

Abstract
Abnormal structure and function of astrocytes have been observed within the lamina cribrosa region of the optic nerve head (ONH) in glaucomatous neurodegeneration. Glutamate excitotoxicity-mediated mitochondrial alteration has been implicated in experimental glaucoma. However, the relationships among glutamate excitotoxicity, mitochondrial alteration and ONH astrocytes in the pathogenesis of glaucoma remain unknown. We found that functional N-methyl-d-aspartate (NMDA) receptors (NRs) are present in human ONH astrocytes and that glaucomatous human ONH astrocytes have increased expression levels of NRs and the glutamate aspartate transporter. Glaucomatous human ONH astrocytes exhibit mitochondrial fission that is linked to increased expression of dynamin-related protein 1 and its phosphorylation at Serine 616. In BAC ALDH1L1 eGFP or Thy1-CFP transgenic mice, NMDA treatment induced axon loss as well as hypertrophic morphology and mitochondrial fission in astrocytes of the glial lamina. In human ONH astrocytes, NMDA treatment in vitro triggered mitochondrial fission by decreasing mitochondrial length and number, thereby reducing mitochondrial volume density. However, blocking excitotoxicity by memantine (MEM) prevented these alterations by increasing mitochondrial length, number and volume density. In glaucomatous DBA/2J (D2) mice, blocking excitotoxicity by MEM inhibited the morphological alteration as well as increased mitochondrial number and volume density in astrocytes of the glial lamina. However, blocking excitotoxicity decreased autophagosome/autolysosome volume density in both astrocytes and axons in the glial lamina of glaucomatous D2 mice. These findings provide evidence that blocking excitotoxicity prevents ONH astrocyte dysfunction in glaucomatous neurodegeneration by increasing mitochondrial fission, increasing mitochondrial volume density and length, and decreasing autophagosome/autolysosome formation.
Keywords: optic nerve head astrocyte, glaucoma, excitotoxicity, mitochondrial fission
Introduction
Primary open angle glaucoma (POAG), the most common form of glaucoma in the United States, has been characterized by a slow and progressive degeneration of retinal ganglion cells (RGCs) and their axons, leading to loss of visual function (Weinreb and Khaw, 2004; Weinreb et al., 2014). Regardless, the biological basis of glaucoma is not yet fully understood, and the factors contributing to its progression are currently not well characterized. Intraocular pressure (IOP) is the only proven treatable risk factor. However, lowering IOP is not enough for reducing disease progression (Weinreb and Khaw, 2004; Weinreb et al., 2014; Zhang et al., 2012).
Astrocytes in the central nervous system (CNS) contribute to many functional roles including regulation of the blood–brain barrier, modulation of synaptic function and plasticity, regulation of energy metabolism, as well as maintenance of extracellular balance of ions and neurotransmitters (Barres, 2008; Brown and Ransom, 2007; Nedergaard et al., 2003; Pellerin et al., 2007; Ransom, 2000; Waxman et al., 1993). Of interest, astrocyte alterations that are accompanied by RGC axon loss has been implicated as important pathophysiological mechanisms in the pathogenesis of glaucomatous optic nerve head (ONH) degeneration (Hernandez et al., 2008; Tezel, 2006). Indeed, structural and functional abnormalities of astrocytes within the lamina cribrosa region of the ONH have been reported in experimental rodent models of glaucoma and in patients with POAG (Dai et al., 2012; Hernandez et al., 2008; Son et al., 2010; Sun et al., 2010). However, the biological basis of the pathophysiological mechanisms within glaucomatous ONH astrocytes is not well understood.
Increasing evidence indicates that alterations in the regulation of mitochondrial dynamics, fusion and fission, can trigger neurodegeneration (Chen and Chan, 2005; Karbowski and Youle, 2003; Song et al., 2011). It is noteworthy that since mitochondrial respiration-mediated dysfunction has been observed in patients with POAG (Abu-Amero et al., 2006; He et al., 2008; Izzotti et al., 2011), our previous studies have shown that alteration of mitochondrial dynamics is linked to mitochondrial dysfunction-mediated ONH degeneration and RGC death in a mouse model of glaucoma (Ju et al., ,,). These observations strongly suggest the presence of a distinct mitochondrial dysfunction-mediated degenerative pathway in the ONH of glaucoma.
Glutamate excitotoxicity triggers mitochondrial dysfunction in the CNS in both acute and chronic neurodegenerative disorders including glaucoma (Beal, 1995; Ju et al., 2009; Nicholls and Ward, 2000; Nguyen et al., 2011a, b,). There is growing evidence that glutamate excitotoxicity contributes to alteration of mitochondrial dynamics, leading to mitochondrial dysfunction and cell death (Grohm et al., 2012; Jahani-Asl et al., 2011; Nguyen et al., ,). Of interest, recent studies in mouse, rat, and humans indicate that CNS astrocytes express functional glutamate N-methyl-d-aspartate (NMDA) receptors (NRs; Krebs et al., 2003; Lee et al., 2010; Palygin et al., 2011), suggesting that there is a direct response of CNS astrocytes to extracellular glutamate. Moreover, blocking glutamate excitotoxicity by selective NR antagonists such as memantine (MEM) and MK801 partially promotes cell survival in human primary astrocyte cultures (Lee et al., 2010). However, it is unknown whether there are functional NRs on human ONH astrocytes. Also unknown is the potential relationship between NR activation and mitochondrial alteration in rodent and human ONH astrocytes.
Here, we report that glaucomatous human ONH astrocytes that have functional NRs upregulate the expression levels of NRs and glutamate aspartate transporter (GLAST), as well as increase dynamin-related protein 1 (DRP1) and its phosphorylation at Serine 616 (pDRP1) that are associated with mitochondrial fission. Moreover, blockade of glutamate excitotoxicity by MEM not only prevents morphological alteration of ONH astrocytes, but also protects ONH astrocytes by increasing mitochondrial fission, increasing mitochondrial volume density and length, and decreasing autophagosome/autolysosome formation against glaucomatous neurodegeneration.
Materials and Methods
Human Eyes
Thirteen eyes from eleven Caucasian American (CA) donors (age 73 ± 9 years) with POAG (referred to as CAG) and six eyes from three African American donors (age 62 ± 13 years) with POAG (referred to as AAG) were used to generate ONH astrocyte cultures as described previously (Lukas et al., 2008). Normal eyes were from 12 CA donors (age 60 + 11 years) and 12 AA donors (age 58 + 12 years) with no history of eye disease, diabetes, or chronic central nervous system disease as described previously (Lukas et al., 2008).
Human ONH Astrocyte Culture
Cultures of human ONH astrocytes were generated as described previously (Yang and Hernandez, 2003). Briefly, explants from each lamina cribrosa were dissected and maintained in DMEM/F-12 supplemented with 10% fetal bovine serum (FBS; BioWhittaker, Walkersville, MD) in a 37°C, 5% CO2 incubator. After 2–4 weeks, primary human ONH astrocytes were purified using a modified immunopanning procedure (Yang and Hernandez, 2003).
Animals
Bacterial artificial chromosome (BAC) aldehyde dehydrogenase 1 family, member L1 (ALDH1L1) enhanced green fluorescent protein (eGFP) reporter (BAC ALDH1L1 eGFP, GENESAT project) and B6.Cg-Tg [Thy1-cyan fluorescent protein (CFP)] 23Jrs (Thy1-CFP, The Jackson Laboratory, Bar Harbor, Maine) mice have been described in detail previously (Doyle et al., 2008; Feng et al., 2000; Heiman et al., 2008; Leung et al., 2008; Yang et al., 2011). Female DBA/2J (D2, The Jackson Laboratory, Bar Harbor, Maine) and C57BL/6 mice (C57BL, Harlan Sprague Dawley, Indianapolis, IN) were used. All animals were housed in covered cages, fed with a standard rodent diet ad libitum, and kept on a 12 h light/12 h dark cycle. All procedures described were performed in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, and under protocols approved by Institutional Animal Care and Use Committee at the University of California, San Diego.
NMDA Injection
The 4-month-old ALDH1L1 and Thy1-CFP mice were anesthetized with intraperitoneal injection of a mixture of ketamine (100 mg/kg, Ketaset; Fort Dodge Animal Health, Fort Dodge, IA) and xylazine (9 mg/kg, TranquiVed; VEDCO, St. Joseph, MO). A glass needle was used to inject a total 1 µL of 40 mM NMDA (Sigma, St. Louis, MO) in balanced saline solution into the vitreous humor (Libby et al., 2005).
IOP Measurement
IOP measurement was performed as described previously (Ju et al., 2008). C57BL and D2 mice used in this study had IOP measurement at 7 and/or 9 months of age (to confirm development of spontaneous IOP elevation exceeding 20 mmHg) after anesthesia with a mixture of ketamine (100 mg/kg, Ketaset; Fort Dodge Animal Health) and xylazine (9 mg/kg, TranquiVed, Vedeco).
Pharmacological Treatment of ONH Astrocytes
Three groups of human ONH astrocytes were studied following treatment of NMDA alone (Sigma) or NMDA plus MEM, an uncompetitive NMDA glutamate receptor antagonist (Sigma): a group treated with vehicle (1 mM glycine and 1.8 mM CaCl2 in EBSS), a group treated with NMDA alone (100 μM with 1 mM glycine and 1.8 mM CaCl2 in EBSS) and a group treated with NMDA (100 μM)/MEM (200 μM). NMDA alone or NMDA/MEM were treated in 1% FBS/DMEM/Ham's F-12 50/50 (DMEM/F12) for 1 h in a 5% CO2 incubator at 37°C and postincubated for 10 h. For the in vivo study, both MEM (5 mg/kg in 0.9% saline) and vehicle (0.9% Saline) were treated in D2 mice twice daily for 3 months by IP injection as described previously (Ju et al., 2009).
Tissue Preparation
Mice were anesthetized with a mixture of ketamine (100 mg/kg, Ketaset; Fort Dodge Animal Health) and xylazine (9 mg/kg, TranquiVed, Vedeco), and perfused transcardially with oxygenated Ringer's solution (0.79% NaCl, 0.038% KCl, 0.02% MgCl2·6H2O, 0.018% Na2HPO4, 0.125% NaHCO3, 0.03% CaCl2·2H2O, 0.2% dextrose, and 0.02% xylocaine) at 37°C for 30 sec, followed by 0.1 M phosphate-buffered saline (PBS), pH 7.4, containing 4% paraformaldehyde. For immunohistochemistry, the ONHs were dissected from the choroids and postfixed with 4% paraformaldehyde in PBS, pH 7.4 for 4 h at 4°C. After several washes in PBS, the retinas were dehydrated through graded ethanol solutions and embedded in polyester wax as described previously (Ju et al., 2008).
Immunohistochemistry and Immunocytochemistry
Immunohistochemical or immunocytochemical staining for 7 µm wax sections of ONHs or cultured ONH astrocytes were performed as described previously (Ju et al., 2008). Five sections per wax block from each group (n = 4 ONHs/group) were used for immunohistochemical analysis. The primary antibodies included mouse monoclonal anti-glial fibrillary acidic protein (GFAP) antibody (1:300; Sigma), guinea pig polyclonal anti-GFAP antibody (1:500; Advanced ImmunoChemical. Long Beach, CA), mouse monoclonal antineurofilament antibody (1:500; Sigma), mouse monoclonal anti-NR1 antibody (1:1,000; BD Pharmingen, San Diego, CA), rabbit monoclonal anti-NR2A antibody (1:100; Millipore, Billerica, MA), rabbit polyclonal anti-NR2B antibody (1:5,000; Millipore), and mouse monoclonal anti-DRP1 antibody (1:1,000; BD Transduction Laboratories, San Diego, CA). The images were acquired with a FluoView1000 confocal microscope (Olympus, Tokyo, Japan).
Western Blot Analysis
Cultured human ONH astrocytes were lysed with lysis buffer as described previously (Noh et al., 2013). The primary antibodies included rabbit polyclonal anti-NR1 and NR2A antibody (1:1,000; Cell Signaling Technology, Danvers, MA), rabbit polyclonal anti-GLAST (EAAT1, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal anti-DRP1 antibody (1:1,000; BD Transduction Laboratories), rabbit polyclonal anti-phospho-DRP1 at Ser616 antibody (1:1,000; Cell Signaling), and mouse monoclonal anti-actin antibody (1:5,000; Millipore). The scanned film images were analyzed with ImageJ (National Institute of Health, MD) and band densities were normalized to the band densities for actin.
Ca2+ Measurements
Cells were incubated with Buffer A supplemented with 2 µM Fluo4-AM (Invitrogen) for 15 min in a humidified 5% CO2 incubator at 37°C. After washing with Buffer A, cells were incubated in F12/DMEM containing 1% FBS for 30 min in a humidified 5% CO2 incubator at 37°C to complete hydrolysis of the Fluo4-AM before making the Ca2+ measurements. Fluorescent signals were recorded for 5–10 min at 37°C, the perfusion medium (Buffer A) was replaced by Buffer A containing 100 µM NMDA with 5 µM Glycine. The recording continued for up to 5 min. Fluo4 Ca2+ signals were recorded in 3–5 cells in each experiment. The response (ΔF/F) was 1.01 ± 0.17 (mean ± SEM, n = 3). ΔF/F was calculated by the following formula. ΔF/F = (FNMDA − Frest)/(Frest − Fmin), where Fmin was determined by saturating the indicator by adding 2 mM MnCl2. The experiment was repeated three times. Ca2+ imaging was carried out on a FlowView 1000 confocal microscope (Olympus) equipped with a 40× objective (NA 1.30, oil) and the dye-loaded cells were excited at 488 nm. Transmitted light differential interference contrast imaging guided the drawing of circular regions of interest (ROIs) in the cytoplasmic area of cells. The fluorescent signal intensity (em 500–600 nm) of each ROI was averaged and plotted every 5 s. After signal stabilization of fluorescent signals, cells were stimulated by 100 µM NMDA and 5 µM glycine.
Whole-Mount Preparation and Quantitative Analysis for RGC Counting
Retinas from enucleated eyes in Thy1-CFP mice (Feng et al., 2000; Leung et al., 2008) were dissected as flattened whole-mounts (Ju et al., 2009). Images were captured with a spinning-disc confocal microscope (Olympus America, Center Valley, PA). RGC densities were measured in the middle area for each condition and the scores were averaged (n = 2 retinal flatmounts/group).
Morphology Analysis of Mitochondria
Mitochondria in the ONH astrocytes were labeled by the addition of a red fluorescent mitochondrial dye (100 nM final concentration; MitoTracker Red CMXRos; Invitrogen) to the cultures and maintaining it for 20 min in a CO2 incubator. The cultures were subsequently fixed with 4% paraformaldehyde (Sigma) in DPBS for 30 min at 4°C. For three-dimensional (3D) reconstruction, images were obtained with an optical section separation (z-interval) of 0.25 μm by an Olympus FluoView1000 (Olympus). Iso-surface rendition was obtained from the stack using Imaris 6.4.2 (Bitplane AG, Zurich, Switzerland).
Electron Microscopy
Following fixation as previously described (Ju et al., 2008; Noh et al., 2013), the cells and tissues were embedded in Durcupan ACM resin (Fluka, St. Louis, MO). Ultrathin (70 nm) sections were poststained with uranyl acetate and lead salts and evaluated with a JEOL 1200FX transmission EM operated at 80kV. Images were recorded on film at 8,000× magnification. For quantitative analysis, the number of astrocytic mitochondria was normalized to the total area occupied by astrocytes in each image, which was measured using ImageJ (National Institute of Health, Bethesda, MD). Mitochondrial lengths were measured with ImageJ. The mitochondrial volume density, defined as the volume occupied by mitochondria divided by the volume occupied by the cytoplasm, was estimated using stereology as follows. A 112 × 112 square grid (112 × 112 chosen for ease of use with Photoshop) was overlaid on each image loaded in Photoshop (Adobe), and mitochondria and cytoplasm lying under intercepts were counted. The relative volume occupied by mitochondria was expressed as the ratio of intercepts coinciding with this organelle relative to the intercepts coinciding with cytoplasm.
Electron Microscope Tomography
For each reconstruction, a series of images at regular tilt increments was collected with a JEOL 4000EX intermediate-voltage electron microscope operated at 400 kV. The IMOD package was used for rough alignment with the fine alignment and reconstruction performed using the TxBR package. Volume segmentation was performed by manual tracing in the planes of highest resolution with the program Xvoxtrace (Perkins et al., 1997). The mitochondrial reconstructions were visualized using Analyze (Mayo Foundation, Rochester, MN) or the surface-rendering graphics of Synu (National Center for Microscopy and Imaging Research, San Diego, CA) as previously described (Perkins et al., 1997). Movies of the tomographic volume were made using Amira (Ju et al., 2008; Kushnareva et al., 2013).
Intracellular Dye Injection
The method for filling cells in fixed tissue slices was adapted from previously reported protocols (Belichenko and Dahlstrom, 1995; Buhl, 1993). The ONH slices were viewed with an Olympus Optical (Melville, NY) BX50WI infrared differential interference contrast (DIC)/epifluorescent microscope. Sharp glass micropipettes were backfilled with 10 mM Alexa Fluor 568 in 200 mM KCl, 10 mM Alexa Fluor 488 in 200 mM KCl (Molecular Probes, Eugene, OR), or 5% aqueous dilithium Lucifer yellow CH (Calbiochem, La Jolla, CA). The astrocytes were identified by the distinctive size and shape of their soma. The somata were impaled, and the dye was injected into the cells by applying a 0.5 sec negative current pulse (1 Hz) until the processes were completely filled. After several cells were filled in a tissue slice, the slice was placed in cold 4% paraformaldehyde/PBS for 30 min. Images were acquired with a FluoView1000 confocal microscope (Olympus).
Serial Block-Face Scanning Electron Microcopy
The ONHs were washed in cacodylate buffer for 2 h at 4°C and placed in cacodylate buffer containing 2% OsO4/1.5% potassium ferrocyanide for 3 h at RT. After several washing steps, the ONHs were dehydrated in a series of ice-cold ethanol solutions followed by ice-cold dry acetone for 10 min. The ONHs were placed in acetone at RT for 10 min and then infiltrated with an ascending series of Durcupan:acetone solutions. The ONHs were infiltrated with 100% Durcupan and then cured at 60°C for 2 days. The ONHs were trimmed to remove excess plastic and attached to an aluminum pin, grounded with silver paint, and sputter coated with gold–palladium before imaging. Specimens were imaged on a FEI Quanta FEG equipped with a 3View serial block-face scanning electron microscopy (SBEM) system (Gatan, Pleasanton, CA). Specimens were imaged at high vacuum with 2.5-kV beam current and 70-nm sectioning thickness. A 2D montage was collected at each Z plane to increase field of view. Volumes were processed and analyzed using IMOD software (http://bio3d.colorado.edu/imod/; Kremer et al., 1996) and stereology was performed using a custom plug-in for IMOD (Hatori et al., 2012).
Statistical Analysis
Data were presented as the mean ± standard deviation (SD). Comparison of two or three experimental conditions was evaluated using the unpaired, two-tailed Student's t-test or one-way analysis of variance and the Bonferroni t-test. P < 0.05 was considered to be statistically significant.
Results
Mitochondrial Fission in Glaucomatous Human ONH Astrocytes
To test whether glaucomatous damage is associated with alteration of mitochondrial dynamics in human astrocytes of lamina cribrosa region, we performed Western blot analysis using antibodies raised against GFAP, as well as DRP1and pDRP1 that regulate mitochondrial fission. We also performed MitoTracker Red staining and TEM analysis for measuring mitochondrial length, volume density, number and cristae surface area. In the current study, we observed that glaucomatous human ONH astrocytes from patients showed significant increases of GFAP, DRP1 and pDRP1 protein expression compared with age-matched normal human ONH astrocytes (Fig. 1A), suggesting the induction of mitochondrial fission in glaucomatous human ONH astrocytes. Consistent with these results, immunocytochemical analysis showed that glaucomatous human ONH astrocytes increased GFAP and DRP1 protein expression in vitro (Fig. 1B). Interestingly, we found that there were accumulated DRP1 immunoreactivities around the nucleus of glaucomatous human ONH astrocytes (Fig. 1B). Representative images from both MitoTracker Red staining and TEM analysis showed that mitochondria from normal human ONH astrocytes appeared as long tubular forms of mitochondria (Fig. 2A,B). In contrast, glaucomatous human ONH astrocytes showed shorter and fragmented mitochondria that were accumulated around the nucleus (Fig. 2A,B). These findings suggest the possibility that glaucomatous stress might induce an altered distribution of mitochondria in human ONH astrocytes. In addition, our previous study showed that DRP1 could translocate from the cytosol to mitochondria in stress conditions such as elevated pressure (Ju et al., 2007).
Figure 1.
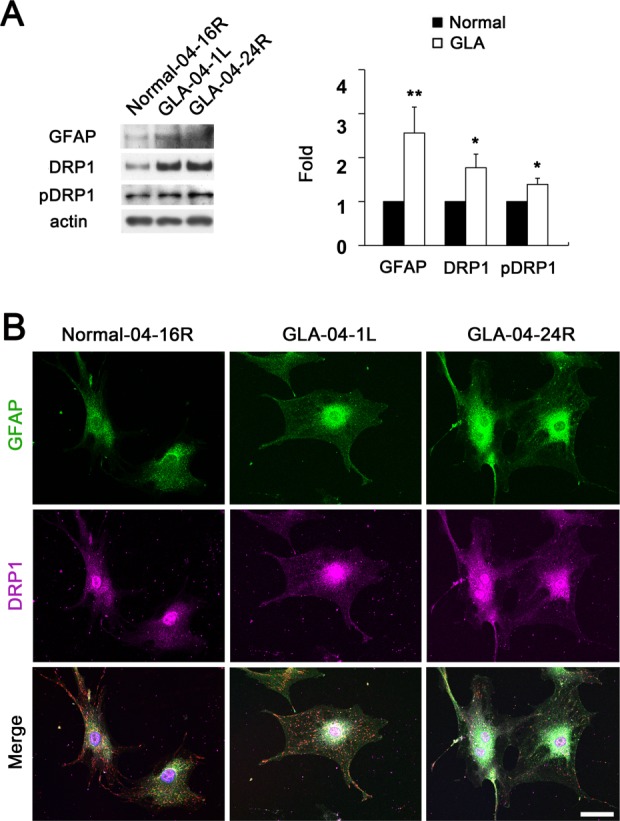
Upregulation of DRP1 and pDRP1 protein expression in glaucomatous human ONH astrocytes. (A) Glaucomatous ONH astrocytes (Patients ID# 04-1L, age 79 years and 04–24R, age 53 years) showed significant upregulation of GFAP, DRP1 and pDRP1 protein expression compared with normal ONH astrocytes (Patient ID# 04–16R, age 80 year). Data are presented as mean ± SD (* and ** denote P < 0.05 and P < 0.01, respectively). (B) Representative fluorescent photomicrographs on the expression of GFAP (green) and DRP1 (pink) in the normal and glaucomatous ONH astrocytes in vitro. Scale bars = 20 μm.
Figure 2.

Mitochondrial fission and 3D reconstructions of mitochondrial cristae in glaucomatous human ONH astrocytes. (A) MitoTracker Red staining of mitochondria in normal (Patient ID# 04–16R) and glaucomatous ONH astrocytes (Patients ID# 04-1L and 1D# 04–24R) in vitro. Nuclei were counterstained with Hoechst (blue). Scale bars = 20 μm. (B) Normal ONH astrocytes typically had elongated mitochondria profiles (arrowhead). In contrast, glaucomatous ONH astrocytes typically had small fragmented organelle profiles (arrowheads) as shown in astrocytes from two different glaucoma patients. (C) Quantitative analysis of mitochondrial lengths, volume density and number in normal and glaucomatous ONH astrocytes. Data represent mean ± SEM (* and *** denote P < 0.05 and P < 0.001, respectively). Scale bar = 500 nm. (D) Electron tomography generated high-resolution, 3D reconstructions of mitochondria from normal and glaucomatous ONH astrocytes. Surface-rendered volumes of the segmented mitochondria provide information concerning shape and cristae architecture. The outer mitochondrial membrane is shown in gray (made translucent be better visualize the cristae) and cristae are in various colors. The sites of mitochondrial fission were indicated in the mitochondrion from a glaucomatous ONH astrocyte (arrowheads). The graph shows the measurement of cristae membrane surface area in representative mitochondria. Scale bar = 500 nm.
In good agreement with these results, quantitative TEM analysis showed that mitochondrial lengths were significantly decreased in glaucomatous human ONH astrocytes (1.17 ± 0.09 µm, P < 0.001) compared with normal human ONH astrocytes (1.80 ± 0.16 µm; Fig. 2C). However, there was no difference in mitochondrial volume density, defined as the volume occupied by mitochondria divided by the volume occupied by the cytoplasm in terms of a percentage, between normal and glaucomatous human ONH astrocytes (Fig. 2C). The number of astrocytic mitochondria, normalized to the total area occupied by astrocytes in each image, was significantly increased in the glaucomatous human ONH astrocytes (0.12 ± 0.13/µm2, P < 0.05) compared with normal human ONH astrocytes (0.07 ± 0.13/µm2; Fig. 2C). Further studies for 3D tomographic reconstructions showed the reduction of the surface area of mitochondrial cristae in a glaucomatous human ONH astrocyte (0.12 µm2) compared with a normal human ONH astrocyte (0.18 µm2; Fig. 2D). Collectively, these results indicate that glaucomatous damage might trigger mitochondrial fission, as well as alters cristae formation and architecture in human ONH astrocytes of lamina cribrosa region. Of interest, in comparison with a normal patient (age 80 years), we found increased DRP1, pDRP1 and mitochondrial fission in both glaucoma patients (ages 79 and 53 years).
Upregulation of NRs and GLAST Protein Expression in Glaucomatous Human ONH Astrocytes
We next investigate whether human ONH astrocytes express NRs. We performed immunofluorescent staining using antibodies raised against NRs (NR1, 2A and 2B subunits) in the lamina cribrosa region of the human ONH tissue (Supp. Info. Fig. 1) and in cultured human ONH astrocytes in vitro. Of note, we found that NR1, 2A and 2B subunits were co-localized with the astrocyte marker, GFAP, in the lamina cribrosa region of normal human ONH (Fig. 3A,B). Furthermore, cultured human ONH astrocytes showed strong immunoreactivities for NR1, 2A and 2B subunits (Fig. 4A,B). Consistently, these immunoreactivities were correlated with GFAP immunoreactivity (Fig. 4A), indicating that human ONH astrocytes express NRs.
Figure 3.
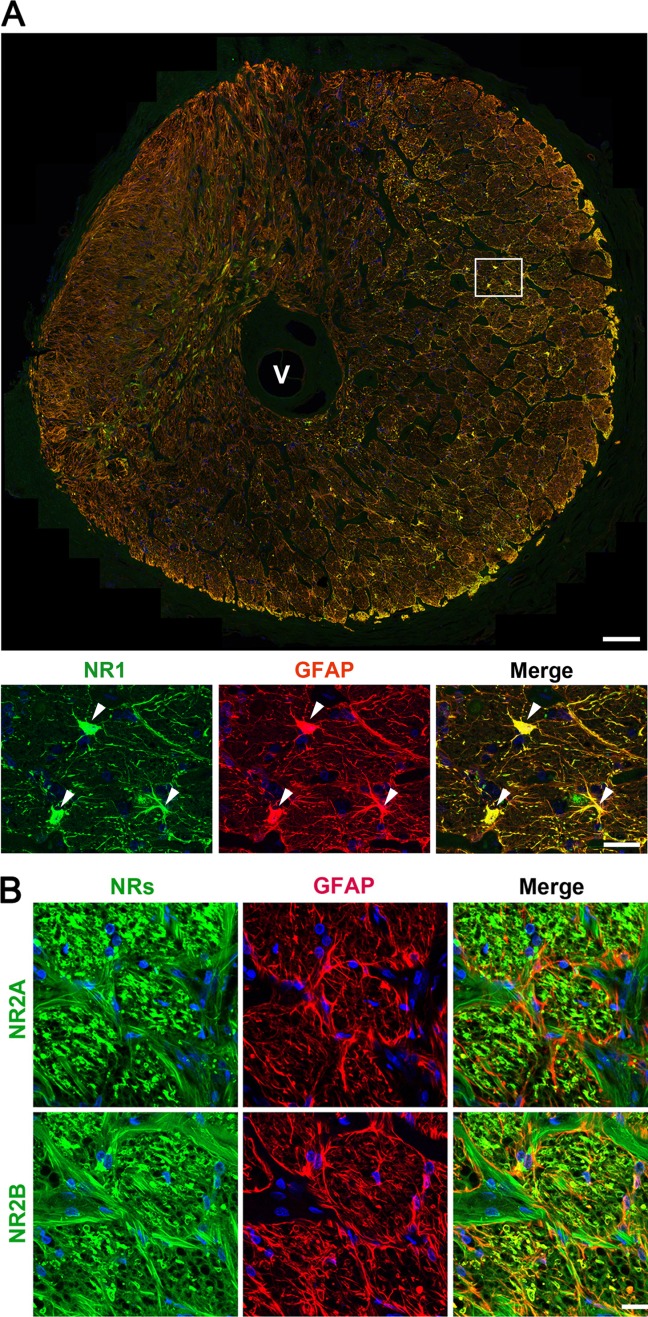
Expression of NRs in normal human ONH astrocytes. (A) Representative fluorescent photomicrographs on the expression of NR1 (green) and GFAP (red) in normal astrocytes in the lamina cribrosa. Scale bars = 100 μm (upper panel) and 20 µm (lower panel). (B) Representative fluorescent photomicrographs on the expression of NR2A and 2B (green) and GFAP (red) in normal human astrocytes from lamina cribrosa tissue sections. Scale bars = 20 μm.
Figure 4.
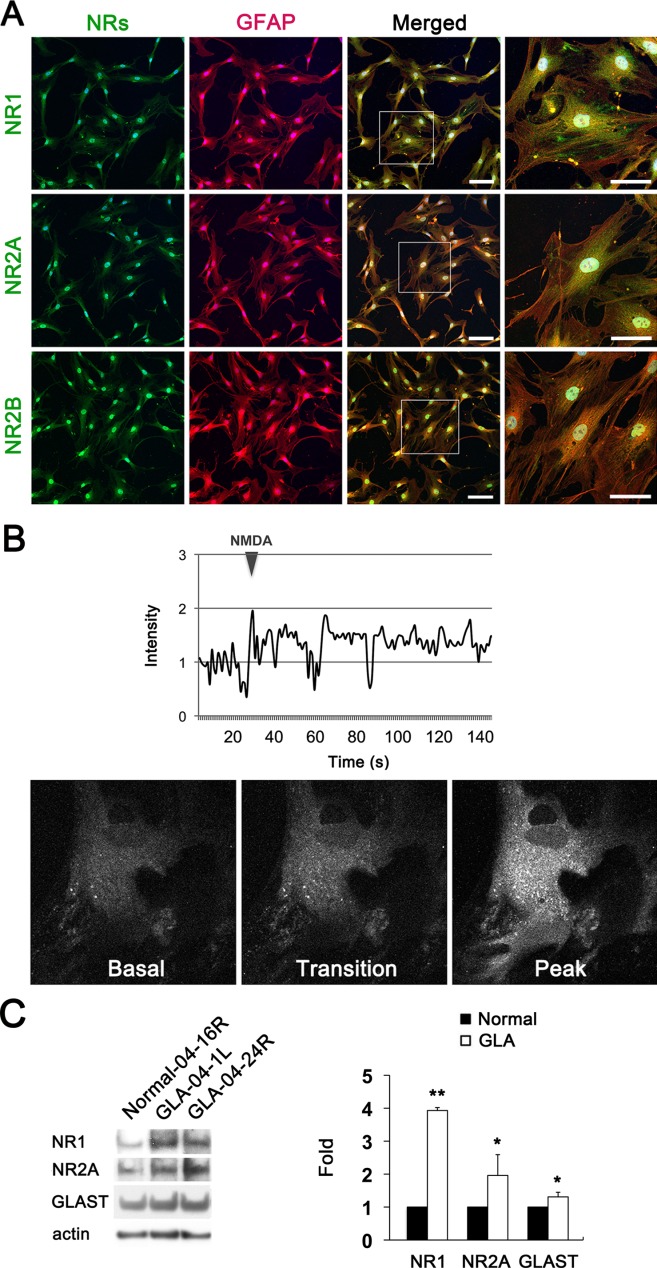
Upregulation of functional NRs and GLAST protein expression in glaucomatous human ONH astrocytes. (A) Representative fluorescent photomicrographs on the expression of NR1, 2A and 2B (green) and GFAP (red) in cultured normal ONH astrocytes in vitro. Scale bars = 20 μm. (B) Ca2+ measurements in normal human ONH astrocytes in vitro. Cultured normal ONH astrocytes were exposed to 100 μM NMDA with 5 μM glycine and measured cytoplasmic Ca2+ level ([Ca2+]cyto) using a fluorescent Ca2+-sensitive dye, Fluo-4. (C) Representative immunoblots of NR1 and 2A, and GLAST in normal and glaucomatous ONH astrocytes. Data are presented as mean ± SD (* and ** denote P < 0.05 and P < 0.01, respectively).
To further determine whether NRs expressed on human ONH astrocytes are functional, we exposed cultured human ONH astrocytes to 100 µM NMDA with 5 µM glycine and observed in cytoplasmic Ca2+ level ([Ca2+]cyto) using the fluorescent Ca2+-indicator, Fluo-4. Consistent with our immunostaining observations, we found that NMDA with glycine induced an oscillatory increase in [Ca2+]cyto in human ONH astrocytes (Fig. 4B). Based on our findings for intracellular Ca2+ response to NMDA in ONH astrocytes, these results indicate that human ONH astrocytes could express functional NRs. In view of the expression of functional NRs in human ONH astrocytes, we determined whether glaucomatous damage alters NRs and the glutamate transporter (GT) system in ONH astrocytes that may reflect glial toxicity. We performed Western blot analysis using antibodies raised against NRs (NR1 and NR2A subunits) and GLAST, one of the main glial GTs. In comparison with normal human ONH astrocytes, glaucomatous human ONH astrocytes showed significant increases of NR1, NR2A and GLAST protein expression (P < 0.05; Fig. 4C).
Glutamate Excitotoxicity Triggers Mitochondrial Fission in Mouse ONH Astrocytes
To verify whether NMDA-induced glutamate excitotoxicity can directly induce morphological change and mitochondrial fission in astrocytes of the glial lamina in vivo, we injected NMDA into the vitreous of the eyes in BAC ALDH1L1 eGFP mice to examine morphological change and Thy1-CFP mice to examine mitochondrial fission. ALDH1L1 is known as the new astroglial marker that selectively labels adult cortical and spinal cord astrocytes (Cahoy et al., 2008; Yang et al., 2011). Examination of changes in astrocyte shape in situ has been made possible by the development of BAC ALDH1L1 eGFP mice (Cahoy et al., 2008; Doyle et al., 2008; Heiman et al., 2008; Yang et al., 2011). Using BAC ALDH1L1 eGFP mice, we first found that astrocytes in the glial lamina showed eGFP expression (Fig. 5A,B). Second, NMDA induced activation of astrocytes by increasing eGFP and GFAP protein expression, and by inducing hypertrophic morphology in the glial lamina at 1 and 3 days after injection (Fig. 5A,B). Third, NMDA triggered axon damage as evidenced by accumulating increased neurofilament protein expression in the glial lamina (Fig. 5A).
Figure 5.
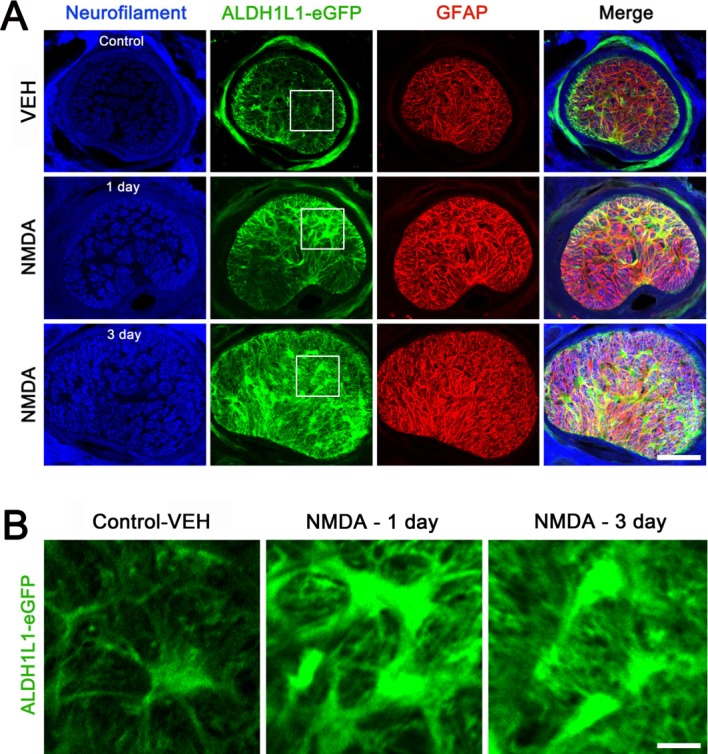
NMDA-mediated glutamate excitotoxicity induces abnormal hypertrophic morphology in astrocytes of the glial lamina in BAC ALDH1L1 eGFP mice. (A and B) Representative fluorescent photomicrographs on the expression of neurofilament (blue), ALDH1L1 (green) and GFAP (red) in the glial lamina of BAC ALDH1L1 eGFP mice treated with vehicle (VEH) or NMDA (40 mM) for 1 and 3 days. (B) Higher magnification showed hypertrophic ONH astrocytes in BAC ALDH1L1 eGFP mice treated with NMDA at 1 and 3 days. Scale bars = 50 μm.
In contrast, Thy-1 promoter-driven CFP expression is visualized on the cell bodies of adult RGCs in Thy1-CFP mice (Leung et al., 2008). We found that retinal flatmounts confirmed approximately 90% loss of CFP-expressing retinal neurons in Thy1-CFP mice at 3 days after NMDA injection (P < 0.001; Fig. 6). Furthermore, axonal damage and loss had spread into the glial lamina of Thy1-CFP mice (Fig. 7A). Together with these data confirming NMDA-induced ONH degeneration and RGC death, 3D tomographic reconstructions showed a tubular form of mitochondrial morphology in astrocytes of the glial lamina in control Thy1-CFP mice. In addition, the mitochondrion showed the cristae that have both tubular and lamellar compartments, which is the most common archetype of brain mitochondria (Fig. 7B). In contrast, mitochondria from NMDA-treated astrocytes of the glial lamina in Thy1-CFP mice had shorter and rounder structures. However, there was no difference in cristae architecture compared with control (Fig. 7B). See the Supporting Information Movies 1 and 2 for presentations of the 3D tomography data.
Figure 6.
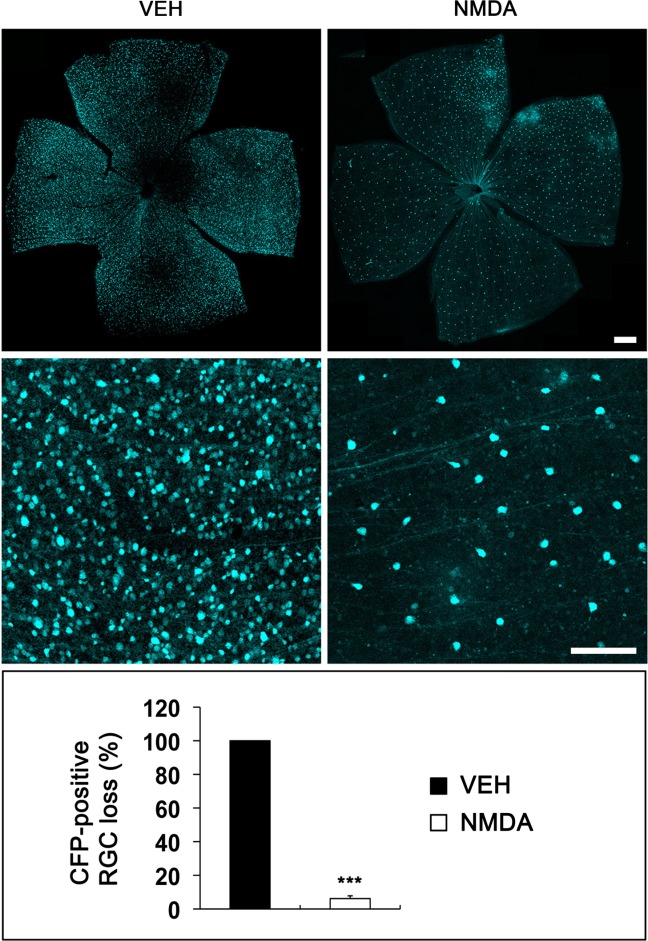
NMDA-induced RGC loss in Thy1-CFP mice. Representative photomicrographs on RGC loss in Thy1-CFP mice exposed to vehicle (VEH) or NMDA (40 mM) for 3 days after injection in vivo. NMDA induced a massive loss of CFP-expressing RGCs in Thy1-CFP mice compared with VEH-treated mice. Data are presented as mean ± SD (*** denotes P < 0.001). Scale bars = 50 μm.
Figure 7.
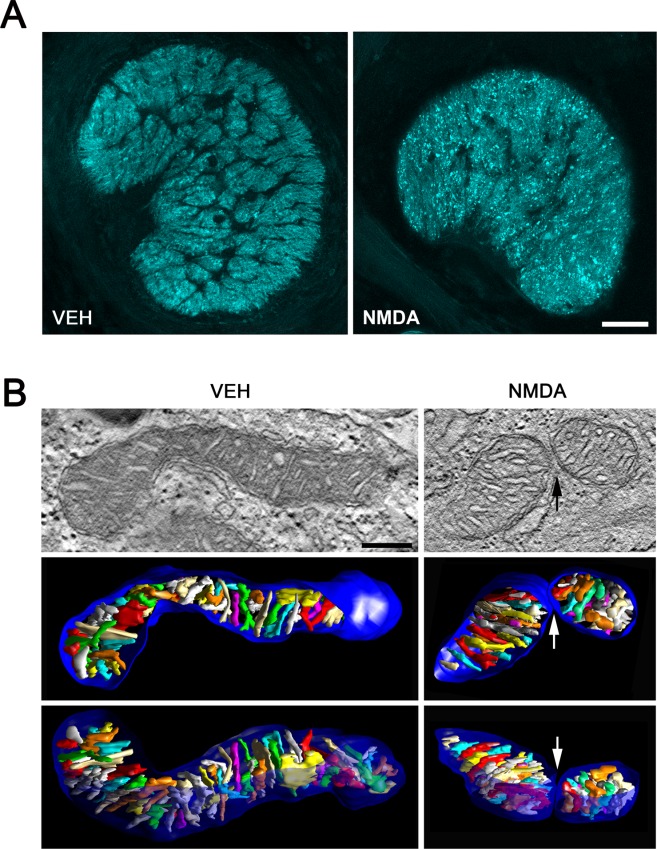
NMDA-mediated glutamate excitotoxicity triggers mitochondrial fission in astrocytes of the glial lamina in Thy1-CFP mice. (A) Representative fluorescent photomicrographs on the dispersed axon degeneration (cyan) and mitochondrial fission in astrocytes of the glial lamina of Thy1-CFP mice treated with vehicle (VEH) or NMDA (40 mM) for 3 days. Scale bars = 100 μm. (B) Typical 3D tomographic reconstructions of mitochondria from mouse ONH astrocytes treated with VEH or NMDA for 3 days. Left: Top, A 2.4 nm slice through the center of a tomographic volume of a mouse ONH astrocyte treated with vehicle shows the mitochondrial outer and cristae membranes in a long mitochondrion with many cristae. Middle (top view) and Bottom, side view of the segmented and surface-rendered volume showing the outer membrane (blue) and the entire complement of cristae (various colors) provide a 3D snapshot of the packing arrangement, shape, size and density of cristae. Right: Top, A 2.4 nm slice through the center of a tomographic volume of a mouse ONH astrocyte treated with NMDA shows the mitochondrial outer and cristae membranes at the site of mitochondrial fission. Middle (top view) and Bottom, side view of the segmented and surface-rendered volume show the arrangement of the two mitochondrial fragments and their membranes. Scale bar = 200 nm.
Blockade of Glutamate Excitotoxicity Increases the Number of Mitochondria as well as Mitochondrial Length and Volume Density in Human ONH Astrocytes
We next determined whether blockade of glutamate excitotoxicity by MEM prevents alteration of the intracellular mitochondrial network in human ONH astrocytes in vitro. Consistently, MitoTracker Red staining and representative 2D images from TEM showed that control human ONH astrocyte treated with vehicle contained long tubular forms of mitochondria (Fig. 8A,B). In contrast, human ONH astrocytes treated with NMDA contained shorter and fragmented mitochondria (Fig. 8A,B). However, MEM treatment preserved mitochondrial morphology in human ONH astrocyte against NMDA-induced glutamate excitotoxicity (Fig. 8A,B). Of interest, quantitative TEM analysis showed significant decreases of mitochondrial length (1.00 ± 0.03 µm, n = 133, P < 0.01), volume density (2.30 ± 0.38%, n = 13 mitochondria, P < 0.01) and number (0.08 ± 0.02/µm2, n = 16, P < 0.05) in NMDA-treated human ONH astrocytes compared with vehicle-treated control human ONH astrocytes (1.35 ± 0.02 µm of lengths, n = 138; 5.10 ± 0.63% of volume density, n = 11; 0.17 ± 0.03/µm2 of number, n = 11; Fig. 8B). More importantly, MEM treatment significantly increased mitochondrial length (1.44 ± 0.03 µm, n = 90, P < 0.001), volume density (3.50 ± 0.34%, n = 11, P < 0.05) and number (0.14 ± 0.02/µm2, n = 23, P < 0.01) in human ONH astrocytes against NMDA-induced glutamate excitotoxicity (Fig. 8B).
Figure 8.
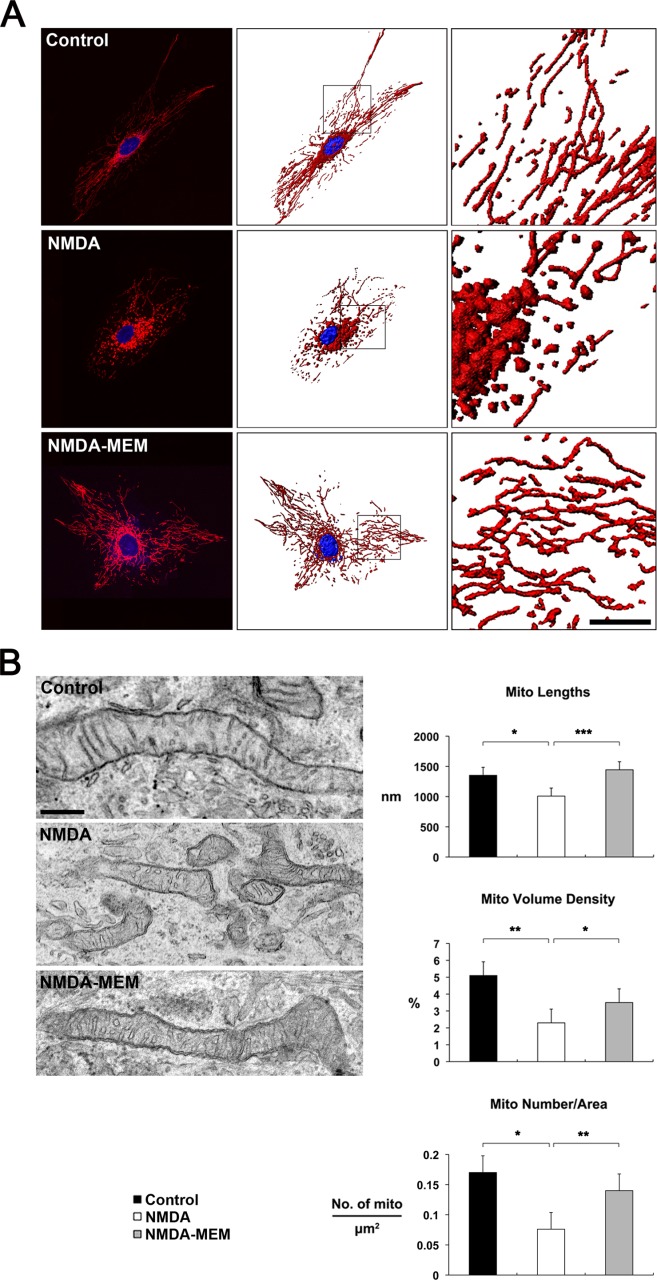
Blockade of NMDA-mediated glutamate excitotoxicity increases the number of mitochondria as well as mitochondrial length and volume density in human ONH astrocytes in vitro. (A) MitoTracker Red staining of mitochondria in normal human ONH astrocytes exposed to control vehicle, NMDA alone or NMDA/MEM. Nuclei were counterstained with Hoechst (blue). Scale bars = 20 μm. (B) Quantitative analysis of mitochondrial lengths, volume density and number in ONH astrocytes exposed to NMDA alone or NMDA/MEM. Data represent mean ± SEM (*, ** and *** denote P < 0.05, P < 0.01 and P < 0.001, respectively). Scale bar = 500 nm.
Blockade of Glutamate Excitotoxicity Prevents Morphological Alteration in Astrocytes of the Glial Lamina in Glaucomatous D2 Mice
Reactive astrocytes are a pathological hallmark of axonal degeneration and contribute to glial scar formation in glaucoma. We determined whether reactive astrocytes express NRs in the glial lamina of glaucomatous D2 mice. In good agreement with data from human ONH astrocytes, astrocytes expressing GFAP were positive for NRs (NR1 and NR2A) in the glial lamina of age-matched control C57BL and glaucomatous D2 mice (Fig. 9A). As previously reported (Sun et al., 2010), we found that there was a glial scar formation that had a dense and disorganized meshwork of thickened processes in the glial lamina of glaucomatous D2 mice treated with vehicle. In addition, glaucomatous astrocytes showed a greater density of immunoreactivities for GFAP and NRs, and hypertrophic morphology in the glial lamina of glaucomatous D2 mice (Fig. 9A).
Figure 9.
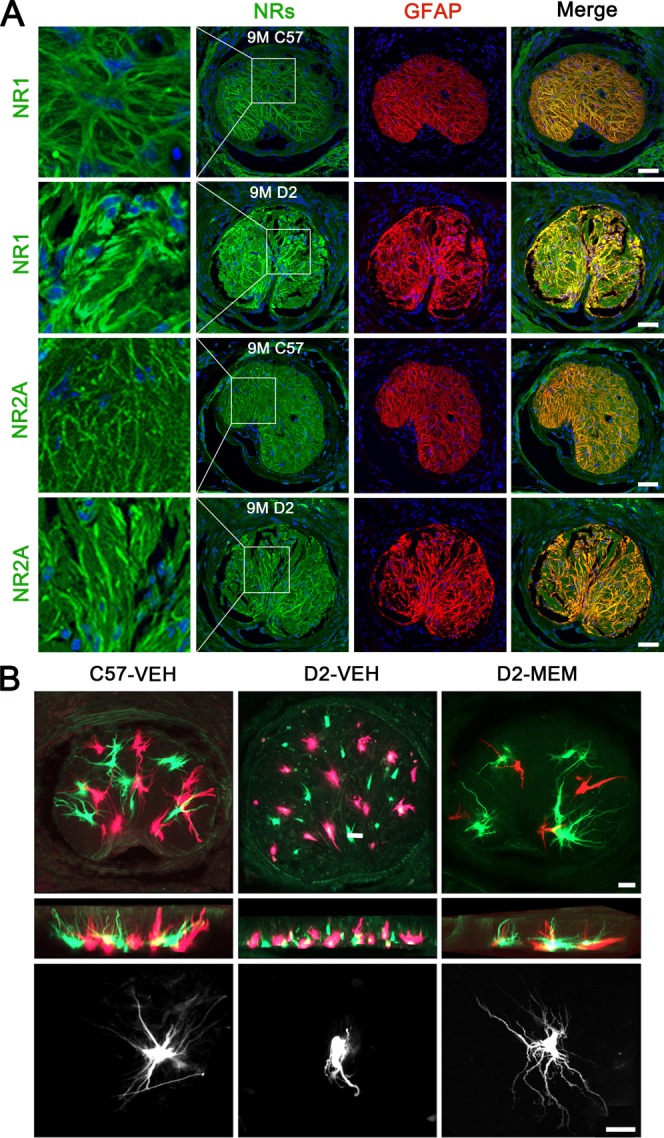
Blockade of glutamate excitotoxicity prevents morphological alteration in astrocytes of the glial lamina in glaucomatous D2 mice. (A) NRs and GFAP double-immunohistochemistry in astrocytes of the glial lamina of glaucomatous D2 mice. Scale bars = 50 μm. (B) Dye injections into astrocytes of the glial lamina region. Astrocytes of the glial lamina in lightly fixed slices from age-matched control C57BL and glaucomatous D2 mice were iontophoretically filled with Lucifer Yellow or Alex 568. Scale bars = 20 μm. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
To further confirm whether blockade of glutamate excitotoxicity by MEM treatment restores astrocyte morphology in their entirety of the glial lamina in glaucomatous D2 mice, astrocytes of the glial lamina in lightly fixed slices were iontophoretically filled with Lucifer Yellow or Alex 568. Because the glial lamina contains no oligodendrocytes or NG2 glial cells (Sun et al., 2010), the majority of cells found in the glial lamina region are astrocytes in nature, being very distinctive in their morphology. We observed normal shapes of astrocytes, showing elongated cell bodies and processes in the glial lamina of age-matched control C57BL mice (Fig. 9B). In contrast, glaucomatous D2 mouse treated with vehicle contained abnormal shapes of astrocytes, showing hypertrophic cell bodies and retracted processes in the glial lamina. Interestingly, however, MEM treatment partially restored the morphology of astrocytes in the glial lamina of glaucomatous D2 mice (Fig. 9B).
Blockade of Glutamate Excitotoxicity Increases Mitochondrial Fission and Volume Density in ONH Astrocytes in Glaucomatous D2 Mice
We next determined whether blockade of glutamate excitotoxicity by MEM treatment prevents alteration of the mitochondrial network in astrocytes of the glial lamina in glaucomatous D2 mice. Representative 2D images from TEM showed that control astrocytes contained a long tubular form of mitochondrion in the glial lamina of age-matched C57BL mice treated with vehicle (Fig. 10A). However, glaucomatous astrocytes contained several fragmented mitochondria in the glial lamina of glaucomatous D2 mice treated with vehicle (Fig. 10A). Interestingly, MEM treatment also contained several fragmented mitochondria in astrocytes of the glial lamina of glaucomatous D2 mice (Fig. 10A). Quantitative TEM analysis showed that the number of astrocytic mitochondria did not show a difference in the glial lamina of glaucomatous D2 mice (0.67 ± 0.09 µm2) compared with age-matched control C57BL mice (0.69 ± 0.19 µm2). However, MEM treatment significantly increased mitochondrial number in astrocytes of the glial lamina in glaucomatous D2 mice (1.60 ± 0.36 µm2; Fig. 10A). Mitochondrial lengths were significantly decreased in astrocytes of the glial lamina in glaucomatous D2 mice (0.30 ± 0.01 µm) compared with age-matched control C57BL mice (0.41 ± 0.02 µm). However, MEM treatment did not show a difference in mitochondrial lengths in astrocytes of the glial lamina in glaucomatous D2 mice (0.31 ± 0.01 µm; Fig. 10A). Mitochondrial volume density was significantly decreased in astrocytes of the glial lamina in glaucomatous D2 mice (0.025 ± 0.003%) compared with compared with age-matched control C57BL mice (0.058 ± 0.007%). However, MEM treatment significantly increased in mitochondrial volume density in astrocytes of the glial lamina in glaucomatous D2 mice (0.050 ± 0.004%; Fig. 10A). Three-dimensional tomographic reconstructions showed an example of mitochondrial fission in astrocyte of the glial lamina in glaucomatous D2 mice treated with vehicle (Fig. 10B). A representative mitochondrion showed the cristae having both tubular and lamellar compartments, which is the most common archetype of brain mitochondria (Fig. 10C). See the Supporting Information Movies 3 and 4 for presentations of the 3D tomography data.
Figure 10.
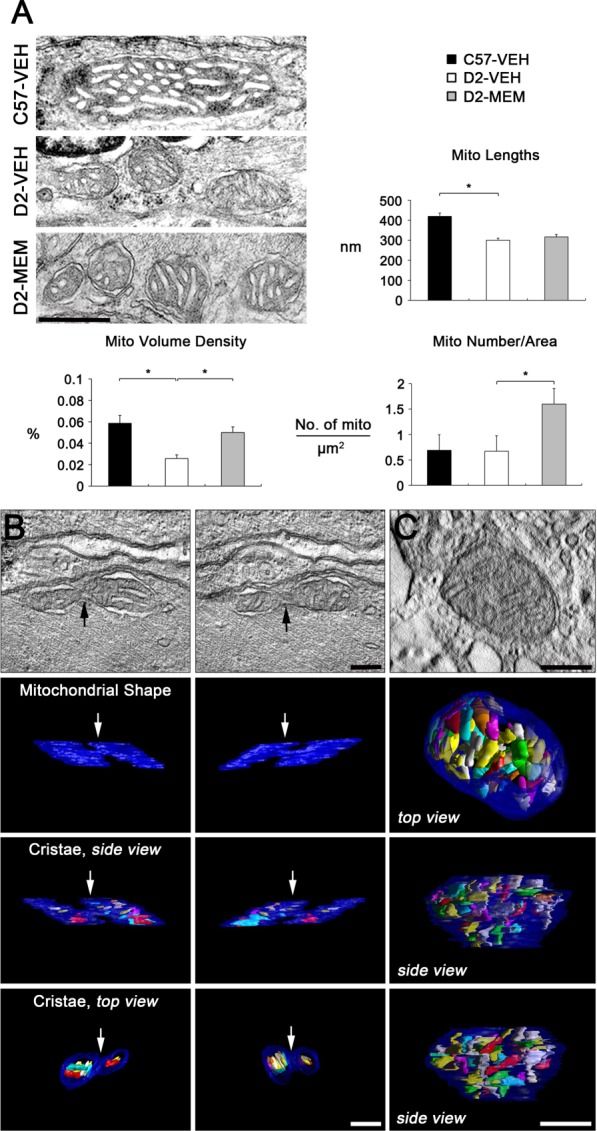
Blockade of glutamate excitotoxicity increases mitochondrial fission and volume density in astrocytes of the glial lamina in glaucomatous D2 mice. (A) Quantitative analysis of mitochondrial lengths, volume density and number in astrocytes of the glial lamina region in glaucomatous D2 mice. Data represent mean ± SEM (* denotes P < 0.05). Scale bar = 500 nm. (B and C) Three-dimensional tomographic reconstructions of mitochondria from an astrocyte in the glial lamina of glaucomatous D2 mice treated with VEH. Representative micrographs showed typical mitochondrial fission (B) and a single mitochondrion, showing cristae structure (C) in astrocyte of the glial lamina in glaucomatous D2 mice treated with VEH. Top: A 1.4 nm slice through the center of a tomographic volume of an astrocyte shows the site of mitochondrial fission (arrows). Top and side view of the segmented and surface-rendered volume showing the outer membrane (blue) and the entire complement of cristae (various colors) provide a 3D snapshot of the packing arrangement, shape, size and density of cristae. Scale bar = 200 nm. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Our previous study demonstrated that glaucomatous damage triggered mitochondrial fragmentation in the axons of the glial lamina in glaucomatous D2 mice (Ju et al., 2008). Hence, we further confirmed whether blockade of glutamate excitotoxicity prevents alterations of axonal and/or astrocytic mitochondria in glaucomatous D2 mice. Representative SBEM images showed abnormal accumulation of autophagosomes/autolysosomes in astrocyte soma and axons of the glial lamina in glaucomatous D2 mice treated with vehicle compared with age-matched control C57BL mice treated with vehicle (Fig. 11A). However, MEM treatment prevented these alterations in both astrocytes and axons of the glial lamina in glaucomatous D2 mice (Fig. 11A). Most importantly, the volume density analyses of SBEM images showed that MEM treatment increased mitochondrial volume density in both astrocyte and axons, but decreased autophagosome/autolysosome volume density in both astrocyte and axons in the glial lamina of glaucomatous D2 mice (Fig. 11B and Supp. Info. Table 1).
Figure 11.
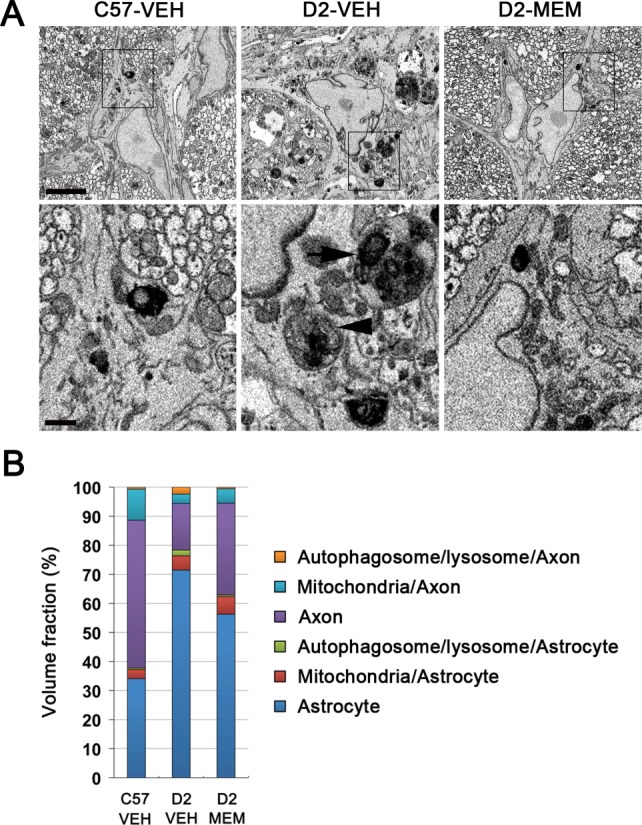
Blockade of glutamate excitotoxicity increases mitochondrial volume density and prevents lysosomal accumulation in both astrocytes and axons of the glial lamina in glaucomatous D2 mice. (A) Representative SBEM micrographs showed abnormal accumulation of autophagosomes (arrowhead)/autolysosomes (arrow) in astrocyte somas of the glial lamina in glaucomatous D2 mice treated with VEH. However, MEM treatment prevented these alterations in both astrocytes and axons of the glial lamina in glaucomatous D2 mice. Scale bars = 5 μm (upper panel) and 1 μm (lower panel). (B) Quantitative volume density analysis using SBEM showed that MEM treatment increased mitochondrial volume density in both astrocytes and axons, but decreased autophagosome/autolysosome volume density in both astrocytes and axons in the glial lamina of glaucomatous D2 mice compared with glaucomatous D2 mice treated with VEH. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Discussion
Although there is growing evidence that glaucomatous damage contributes to dysfunction of glial cells or structural loss of astrocytic processes in the ONH of rodent models of glaucoma (Dai et al., 2012; Son et al., 2010), the relationship between mitochondrial dysfunction and ONH astrocytes in glaucomatous neurodegeneration remains unknown. It has been proposed that mitochondrial fission initiates neurodegeneration in the CNS (Knott et al., 2008; Song et al., 2011). Of interest, reports of altered mitochondrial function in patients with POAG (Abu-Amero et al., 2006; Bosley et al., 2011; He et al., 2008; Izzotti et al., 2011) and of increasing excessive mitochondrial fission-mediated dysfunction in an experimental mouse model of glaucoma (Ju et al., ,) suggest a distinct mitochondrial dysfunction-mediated degenerative pathway in glaucomatous neurodegeneration. In the current study, we observed direct evidence that mitochondrial fission is triggered by increasing levels of DRP1 and pDRP in glaucomatous human ONH astrocytes. Emerging evidence from our group demonstrates that oxidative stress triggers mitochondrial fission-mediated dysfunction in primary rat ONH astrocytes in vitro (Noh et al., 2013). Moreover, inhibition of oxidative stress increases mitochondrial mass and improves mitochondrial bioenergetics in rat ONH astrocytes in vitro (Noh et al., 2013). Collectively, these findings reflect the possibility that mitochondrial fission may be associated with dysfunction of ONH astrocytes in glaucomatous ONH degeneration.
In the past decade, research on glaucoma has been focused on IOP elevation, glutamate excitotoxicity, neurotrophic deprivation, abnormal immune response and ischemia/hypoxia as the possible pathophysiological mechanisms, leading to ONH axon degeneration and RGC death (Danesh-Meyer, 2011; Johnson et al., 2011; Seki and Lipton, 2008; Stefansson et al., 2005; Tezel and Wax, 2004; Weinreb and Khaw, 2004). Since the first demonstration that glutamate excitotoxicity has been linked to impaired mitochondrial dynamics and function in RGC degeneration of a rodent model of glaucoma (Ju et al., 2009), increasing evidence supports the hypothesis that glutamate excitotoxicity may induce mitochondrial dysfunction not only in RGCs, but also in ONH astrocytes by altering mitochondrial dynamics in glaucomatous neurodegeneration (Ju et al., 2008; Kushnareva et al., 2013; Nguyen et al., ,; Noh et al., 2013). Functional NR expression has been reported in brain astrocytes of the mouse, rat, or human (Krebs et al., 2003; Lee et al., 2010; Palygin et al., 2011; Uchihori and Puro, 1993). However, it is unknown whether NRs are present and functional in the astrocytes of the lamina cribrosa region in humans. In the current study, we found that the immunoreactivities of NRs (NR1, 2A and 2B subunits) are present in human ONH astrocytes. More importantly, based on our finding of intracellular calcium response to NMDA in human ONH astrocytes, our results provide evidence for the first time that human ONH astrocytes express functional NRs. Therefore, these findings importantly suggest that astrocytes may contribute to modulation of NR-mediated cellular function between glia and axons in the lamina cribrosa region in humans.
Excessive glutamate excitotoxicity can trigger apoptotic cell death in brain astrocytes in vitro and in vivo (Szydlowska et al., 2006). Furthermore, inhibition of reactive gliosis decreases GFAP expression and protects RGCs against glutamate excitotoxicity-mediated cell death (Ganesh and Chintala, 2011). In the current study, we found that ONH astrocytes from human glaucoma patients showed significant increases of GFAP, NR1 and NR2A protein expression. Thus, our findings suggest that the increased expression of GFAP and NRs in glaucomatous human ONH astrocytes may reflect a possible link to glutamate excitotoxicity in response to an excess of glutamate in the lamina cribrosa during glaucomatous neurodegeneration. In contrast, astrocytes regulate the extracellular environment and astrocytic GTs remove excessive glutamate from the extracellular space under pathological conditions (Morgan, 2000; Waniewski and Martin, 1986). In the current study, we also found that ONH astrocytes from human glaucoma patients showed a significant increase in GLAST protein expression, suggesting additional evidence for the possible existence of glutamate excitotoxicity. Although it is still controversial whether glutamate excitotoxicity contributes to glaucomatous neurodegeneration (Kwon et al., 2005; Osborne et al., ,; Salt and Cordeiro, 2006; Vorwerk et al., 1999), our findings importantly suggest the possibility that the increased expression of GLAST might be a compensatory response to remove excessive glutamate levels for ameliorating glutamate excitotoxicity-mediated degeneration in the lamina cribrosa in glaucoma. Collectively, these results suggest that glutamate may directly mediate dysfunction of ONH astrocytes in glaucomatous neurodegeneration.
Glutamate excitotoxicity triggers an increase in DRP1-mediated mitochondrial fission in HT-22 cells derived from immortalized hippocampal neurons (Grohm et al., 2012). Furthermore, inhibition of DRP1 protects HT-22 neuronal cells against glutamate excitotoxicity (Grohm et al., 2012). However, it remains unknown whether NR activation is directly associated with mitochondrial fission-mediated dysfunction in CNS astrocytes including ONH astrocytes. In the current study, we found that NMDA-mediated glutamate excitotoxicity triggered mitochondrial fission in human ONH astrocytes in vitro as well as in hypertrophic astrocytes of the glial lamina accompanied by axonal degeneration in BAC ALDH1L1 eGFP or Thy1-CFP transgenic mice. Furthermore, NMDA-mediated glutamate excitotoxicity reduced mitochondrial volume density in human ONH astrocytes in vitro as well as in astrocytes of the glial lamina in glaucomatous D2 mice. Of interest, this reduction of mitochondrial volume density was accompanied by abnormal accumulation of autophagosome/autolysosome in both ONH astrocytes and axons of glaucomatous D2 mice. Together with these findings, therefore, our results suggest that glutamate excitotoxicity may contribute to fission-mediated mitochondrial loss, induction of autophagosome/autolysosome formation and dysfunction of ONH astrocytes in glaucomatous neurodegeneration.
Our previous study has shown that blocking glutamate excitotoxicity by MEM treatment inhibited mitochondrial alterations and protected RGCs against apoptotic cell death in the retina of glaucomatous D2 mice (Ju et al., 2009). In the current study, we found that blocking glutamate excitotoxicity by MEM treatment not only preserved the morphological integrity of astrocytes by inhibiting glial scar formation, but also increased mitochondrial fission as well as volume density and length in human ONH astrocytes in vitro or in astrocytes of glial lamina in glaucomatous D2 mice. In addition, blocking glutamate excitotoxicity decreased autophagosome/autolysosome formation in both astrocytes and axon in the glial lamina of glaucomatous D2 mice. These findings first showed a good correlation with results from our own and other groups of promoted RGC survival in experimental models of glaucoma by MEM treatment (Hare et al., 2004; Hare and Wheeler, 2009; Ju et al., 2009). Furthermore, because there is increasing evidence that mitochondrial fission-mediated biogenesis may activate neuroprotective intervention against mitochondrial dysfunction (Bastin et al., 2008; Lee et al., 2011; Srivastava et al., 2009; Wenz, 2009), our findings suggest that increases of mitochondrial fission as well as volume density and length by blocking glutamate excitotoxicity may be a new mitochondria-mediated protective mechanism in ONH astrocytes against glutamate excitotoxicity-mediated glaucomatous ONH degeneration. However, we could not rule out the possibility that the preservation of RGCs and their axons by blocking glutamate excitotoxicity may initially avoid glial activation and scar formation.
It has been reported that glutamate is released at discrete sites along axons in white matter of rat brain that has no neurons and nerve terminals (Kukley et al., 2007) and that the internodal axons also have NRs in the ON of rat (Micu et al., ,). Interestingly, several studies have demonstrated that astrocytes can release glutamate into the extracellular space (Malarkey and Parpura, 2008; Zhang et al., 2004). Moreover, a recent study has reported that amyloid beta induces astrocytic glutamate release, extrasynaptic NR activation, and synaptic loss (Talantova et al., 2013). We have demonstrated that elevated IOP-mediated glaucomatous damage is associated with excessive mitochondrial fission-mediated axonal degeneration in the ON of glaucomatous D2 mice (Ju et al., 2008; Lee et al., 2014). Based on our current findings of excitotoxicity-mediated mitochondrial dysfunction in ONH astrocytes, therefore, we collectively suggest the possibility that NR-mediated excitotoxicity may be associated with both astrocytes and internodal axons by autocrine or paracrine manner, and subsequently induce dysfunction not only in astrocytes, but also in internodal axons during glaucomatous ON neurodegeneration. In contrast, however, because there are no internodal axons in the lamiar cribrosa region of the human and in the glial lamina of the mouse, it is possible that glutamate excitotoxicity in the ONH may primarily contribute to dysfunction of astrocytes and this astrocytic dysfunction may in turn accelerate axonal degeneration in glaucomatous neurodegeneration. Thus, we propose an important degenerative mechanism of axon-glia dysfunction involved in glaucomatous neurodegeneration that includes glutamate excitotoxicity, mitochondrial dysfunction, and astrocyte and/or axon degeneration in the ON. Further studies addressing these important concepts may help to understand excitotoxicity-mediated mitochondrial dysfunction and its pathophysiological mechanisms in axon-glia network of the ONH in glaucoma pathogenesis.
In summary, the current findings provide evidence that glutamate excitotoxicity-mediated mitochondrial fission and loss may be important mitochondrial dysfunction-mediated pathophysiological mechanisms in dysfunction of ONH astrocytes during glaucomatous neurodegeneration. In addition, our findings indicate for the first time evidence that astrocytes utilize mitochondrial fission as a protective mechanism against glutamate excitotoxicity-mediated glaucomatous neurodegeneration. This new paradigm for preserving ONH astrocytes by increases of mitochondrial fission as well as volume density and length may lead to potential therapeutic strategies for ameliorating mitochondrial dysfunction-mediated ONH degeneration in glaucomatous optic neuropathy.
Acknowledgments
The late Dr. M.R. Hernandez provided purified human ONH astrocytes and we thank Dr. James D. Lindsey for critical comments on the article.
Additional Supporting Information may be found in the online version of this article.
References
- Abu-Amero KK, Morales J, Bosley TM. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2006;47:2533–2541. doi: 10.1167/iovs.05-1639. [DOI] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Bastin J, Aubey F, Rotig A, Munnich A, Djouadi F. Activation of peroxisome proliferator-activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients' cells lacking its components. J Clin Endocrinol Metab. 2008;93:1433–1441. doi: 10.1210/jc.2007-1701. [DOI] [PubMed] [Google Scholar]
- Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Dahlstrom A. Studies on the 3-dimensional architecture of dendritic spines and varicosities in human cortex by confocal laser scanning microscopy and Lucifer yellow microinjections. J Neurosci Methods. 1995;57:55–61. doi: 10.1016/0165-0270(94)00125-z. [DOI] [PubMed] [Google Scholar]
- TM Bosley, A Hellani, GL Spaeth, J Myers, LJ Katz, MR Moster, B Milcarek, KK Abu-Amero. Down-regulation of OPA1 in patients with primary open angle glaucoma. Mol Vis. 2011;17:1074–1079. [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Ransom BR. 2007;55:1263–1267. doi: 10.1002/glia.20557. Astrocyte glycogen and brain energy metabolosm. [DOI] [PubMed] [Google Scholar]
- Buhl EH. Intracellular injection in fixed slices in combination with neuroanatomical tracing techniques and electron microscopy to determine multisynaptic pathways in the brain. Microsc Res Tech. 1993;24:15–30. doi: 10.1002/jemt.1070240104. [DOI] [PubMed] [Google Scholar]
- JD Cahoy, B Emery, A Kaushal, LC Foo, JL Zamanian, KS Christopherson, Y Xing, JL Lubischer, PA Krieg, SA Krupenko, WJ Thompson, BA Barres. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet 14 Spec No. 2005;2:R283–R289. doi: 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- Dai C, Khaw PT, Yin ZQ, Li D, Raisman G, Li Y. Structural basis of glaucoma: The fortified astrocytes of the optic nerve head are the target of raised intraocular pressure. Glia. 2012;60:13–28. doi: 10.1002/glia.21242. [DOI] [PubMed] [Google Scholar]
- Danesh-Meyer HV. Neuroprotection in glaucoma: Recent and future directions. Curr Opin Ophthalmol. 2011;22:78–86. doi: 10.1097/ICU.0b013e32834372ec. [DOI] [PubMed] [Google Scholar]
- JP Doyle, JD Dougherty, M Heiman, EF Schmidt, TR Stevens, G Ma, S Bupp, P Shrestha, RD Shah, ML Doughty, S Gong, P Greengard, N Heintz. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- G Feng, RH Mellor, M Bernstein, C Keller-Peck, QT Nguyen, M Wallace, JM Nerbonne, JW Lichtman, JR Sanes. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Ganesh BS, Chintala SK. Inhibition of reactive gliosis attenuates excitotoxicity-mediated death of retinal ganglion cells. PLoS One. 2011;6:e18305. doi: 10.1371/journal.pone.0018305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- J Grohm, SW Kim, U Mamrak, S Tobaben, A Cassidy-Stone, J Nunnari, N Plesnila, C Culmsee. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012;19:1446–1458. doi: 10.1038/cdd.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare WA, Wheeler L. Experimental glutamatergic excitotoxicity in rabbit retinal ganglion cells: Block by memantine. Invest Ophthalmol Vis Sci. 2009;50:2940–2948. doi: 10.1167/iovs.08-2103. [DOI] [PubMed] [Google Scholar]
- WA Hare, E WoldeMussie, RK Lai, H Ton, G Ruiz, T Chun, L Wheeler. Efficacy and safety of memantine treatment for reduction of changes associated with experimental glaucoma in monkey, I: Functional measures. Invest Ophthalmol Vis Sci. 2004;45:2625–2639. doi: 10.1167/iovs.03-0566. [DOI] [PubMed] [Google Scholar]
- M Hatori, C Vollmers, A Zarrinpar, L DiTacchio, EA Bushong, S Gill, M Leblanc, A Chaix, M Joens, JA Fitzpatrick, MH Ellisman, S Panda. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15:848–860. doi: 10.1016/j.cmet.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Y He, KW Leung, YH Zhang, S Duan, XF Zhong, RZ Jiang, Z Peng, J Tombran-Tink, J Ge. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: Protection by antioxidants. Invest Ophthalmol Vis Sci. 2008;49:1447–1458. doi: 10.1167/iovs.07-1361. [DOI] [PubMed] [Google Scholar]
- M Heiman, A Schaefer, S Gong, JD Peterson, M Day, KE Ramsey, M Suárez-Fariñas, C Schwarz, DA Stephan, DJ Surmeier, P Greengard, N Heintz. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez MR, Miao H, Lukas T. Astrocytes in glaucomatous optic neuropathy. Prog Brain Res. 2008;173:353–373. doi: 10.1016/S0079-6123(08)01125-4. [DOI] [PubMed] [Google Scholar]
- Izzotti A, Longobardi M, Cartiglia C, Sacca SC. Mitochondrial damage in the trabecular meshwork occurs only in primary open-angle glaucoma and in pseudoexfoliative glaucoma. PLoS One. 2011;6:e14567. doi: 10.1371/journal.pone.0014567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A Jahani-Asl, K Pilon-Larose, W Xu, JG MacLaurin, DS Park, HM McBride, RS Slack. The mitochondrial inner membrane GTPase, optic atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J Biol Chem. 2011;286:4772–4782. doi: 10.1074/jbc.M110.167155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TV, Bull ND, Martin KR. Neurotrophic factor delivery as a protective treatment for glaucoma. Exp Eye Res. 2011;93:196–203. doi: 10.1016/j.exer.2010.05.016. [DOI] [PubMed] [Google Scholar]
- WK Ju, KY Kim, M Angert, KX Duong-Polk, JD Lindsey, MH Ellisman, RN Weinreb. Memantine blocks mitochondrial OPA1 and cytochrome c release and subsequent apoptotic cell death in glaucomatous retina. Invest Ophthalmol Vis Sci. 2009;50:707–716. doi: 10.1167/iovs.08-2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju WK, Kim KY, Duong-Polk KX, Lindsey JD, Ellisman MH, Weinreb RN. Increased optic atrophy type 1 expression protects retinal ganglion cells in a mouse model of glaucoma. Mol Vis. 2010;16:1331–1342. [PMC free article] [PubMed] [Google Scholar]
- WK Ju, KY Kim, JD Lindsey, M Angert, KX Duong-Polk, RT Scott, JJ Kim, I Kukhmazov, MH Ellisman, GA Perkins, RN Weinreb. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Invest Ophthalmol Vis Sci. 2008;49:4903–4911. doi: 10.1167/iovs.07-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju WK, Liu Q, Kim KY, Crowston JG, Lindsey JD, Agarwal N, Ellisman MH, Perkins GA, Weinreb RN. Elevated hydrostatic pressure triggers mitochondrial fission and decreases cellular ATP in differentiated RGC-5 cells. Invest Ophthalmol Vis Sci. 2007;48:2145–2151. doi: 10.1167/iovs.06-0573. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–880. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs C, Fernandes HB, Sheldon C, Raymond LA, Baimbridge KG. Functional NMDA receptor subtype 2B is expressed in astrocytes after ischemia in vivo and anoxia in vitro. J Neurosci. 2003;23:3364–3372. doi: 10.1523/JNEUROSCI.23-08-03364.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Kukley M, Capetillo-Zarate E, Dietrich D. Vesicular glutamate release from axons in white matter. Nat Neurosci. 2007;10:311–320. doi: 10.1038/nn1850. [DOI] [PubMed] [Google Scholar]
- YE Kushnareva, AA Gerencser, B Bossy, WK Ju, AD White, J Waggoner, MH Ellisman, G Perkins, E Bossy-Wetzel. Loss of OPA1 disturbs cellular calcium homeostasis and sensitizes for excitotoxicity. Cell Death Differ. 2013;20:353–365. doi: 10.1038/cdd.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YH Kwon, DW Rickman, S Baruah, MB Zimmerman, CS Kim, HC Boldt, SR Russell, SS Hayreh. Vitreous and retinal amino acid concentrations in experimental central retinal artery occlusion in the primate. Eye (Lond) 2005;19:455–463. doi: 10.1038/sj.eye.6701546. [DOI] [PubMed] [Google Scholar]
- D Lee, MS Shim, KY Kim, YH Noh, H Kim, SY Kim, RN Weinreb, WK Ju. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2014;55:993–1005. doi: 10.1167/iovs.13-12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Ting KK, Adams S, Brew BJ, Chung R, Guillemin GJ. Characterisation of the expression of NMDA receptors in human astrocytes. PLoS One. 2010;5:e14123. doi: 10.1371/journal.pone.0014123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- S Lee, NJ VanBergen, GY Kong, V Chrysostomou, HS Waugh, EC O'Neill, JG Crowston, IA Trounce. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp Eye Res. 2011;93:204–212. doi: 10.1016/j.exer.2010.07.015. [DOI] [PubMed] [Google Scholar]
- CK Leung, JD Lindsey, JG Crowston, WK Ju, Q Liu, DU Bartsch, RN Weinreb. In vivo imaging of murine retinal ganglion cells. J Neurosci Methods. 2008;168:475–478. doi: 10.1016/j.jneumeth.2007.10.018. [DOI] [PubMed] [Google Scholar]
- RT Libby, Y Li, OV Savinova, J Barter, RS Smith, RW Nickells, SW John. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TJ Lukas, H Miao, L Chen, SM Riordan, W Li, AM Crabb, A Wise, P Du, SM Lin, MR Hernandez. Susceptibility to glaucoma: Differential comparison of the astrocyte transcriptome from glaucomatous African American and Caucasian American donors. Genome Biol. 2008;9:R111. doi: 10.1186/gb-2008-9-7-r111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey EB, Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem Int. 2008;52:142–154. doi: 10.1016/j.neuint.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- I Micu, Q Jiang, E Coderre, A Ridsdale, L Zhang, J Woulfe, X Yin, BD Trapp, JE McRory, R Rehak, GW Zamponi, W Wang, PK Stys. NMDA receptors mediate calcium accumulation in myelin during chemical ischemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- I Micu, A Ridsdale, L Zhang, J Woulfe, J McClintock, CA Brantner, SB Andrews, PK Stys. Real-time measurement of free Ca2+ changes in CNS myelin by two-photon microscopy. Nat Med m. 2007;13:874–879. doi: 10.1038/nm1568. [DOI] [PubMed] [Google Scholar]
- Morgan JE. Optic nerve head structure in glaucoma: Astrocytes as mediators of axonal damage. Eye (Lond) 14(Pt. 2000;3B):437–444. doi: 10.1038/eye.2000.128. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- D Nguyen, MV Alavi, KY Kim, T Kang, RT Scott, YH Noh, JD Lindsey, B Wissinger, MH Ellisman, RN Weinreb, GA Perkins, WK Ju. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011a;2:e240. doi: 10.1038/cddis.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JV Nguyen, I Soto, KY Kim, EA Bushong, E Oglesby, FJ Valiente-Soriano, Z Yang, CH Davis, JL Bedont, JL Son, JO Wei, VL Buchman, DJ Zack, M Vidal-Sanz, MH Ellisman, N Marsh-Armstrong. Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci USA. 2011b;108:1176–1181. doi: 10.1073/pnas.1013965108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: Mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- YH Noh, KY Kim, MS Shim, SH Choi, S Choi, MH Ellisman, RN Weinreb, GA Perkins, WK Ju. Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis. 2013;4:e820. doi: 10.1038/cddis.2013.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne NN, Chidlow G, Wood JP. Glutamate excitotoxicity in glaucoma: Truth or fiction? By AJ Lotery. Eye (Lond) 2006;20:1392–1394. doi: 10.1038/sj.eye.6702234. [DOI] [PubMed] [Google Scholar]
- Osborne NN, Melena J, Chidlow G, Wood JP. A hypothesis to explain ganglion cell death caused by vascular insults at the optic nerve head: Possible implication for the treatment of glaucoma. Br J Ophthalmol. 2001;85:1252–1259. doi: 10.1136/bjo.85.10.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palygin O, Lalo U, Pankratov Y. Distinct pharmacological and functional properties of NMDA receptors in mouse cortical astrocytes. Br J Pharmacol. 2011;163:1755–1766. doi: 10.1111/j.1476-5381.2011.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L Pellerin, AK Bouzier-Sore, A Aubert, S Serres, M Merle, R Costalat, PJ Magistretti. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- GA Perkins, GE Sosinsky, S Ghassemzadeh, A Perez, Y Jones, MH Ellisman. Electron tomography of large, multicomponent biological structures. J Struct Biol. 1997;120:219–227. doi: 10.1006/jsbi.1997.3920. [DOI] [PubMed] [Google Scholar]
- Ransom BR. Glial modulation of neural excitability mediated by extracellular pH: A hyposthesis revisited. Prog Brain Res. 2000;125:217–228. doi: 10.1016/S0079-6123(00)25012-7. [DOI] [PubMed] [Google Scholar]
- Salt TE, Cordeiro MF. Glutamate excitotoxicity in glaucoma: Throwing the baby out with the bathwater? Eye (Lond) 2006;20:730–731. doi: 10.1038/sj.eye.6701967. author reply 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, Lipton SA. Targeting excitotoxic/free radical signaling pathways for therapeutic intervention in glaucoma. Prog Brain Res. 2008;173:495–510. doi: 10.1016/S0079-6123(08)01134-5. [DOI] [PubMed] [Google Scholar]
- JL Son, I Soto, E Oglesby, T Lopez-Roca, ME Pease, HA Quigley, N Marsh-Armstrong. Glaucomatous optic nerve injury involves early astrocyte reactivity and late oligodendrocyte loss. Glia. 2010;58:780–789. doi: 10.1002/glia.20962. [DOI] [PubMed] [Google Scholar]
- W Song, J Chen, A Petrilli, G Liot, E Klinglmayr, Y Zhou, P Poquiz, J Tjong, MA Pouladi, MR Hayden, E Masliah, M Ellisman, I Rouiller, R Schwarzenbacher, B Bossy, G Perkins, E Bossy-Wetzel. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med. 2011;17:377–382. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Diaz F, Iommarini L, Aure K, Lombes A, Moraes CT. PGC-1alpha/beta induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum Mol Genet. 2009;18:1805–1812. doi: 10.1093/hmg/ddp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E Stefánsson, DB Pedersen, PK Jensen, M laCour, JF Kiilgaard, K Bang, T Eysteinsson. Optic nerve oxygenation. Prog Retin Eye Res. 2005;24:307–332. doi: 10.1016/j.preteyeres.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Sun D, Lye-Barthel M, Masland RH, Jakobs TC. Structural remodeling of fibrous astrocytes after axonal injury. J Neurosci. 2010;30:14008–14019. doi: 10.1523/JNEUROSCI.3605-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szydlowska K, Zawadzka M, Kaminska B. Neuroprotectant FK506 inhibits glutamate-induced apoptosis of astrocytes in vitro and in vivo. J Neurochem. 2006;99:965–975. doi: 10.1111/j.1471-4159.2006.04136.x. [DOI] [PubMed] [Google Scholar]
- M Talantova, S Sanz-Blasco, X Zhang, P Xia, MW Akhtar, S Okamoto, G Dziewczapolski, T Nakamura, G Cao, AE Pratt, YJ Kang, S Tu, E Molokanova, SR McKercher, SA Hires, H Sason, DG Stouffer, MW Buczynski, JP Solomon, S Michael, ET Powers, JW Kelly, A Roberts, G Tong, T Fang-Newmeyer, J Parker, EA Holland, D Zhang, N Nakanishi, HS Chen, H Wolosker, Y Wang, LH Parsons, R Ambasudhan, E Masliah, SF Heinemann, JC Piña-Crespo, SA Lipton. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci USA. 2013;110:E2518–E2527. doi: 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog Retin Eye Res. 2006;25:490–513. doi: 10.1016/j.preteyeres.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel G, Wax MB. The immune system and glaucoma. Curr Opin Ophthalmol. 2004;15:80–84. doi: 10.1097/00055735-200404000-00003. [DOI] [PubMed] [Google Scholar]
- Uchihori Y, Puro DG. Glutamate as a neuron-to-glial signal for mitogenesis: Role of glial N-methyl-d-aspartate receptors. Brain Res. 1993;613:212–220. doi: 10.1016/0006-8993(93)90901-x. [DOI] [PubMed] [Google Scholar]
- Vorwerk CK, Gorla MS, Dreyer EB. An experimental basis for implicating excitotoxicity in glaucomatous optic neuropathy. Surv Ophthalmol 43 Suppl. 1999;1:S142–S150. doi: 10.1016/s0039-6257(99)00017-x. [DOI] [PubMed] [Google Scholar]
- Waniewski RA, Martin DL. Exogenous glutamate is metabolized to glutamine and exported by rat primary astrocyte cultures. J Neurochem. 1986;47:304–313. doi: 10.1111/j.1471-4159.1986.tb02863.x. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Sontheimer H, Black JA, Minturn JE, Ransom BR. Dynamic aspects of sodium channel expression in astrocytes. Adv Neurol. 1993;59:135–155. [PubMed] [Google Scholar]
- Weinreb RN, Aung T, Medeiros FA. The pathophysiology, genetics and treatment of glaucoma: A review. JAMA. 2014;311:1901–1911. doi: 10.1001/jama.2014.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb RN, Khaw PT. Primary open-angle glaucoma. Lancet. 2004;363:1711–1720. doi: 10.1016/S0140-6736(04)16257-0. [DOI] [PubMed] [Google Scholar]
- Wenz T. PGC-1alpha activation as a therapeutic approach in mitochondrial disease. IUBMB Life. 2009;61:1051–1062. doi: 10.1002/iub.261. [DOI] [PubMed] [Google Scholar]
- Yang P, Hernandez MR. Purification of astrocytes from adult human optic nerve heads by immunopanning. Brain Res Brain Res Protoc. 2003;12:67–76. doi: 10.1016/s1385-299x(03)00073-4. [DOI] [PubMed] [Google Scholar]
- Y Yang, S Vidensky, L Jin, C Jie, I Lorenzini, M Frankl, JD Rothstein. Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia. 2011;59:200–207. doi: 10.1002/glia.21089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Fukuda M, Van Bockstaele E, Pascual O, Haydon PG. Synaptotagmin IV regulates glial glutamate release. Proc Natl Acad Sci USA. 2004;101:9441–9446. doi: 10.1073/pnas.0401960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Zhang L, Weinreb RN. Ophthalmic drug discovery: Novel targets and mechanisms for retinal diseases and glaucoma. Nat Rev Drug Discov. 2012;11:541–559. doi: 10.1038/nrd3745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.