficiency (Fig. S2 in the Supplementary Appendix). In the standard Beutler test, the activity of the GALT pathway was significantly lower in eight study patients with phosphoglucomutase 1 deficiency than in healthy newborn controls (Fig. S13 and Table S4 in the Supplementary Appendix). The addition of phosphoglucomutase 1 from rabbit heart restored the activity of the GALT pathway in the patients to normal levels.
In a modified Beutler test with the use of glucose-6-phosphate, which is dependent on the activity of glucose-6-phosphate dehydrogenase, there was no significant difference between study patients and controls. However, in a modified Beutler test with the use of glucose-1-phosphate, which is dependent on the activity of phosphoglucomutase 1, there was a significant reduction in enzyme activity, as assessed by means of NADPH fluorescence, in the patients (Fig. S13 and Table S4 in the Supplementary Appendix).
DISCUSSION
In our study, we found that phosphoglucomutase 1 deficiency, which has been shown to be a glycogen storage disorder, is also a mixed-type congenital disorder of glycosylation. We identified 21 different mutations in PGM1 in 19 patients. Although all the patients had multisystem disease at the time of the study, the only apparent clinical feature at birth was a bifid uvula (in 16 of the 19 patients).
Extensive use of common protein polymorphisms of phosphoglucomutase 1 in paternity testing led to the discovery of a case of reduced enzyme activity in 1963, and since then additional cases of reduced phosphoglucomutase 1 activity have been described sporadically.8,17–22 In 2009, data on an adult with exercise-induced muscle cramps and episodes of rhabdomyolysis (Patient 8 in the current study) were reported.2 Abnormal glycogen accumulation was noted in the muscles, the level of phosphoglucomutase 1 activity was only 1% of the normal level, and mutations in PGM1 were identified. The disease was designated as glycogenosis type XIV. However, the broad clinical and biochemical spectrum of phosphoglucomutase 1 deficiency remained unrecognized.
In contrast to many other congenital disorders of glycosylation, in which patients have psychomotor retardation, the brains of our patients were not affected. This is probably explained by the fact that other isoenzymes substitute for phosphoglucomutase 1 in the brain (see the Supplementary Appendix).
We did not attempt to show experimentally the mechanisms that account for the clinical phenotypes of phosphoglucomutase 1 deficiency. However, analysis of the literature provides at least three different mechanisms. Hypoglycemia, lactic acidosis, and exercise intolerance are presumably consequences of disordered glucose metabolism. After a dietary glucose load, large amounts of glucose-6-phosphate are generated, which cannot be converted into glycogen. Thus, a substantial portion is diverted to lactate and fatty acid production (Fig. 2). During fasting, liver glycogen cannot be converted into glucose via glucose-6-phosphate, and hypoglycemia may occur. The inability of skeletal muscles to quickly use glycogen for the anaerobic production of energy leads to exercise intolerance and potentially to rhabdomyolysis. These characteristics are also seen with glycogen storage disease Ia, in which there is a defect in hepatic glucose-6-phosphatase. Stabilizing glucose homeostasis by means of dietary intake of complex carbohydrates23 and restricting exercise to a level below the aerobic threshold might be beneficial.
Two endocrinologic features of phosphoglucomutase 1 deficiency may be directly linked to defective glycosylation. First, Patients 2 and 6 had hypogonadotropic hypogonadism without clinical manifestations of puberty. The gonadotropins and their receptors are glycoproteins, and their function has been shown to be reduced by inadequate glycosylation.24 Second, 15 patients had short stature, although the growth hormone (somatotropin) level was normal to high in the patients in whom it was measured. However, levels of insulin-like growth factor 1 (IGF-1) and IGF-binding protein 3 were low, as they are in patients with CDG-Ib, a glycosylation disorder in which dietary supplementation with mannose improves glycosylation and growth.3,25 A detailed discussion of the literature concerning the role of glycosylation in the gonadotropin and IGF-1 systems is provided in the Supplementary Appendix.
A third mechanism relates PGM1 mutations to dilated cardiomyopathy. Phosphoglucomutase 1 binds to the heart-muscle-cell–specific splice variant of ZASP (Z-band alternatively spliced PDZ-motif protein), and ZASP mutations that affect the binding of phosphoglucomutase 1 are associated with dilated cardiomyopathy.26,27 Our study shows that defects in PGM1 are also associated with dilated cardiomyopathy. A detailed discussion of this mechanism is provided in the Supplementary Appendix.
We also found that galactose supplementation can improve glycosylation both in fibroblast-cell culture and in patients. The extent to which this improvement in glycosylation mitigates the clinical features of phosphoglucomutase 1 deficiency was not investigated systematically in our study. In the six patients in this study who received dietary supplementation with galactose, some potentially important clinical changes were seen. In the two girls with hypogonadotropic hypogonadism, the levels of luteinizing hormone increased markedly, FSH levels normalized, and clinical signs of puberty appeared. However, a systematic clinical study of the effects of galactose supplementation will be necessary to determine the extent to which such therapy can correct the clinical features of this condition; such studies will also incidentally help to define the pathogenesis of the individual elements of the clinical syndrome of phosphoglucomutase 1 deficiency.
We devised a clinical test for the diagnosis of phosphoglucomutase 1 deficiency, which was based on the Beutler test for galactosemia. On initial evaluation, this modified Beutler test appeared to discriminate effectively between persons with phosphoglucomutase 1 deficiency and controls. However, validation is required before the assay can be recommended for routine clinical use.
In conclusion, we found that phosphoglucomutase 1 deficiency, previously identified as a glycogen storage disorder, is also a mixed-type congenital disorder of protein N-glycosylation. The presence of a bifid uvula at birth may be an early clinical clue to the presence of this syndrome. Supplementation with galactose leads to biochemical improvement in indexes of glycosylation. The degree to which galactose supplementation may lead to clinical improvement in the disease syndrome is not yet established.
Supplementary Material
Acknowledgments
Supported by grants from the Netherlands Organization for Scientific Research (40-00506-98-9001 and 91713359, to Dr. Lefeber), the Institute for Genetic and Metabolic Disease (to Drs. Veltman and Lefeber), the Rocket Fund and the National Institutes of Health (R01DK55615, to Dr. Freeze), and ERA-NET E-Rare-2. Dr. Vanderschaeghe is a postdoctoral fellow of Fonds Wetenschappelijk Onderzoek Vlaanderen. Galactose was supplied by Falcento.
We thank Dr. S. Anghelescu for pictures of the oral cavity, Ms. I. Du Chesne for help with DNA sequencing, Ms. M. Herting for help with isoelectric focusing and sodium dodecyl sulfate–poly-acrylamide-gel electrophoresis, Ms. M. Jansen-Rust and Ms. T. Seehafer for library preparation (Leibniz-Institut für Arterioskleroseforschung), Ms. U. Mangels for determinations of galactose levels, Ms. M. Schreiner for data analysis, Drs. M. Gahr and W. Schröter for helpful discussions about their patient described in 1981, and Dr. A. Küçükçongar for discussing patient information.
APPENDIX
The authors’ full names and degrees are as follows: Laura C. Tegtmeyer, Stephan Rust, Ph.D., Monique van Scherpenzeel, Ph.D., Bobby G. Ng, B.S., Marie-Estelle Losfeld, Ph.D., Sharita Timal, B.S., Kimiyo Raymond, M.D., Ping He, Ph.D., M.D., Mie Ichikawa, M.S., Joris Veltman, Ph.D., Karin Huijben, B.Sc., Yoon S. Shin, Ph.D., Vandana Sharma, Ph.D., Maciej Adamowicz, Ph.D., Martin Lammens, Ph.D., Janine Reunert, Ph.D., Anika Witten, Ph.D., Esther Schrapers, Gert Matthijs, Ph.D., Jaak Jaeken, M.D., Ph.D., Daisy Rymen, M.D., Tanya Stojkovic, M.D., Ph.D., Pascal Laforet, M.D., François Petit, Ph.D., Olivier Aumaître, M.D., Ph.D., Elzbieta Czarnowska, Ph.D., Monique Piraud, Ph.D., Teodor Podskarbi, M.D., Charles A. Stanley, M.D., Reuben Matalon, M.D., Ph.D., Patricie Burda, Ph.D., Soraya Seyyedi, M.D., Volker Debus, M.D., Piotr Socha, M.D., Ph.D., Jolanta Sykut-Cegielska, M.D., Ph.D., Francjan van Spronsen, M.D., Ph.D., Linda de Meirleir, M.D., Pietro Vajro, Ph.D., M.D., Terry DeClue, M.D., Can Ficicioglu, M.D., Ph.D., Yoshinao Wada, M.D., Ph.D., Ron A. Wevers, Ph.D., Dieter Vanderschaeghe, Ph.D., Nico Callewaert, Ph.D., Ralph Fingerhut, Ph.D., Emile van Schaftingen, Ph.D., M.D., Hudson H. Freeze, Ph.D., Eva Morava, M.D., Ph.D., Dirk J. Lefeber, Ph.D., and Thorsten Marquardt, M.D.
The authors’ affiliations are as follows: Departments of General Pediatrics (L.C.T., J.R., E. Schrapers, T.M.) and Pediatric Cardiology (V.D.), University Children’s Hospital Münster, and Leibniz-Institut für Arterioskleroseforschung (S.R., A.W.), Münster, Molecular Genetics and Metabolism Laboratory, Munich (Y.S.S., T.P.), and City Medical Center, Wiesbaden (S.S.) — all in Germany; the Departments of Laboratory Medicine (M.S., S.T, K.H., R.A.W., D.J.L.), Neurology (S.T., D.J.L.), Pathology (M.L.), and Human Genetics (J.V.), Institute for Genetic and Metabolic Disease, Radboud University Nijmegen Medical Center, Nijmegen, and Beatrix Children’s Hospital, University Medical Center of Groningen, University of Groningen, Groningen (F.S.) — both in the Netherlands; Sanford–Burnham Medical Research Institute, La Jolla, CA (B.G.N., M.-E.L., P.H., M.I., V.S., H.H.F.); the Biochemical Genetics Laboratory, Mayo Clinic College of Medicine, Rochester, MN (K.R.); the Departments of Biochemistry and Experimental Medicine (M.A.), Pathology (E.C.), Gastroenterology, Hepatology, and Immunology (P.S.), and Metabolic Diseases (J.S.-C.), Children’s Memorial Health Institute, Warsaw, Poland; University Hospital Gasthuisberg, Leuven (G.M., J.J., D.R.), the Department of Biochemistry and Microbiology, Ghent University (D.V., N.C.), and the Unit for Medical Biotechnology, Department for Molecular Biomedical Research, Vlaams Instituut voor Bio-technologie (D.V., N.C.), Ghent, and the University of Brussels (L.M.) and De Duve Institute, Université Catholique de Louvain, Brussels (E. van Schaftingen) — all in Belgium; Centre de Référence de Pathologie Neuromusculaire Paris-Est, Institut de Myologie, Groupe Hospitalier Pitié–Salpêtrière, Assistance Publique–Hôpitaux de Paris, Paris (T.S., P.L.), Hôpital Antoine Béclère, Laboratoire de Génétique Moléculaire des Maladies Métaboliques, Clamart (F.P.), Centre Hospitalier Universitaire (CHU) Gabriel Montpied, Service de Médecine Interne, Clermont-Ferrand (O.A.), and CHU de Lyon, Service des Maladies Héréditaires du Métabolisme, Bron (M.P.) — all in France; the Divisions of Endocrinology (C.A.S.) and Pediatrics (C.F.), Children’s Hospital of Philadelphia, Philadelphia; University of Texas Medical Branch, Galveston (R.M.); Division of Metabolism (P.B.) and Swiss Newborn Screening Laboratory (R.F.), Children’s Research Center, University Children’s Hospital, Zurich, Switzerland; Department of Medicine and Surgery, Division of Pediatrics, University of Salerno, Baronissi, Italy (P.V.); Department of Pediatrics, University of South Florida Morsani College of Medicine, Tampa (T.D.); Osaka Medical Center and Research Institute for Maternal and Child Health, Osaka, Japan (Y.W.); and the Department of Pediatrics, Hayward Genetics Center, Tulane University Medical School, New Orleans (E.M.).
Footnotes
The authors’ full names, degrees, and affiliations are listed in the Appendix.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Quick CB, Fisher RA, Harris H. A kinetic study of the isozymes determined by the three human phosphoglucomutase loci PGM1, PGM2, and PGM3. Eur J Biochem. 1974;42:511–7. doi: 10.1111/j.1432-1033.1974.tb03366.x. [DOI] [PubMed] [Google Scholar]
- 2.Stojkovic T, Vissing J, Petit F, et al. Muscle glycogenosis due to phosphoglucomutase 1 deficiency. N Engl J Med. 2009;361:425–7. doi: 10.1056/NEJMc0901158. [DOI] [PubMed] [Google Scholar]
- 3.Niehues R, Hasilik M, Alton G, et al. Carbohydrate-deficient glycoprotein syndrome type Ib: phosphomannose isomerase deficiency and mannose therapy. J Clin Invest. 1998;101:1414–20. doi: 10.1172/JCI2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Jong G, van Noort WL, van Eijk HG. Optimized separation and quantitation of serum and cerebrospinal fluid transferrin subfractions defined by differences in iron saturation or glycan composition. Adv Exp Med Biol. 1994;356:51–9. doi: 10.1007/978-1-4615-2554-7_6. [DOI] [PubMed] [Google Scholar]
- 5.Mandato C, Brive L, Miura Y, et al. Cryptogenic liver disease in four children: a novel congenital disorder of glycosylation. Pediatr Res. 2006;59:293–8. doi: 10.1203/01.pdr.0000196378.30165.26. [DOI] [PubMed] [Google Scholar]
- 6.Mohamed M, Guillard M, Wortmann SB, et al. Clinical and diagnostic approach in unsolved CDG patients with a type 2 transferrin pattern. Biochim Bio-phys Acta. 2011;1812:691–8. doi: 10.1016/j.bbadis.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 7.Gehrmann J, Sohlbach K, Linnebank M, et al. Cardiomyopathy in congenital disorders of glycosylation. Cardiol Young. 2003;13:345–51. [PubMed] [Google Scholar]
- 8.Timal S, Hoischen A, Lehle L, et al. Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum Mol Genet. 2012;21:4151–61. doi: 10.1093/hmg/dds123. [DOI] [PubMed] [Google Scholar]
- 9.Biffi S, Tamaro G, Bortot B, Zamberlan S, Severini GM, Carrozzi M. Carbohydrate-deficient transferrin (CDT) as a biochemical tool for the screening of congenital disorders of glycosylation (CDGs) Clin Biochem. 2007;40:1431–4. doi: 10.1016/j.clinbiochem.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 10.He P, Ng BG, Losfeld ME, Zhu W, Freeze HH. Identification of intercellular cell adhesion molecule 1 (ICAM-1) as a hypoglycosylation marker in congenital disorders of glycosylation cells. J Biol Chem. 2012;287:18210–7. doi: 10.1074/jbc.M112.355677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Losfeld ME, Soncin F, Ng BG, Singec I, Freeze HH. A sensitive green fluorescent protein biomarker of N-glycosylation site occupancy. FASEB J. 2012;26:4210–7. doi: 10.1096/fj.12-211656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Räbinä J, Mäki M, Savilahti EM, Järvinen N, Penttilä L, Renkonen R. Analysis of nucleotide sugars from cell lysates by ion-pair solid-phase extraction and reversed-phase high-performance liquid chromatography. Glycoconj J. 2001;18:799–805. doi: 10.1023/a:1021107602535. [DOI] [PubMed] [Google Scholar]
- 13.Nakajima K, Kitazume S, Angata T, et al. Simultaneous determination of nucleotide sugars with ion-pair reversed-phase HPLC. Glycobiology. 2010;20:865–71. doi: 10.1093/glycob/cwq044. [DOI] [PubMed] [Google Scholar]
- 14.Shin YS. Galactose metabolites and disorders of galactose metabolism. In: Hommes FA, editor. Techniques in diagnostic human biochemical genetics. New York: Wiley-Liss; 1991. pp. 267–83. [Google Scholar]
- 15.Passonneau JV, Lauderdale VR. A comparison of three methods of glycogen measurement in tissues. Anal Biochem. 1974;60:405–12. doi: 10.1016/0003-2697(74)90248-6. [DOI] [PubMed] [Google Scholar]
- 16.Sickmann HM, Schousboe A, Fosgerau K, Waagepetersen HS. Compartmentation of lactate originating from glycogen and glucose in cultured astrocytes. Neurochem Res. 2005;30:1295–304. doi: 10.1007/s11064-005-8801-4. [DOI] [PubMed] [Google Scholar]
- 17.Pérez B, Medrano C, Ecay MJ, et al. A novel congenital disorder of glycosylation type without central nervous system involvement caused by mutations in the phosphoglucomutase 1 gene. J Inherit Metab Dis. 2013;36:535–42. doi: 10.1007/s10545-012-9525-7. [DOI] [PubMed] [Google Scholar]
- 18.Thomson WH, MacLaurin JC, Prineas JW. Skeletal muscle glycogenosis: an investigation of two dissimilar cases. J Neurol Neurosurg Psychiatry. 1963;26:60–8. doi: 10.1136/jnnp.26.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugie H, Kobayashi J, Sugie Y, et al. Infantile muscle glycogen storage disease: phosphoglucomutase deficiency with decreased muscle and serum carnitine levels. Neurology. 1988;38:602–5. doi: 10.1212/wnl.38.4.602. [DOI] [PubMed] [Google Scholar]
- 20.Illingworth B, Brown DH. Glycogen storage diseases, types III, IV, and VI. In: Whelan WJ, Cameron MP, editors. Control of glycogen metabolism (Ciba Foundation Symposium) London: J. & A. Churchill; 1964. pp. 336–53. [Google Scholar]
- 21.Illingworth Brown B, Brown DH. Phosphoglucomutase deficiency as a possible cause of glycogenosis. In: Dickens F, Randle PJ, Whelan WJ, editors. Carbohydrate metabolism and its disorders. New York: Academic Press; 1968. pp. 144–6. [Google Scholar]
- 22.Nakashima H, Suo H, Ochiai J, Sugie H, Kawamura Y. A case of adult onset phosphoglucomutase deficiency. Rinsho Shinkeigaku. 1992;32:42–7. (In Japanese.) [PubMed] [Google Scholar]
- 23.Weinstein DA, Wolfsdorf JI. Effect of continuous glucose therapy with uncooked cornstarch on the long-term clinical course of type 1a glycogen storage disease. Eur J Pediatr. 2002;161(Suppl 1):S35–S39. doi: 10.1007/s00431-002-1000-2. [DOI] [PubMed] [Google Scholar]
- 24.Huhtaniemi IT. Polymorphism of gonadotropin action: clinical implications. Asian J Androl. 2000;2:241–6. [PubMed] [Google Scholar]
- 25.Miller BS, Khosravi MJ, Patterson MC, Conover CA. IGF system in children with congenital disorders of glycosylation. Clin Endocrinol (Oxf) 2009;70:892–7. doi: 10.1111/j.1365-2265.2009.03531.x. [DOI] [PubMed] [Google Scholar]
- 26.Arimura T, Inagaki N, Hayashi T, et al. Impaired binding of ZASP/Cypher with phosphoglucomutase 1 is associated with dilated cardiomyopathy. Cardiovasc Res. 2009;83:80–8. doi: 10.1093/cvr/cvp119. [DOI] [PubMed] [Google Scholar]
- 27.Vatta M, Mohapatra B, Jimenez S, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42:2014–27. doi: 10.1016/j.jacc.2003.10.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.