

Abstract
Acute myeloid leukemia is a clonal malignant disorder derived from a small number of leukemic stem cells (LSCs). Rearrangements of the mixed lineage leukemia (MLL) gene are found in acute myeloid leukemia associated with poor prognosis. The upregulation of Hox genes is critical for LSC induction and maintenance, but is unlikely to support malignancy and the high LSC frequency observed in MLL leukemias. The present study shows that MLL fusion proteins interact with the transcription factor PU.1 to activate the transcription of CSF-1R, which is critical for LSC activity. Acute myeloid leukemia is cured by either deletion of PU.1 or ablation of cells expressing CSF-1R. Kinase inhibitors specific for CSF-1R prolong survival time. These findings indicate that PU.1-mediated upregulation of CSF-1R is a critical effector of MLL leukemogenesis.
Keywords: Acute myeloid leukemia, CSF-1R, mixed lineage leukemia, Spi-1, stem cells
Acute myeloid leukemia (AML) is a clonal malignant disorder derived from a small number of leukemic stem cells (LSCs).1,2 Leukemic stem cells (LSCs) are capable of the limitless self-renewal that is necessary for cancer initiation and maintenance. Conventional chemotherapies are often effective in reducing the total number of leukemia cells, but are not curative in many cases of AML. As LSCs are often resistant to conventional chemotherapies, residual LSCs are a potential cause of AML relapse. Thus, eradication of LSCs is critical to cure the disease.
Chromosome translocations that involve the mixed lineage leukemia gene (MLL) are frequently observed in human AML and often predict a poor prognosis.3–6 More than 60 genes have been identified as MLL fusion partners to date; chromosome rearrangements such as t(9;11), t(11;19), and t(10;11), which express MLL-AF9, MLL-ELL, and MLL-AF10, respectively, are commonly associated with AML.5 The MLL fusion proteins transform non-self-renewing myeloid progenitors into LSCs.7,8 Acute myeloid leukemia with MLL rearrangements consistently express HOX genes such as HOXA7, HOXA9, and MEIS1.9–11 The upregulation of Hox genes is critical for LSC induction and maintenance, but does not recapitulate the entire phenotype and biology of MLL leukemias.12–15 Moreover, it is unlikely to support malignancy and the high LSC levels observed in MLL leukemias.16 These facts suggest that unknown critical mediators of leukemogenesis exist.
The present study shows that the upregulation of macrophage colony-stimulating factor (M-CSF) receptor (CSF-1R, also called M-CSFR/c-FMS/CD115) is critical for LSC activity in MLL leukemia. Acute myeloid leukemia was cured after eradication of cells expressing high levels of Csf-1r in mice. It was found that MLL fusions regulated CSF-1R transcription through a novel mechanism involving interaction with the transcription factor PU.1. These findings indicate that PU.1-mediated upregulation of CSF-1R is a novel therapeutic target for MLL leukemias.
Materials and Methods
Mice
C57BL/6 mice were purchased from CLEA Japan (Tokyo, Japan). NGF-FKBP-Fas transgenic mice17 (Jackson Laboratory, Bar Harbor, ME, USA), CSF-1R-deficient mice18, PU.1-null/conditional deficient mice,19 and CreERT2 mice (TaconicArtemis (Germantown, NY, USA))20 were maintained on a C57BL/6 genetic background. Mouse experiments were carried out in a specific pathogen-free environment at the National Cancer Center (Tokyo, Japan) animal facility according to institutional guidelines and with approval of the National Cancer Center Animal Ethics Committee.
Generation of AML mouse models
MSCV-MLL-AF10-ires-GFP was transfected with PLAT-E21 cells using the FuGENE 6 reagent (Roche Diagnostics Mannheim, Germany), and supernatants containing retrovirus were collected 48 h after transfection. The c-Kit+ cells (1 × 105 cells), which were selected from bone marrow (BM) or fetal liver cells using CD117 MicroBeads (Miltenyi Biotec Bergisch Gladbach, Germany), were incubated with the retrovirus using RetroNectin (Takara Bio Otsu, Japan) for 24 h in StemPro-34 serum-free medium (Invitrogen Waltham, MA, USA) containing cytokines (20 ng/mL stem cell factor [SCF], 10 ng/mL interleukin [IL]-6, and 10 ng/mL IL-3). The infectants were then transplanted together with BM cells (2 × 105) into lethally irradiated (9 Gy) 6- to 8-week-old C57BL/6 mice by i.v. injection. Secondary transplants were carried out by i.v. injection of BM cells from the primary AML mice into sublethally irradiated (6 Gy) C57BL/6 mice.
Treatment with AP20187, AraC, or Ki20227
AP20187 (10 mg/kg; gift from Ariad Pharmaceuticals Cambridge, MA, USA) was given daily by i.v. injection for 5 days, then 1 mg/kg AP20187 was given every 3 days thereafter as described previously.17 Ki2022722 (20 mg/kg; gift from KIRIN Pharma) was given orally daily from 7 days after transplantation. AraC (75 mg/kg) was given daily by i.v. injection for 5 days from 7 days after transplantation.
Immunofluorescent staining, flow cytometric analysis, and cell sorting
Bone marrow cells from AML mice were preincubated with rat IgG, and then incubated on ice with anti-CD115(CSF-1R)-PE (eBioscience San Diego, CA, USA) and anti-c-Kit-APC (2B8)-APC (BD Pharmingen San Jose, CA, USA). Flow cytometric analysis and cell sorting were carried out using the cell sorter JSAN (Baybioscience Kobe, Japan), and the results were analyzed using FlowJo software (Tree Star Ashland, OR, USA)).
Reporter analysis
Csf1r-luciferase constructs were generated by ligation of WT and PU.1-lacking Csf1r promoter23 with pGL4. For reporter analysis, SaOS2 cells were transfected with Csf1r-luc and phRL-CMV together with various expression constructs in 24-well plates, and luciferase activity was assayed 24 h after transfection using the microplate luminometer GLOMAX (Promega Madison, WI, USA). Results of reporter assays represent the average values for relative luciferase activity generated from at least three independent experiments that were normalized using the activity of the enzyme from phRL-CMV as an internal control.
Immunoprecipitation and immunoblotting
For immunoprecipitation experiments, cells were lysed in a lysis buffer containing 250 mM NaCl, 20 mM sodium phosphate (pH 7.0), 30 mM sodium pyrophosphate, 10 mM NaF, 0.1% NP-40, 5 mM DTT, 1 mM PMSF, and protease inhibitor. Cell lysates were incubated with anti-FLAG antibody-conjugated agarose beads (Sigma) and gently rotated at 4°C overnight. The absorbed beads were washed six times with lysis buffer. Precipitated proteins were eluted from the beads by FLAG peptide and dissolved with the same volume of 2× SDS sample buffer. When immunoprecipitation was not carried out, total protein lysates were prepared in 2× SDS sample buffer. Antibodies were detected by chemiluminescence using ECL plus Detection Reagents (Amersham Biosciences, Little Chalfont, UK). The primary antibodies used in this study were anti-FLAG (M2; Sigma-Aldrich (St. Louis, MO, USA)), anti-HA (3F10; Roche Diagnostics (Mannheim, Germany)), and anti-MLL-N24 antibodies.
Statistical analyses
We used unpaired two-tailed Student's t-tests for comparisons and a log–rank test for survival data using JMP8 software (SAS Institute (Cary, CA, USA)).
Colony formation assays
Cells were cultured in 1% methylcellulose in Iscove's modified Dulbecco's medium containing 15% FBS, 1% BSA, 10 μg/mL rh-Insulin, 200 μg/mL human transferrin, 100 μM 2-mercaptoethanol, 2 mM l-glutamine, and the following cytokines: 50 ng/mL rm SCF, 10 ng/mL rm IL-3, and 10 ng/mL rh IL-6; or 10 ng/mL mCSF-1. Cultures were maintained at 37°C under humidified conditions with 5% CO2. Colonies containing >50 cells were counted on day 5.
Results
Upregulation of CSF-1R is critical for MLL-AF10-induced AML
Previous results indicated that the expression of CSF-1R was high in MOZ-TIF2-induced AML25 and human AML.26 Expression of Csf-1r was investigated in MLL-AF10-induced AML in mice. Results showed that Csf-1r expression was high in some AML cell populations (Fig.1a). To assess LSC activity, cells expressing high (Csf-1rhigh) and low (Csf-1rlow/−) levels of Csf-1r were purified and transplanted into irradiated mice. Transplantation of 102 flow-sorted Csf-1rhigh cells was sufficient to induce AML in all mice transplanted (Fig.1b). Conversely, no mice developed AML after transplantation of 102 Csf-1rlow/− cells (Fig.1c). Thus, Csf-1rhigh cells displayed stronger LIC activity compared to Csf-1rlow/− cells in MLL-AF10-induced AML.
Figure 1.

Cells expressing high levels of macrophage colony-stimulating factor (M-CSF) receptor (CSF-1R high) show potent leukemia-initiating activity. (a) Bone marrow (BM) cells from mice with mixed lineage leukemia (MLL)-AF10-induced acute myeloid leukemia were analyzed by flow cytometer for expression of GFP and Csf-1r. (b,c) Csf-1rhigh and Csf-1rlow/− cells were sorted by flow cytometry. The indicated numbers of flow-sorted CSF-1Rhigh (b) and Csf-1rlow/− (c) cells were transplanted into sublethally irradiated mice, and leukemia-free survival was investigated. n = 8, P < 0.001. (d) Csf-1rhigh (h) and Csf-1rlow/− (l) cells were analyzed for levels of total and phosphorylated signal transducer and activator of transcription 5 (STAT5 and pSTAT5), phosphorylated ERK p(ERK), and Pu.1 (e,f). Csf-1rhigh and Csf-1rlow/– cells were analyzed for colony-forming activity in methylcellulose medium supplemented with interleukin (IL)-3, stem cell factor (SCF), and IL-6 (e) or with M-CSF (f). (g,h) Levels of HoxA9 (g) and Csf1r (h) mRNAs were measured in Csf-1rhigh and Csf-1rlow/− cells prepared from BM of mice with acute myeloid leukemia.
Signal transducer and activator of transcription 5 (STAT5) and ERK, which are downstream effectors of CSF-1R, are activated in a variety of leukemias and myeloproliferative disorders. The phosphorylation status of these proteins was investigated in Csf-1rhigh and Csf-1rlow/− cells from MLL-AF10-induced AML mice by immunoblot analysis with phospho-specific anti-STAT5 and anti-ERK antibodies. Stat5 was highly phosphorylated in Csf-1rhigh cells but not in Csf-1rlow/− cells (Fig.1d), whereas Erk1/2 were phosphorylated in both Csf-1rhigh and Csf-1rlow/− cells. Further analyses are required to determine the role(s) of Stat5 during leukemogenesis.
As MLL-AF10-induced leukemia cells can form colonies in methylcellulose,27 flow-sorted Csf-1rhigh and Csf-1rlow/− cells were tested for colony formation in the presence of either M-CSF or multiple cytokines. Csf-1rhigh cells and Csf-1rlow/− formed equivalent numbers of colonies when stimulated with multiple cytokines (Fig.1e). However, Csf-1rlow/− cells showed reduced colony formation when stimulated with M-CSF alone (Fig.1f). Quantitative RT-PCR analysis showed that HoxA9 was upregulated in both Csf-1rhigh and Csf-1rlow/− cells (Fig.1g) and that Csf1r mRNA was appropriately differentially expressed (Fig.1h). Csf-1rhigh and Csf-1rlow/− cells were also observed in normal BM and fetal liver (Fig. S1). Populations of Csf-1rhigh were reduced in Mll−/− fetal liver cells, suggesting that Csf-1r expression is regulated by WT Mll as well as by Mll-fusions.
MLL fusions activate CSF-1R transcription through interaction with PU.1
Monocyte-specific expression of CSF-1R is reportedly regulated by transcription factors such as AML1, PU.1, and C/EBP.28 To investigate MLL-mediated regulation of CSF-1R transcription, the interaction of MLL with several hematopoietic transcription factors was tested. Results showed that MLL strongly interacts with PU.1 (Fig.2a). MLL-AF10 also interacted with PU.1 (Fig.2b). Both MLL and MLL fusions very strongly stimulated PU.1-dependent activation of the CSF-1R promoter (Fig.2c). Neither MLL nor MLLAF10 activated a CSF-1R promoter mutant lacking PU.1 binding sites (Fig.2d). Interaction of MLL with AML1/RUNX129 and other factors was less strong, and MLL and MLL fusions did not activate the CSF-1R promoter in the presence of AML1 or C/EBPα (data not shown). Chromatin immunoprecipitation analysis indicated genomic localizations of MLL-AF10 and PU.1 on Csf-1r (Fig.2e). These results suggest that MLL and MLL fusion proteins interact with PU.1 to activate CSF-1R transcription.
Figure 2.

PU.1-dependent upregulation of macrophage colony-stimulating factor receptor (CSF-1R) by mixed lineage leukemia (MLL) and MLL fusions. (a) Interaction of MLL with PU.1. 293T cells were co-transfected with MLL-HA and the indicated FLAG-tagged transcription factors, including FLAG-PU.1. Anti-FLAG antibody immunoprecipitates (IP:FLAG) or cell lysates (Input) were subjected to immunoblotting with anti-HA, anti-MLL-N, or anti-FLAG antibodies. (b) Interaction between MLL-AF10 and PU.1. 293T cells were co-transfected with MLL-AF10 and FLAG-tagged WT PU.1 or PU.1/FR232A. Anti-FLAG antibody immunoprecipitates (IP:FLAG) or cell lysates (Input) were subjected to immunoblotting with anti-MLL-N or anti-PU.1 antibodies. (c) Effects of MLL, and MLL fusions on PU.1-mediated Csf1r promoter-driven transcription. SaOS2 cells were co-transfected with the Csf1r–luciferase construct and the indicated effectors. Luciferase activity was analyzed 24 h after transfection. Error bars represent SD (n = 3). (d) PU.1 binding site-dependence of MLL enhancement of Csf1r promoter-driven transcription. SaOS2 cells were transfected with the WT Csf1r–luciferase construct or its mutant lacking the PU.1-binding site, together with the indicated effectors. (e) ChIP of MLL-AF10 and PU.1. Bone marrow (BM) cells from acute myeloid leukemia mice (AML) induced by Flag-MLL-AF10, were subjected to ChIP analysis using anti-Flag (MLL-AF10), anti-PU.1, and anti-MOZ antibodies. Semiquantitative real-time PCR was carried out on the co-precipitated DNAs.
Immunoprecipitation analysis using MLL deletion mutants indicated that PU.1 interacts with at least two regions in the N-terminus of MLL (Figs3a,S1). The menin and LEGDF-interacting domains30 and the C-terminal SET domain, which is needed for histone methyltransferase activity,31 are not required for interaction with PU.1 (Fig.3b,c) or the PU.1-dependent activation of CSF-1R by MLL (Fig.3d), suggesting that interaction with menin and LEGDF and histone methyltransferase activity are not required for MLL-mediated transactivation of CSF-1R. PU.1 deletion analysis indicated that the ETS domain of PU.1 was required for the interaction of PU.1 with MLL (Fig.4a,b). As the ETS domain is a DNA-binding domain, it is possible that the interaction between MLL/MLL fusions and PU.1 is DNA-dependent. However, this seems unlikely because MLL-AF10 also interacted with PU.1/R232A, which lacks DNA-binding capacity (Fig.2b). Both the DEQ region and the ETS domain of PU.1 were required to activate PU.1-mediated transcription by MLL and MLL-AF10 (Fig.4c).
Figure 3.

Functional domains of mixed lineage leukemia (MLL) required for interaction with PU.1 and for PU.1-mediated activation of macrophage colony-stimulating factor receptor (Csf1r) promoter. (a) PU.1 binding and PU.1-mediated Csf1r promoter activity of MLL deletion mutants. The PU.1-, menin-, and LEDGF-interacting domains and the results for interaction with PU.1 and PU.1-mediated transactivation of Csf1r-luc are indicated. ND, not determined. (b,c) Pu.1 binding. 293T cells were co-transfected with WT or mutants of HA-tagged MLL and FLAG-tagged PU.1 (b), or with WT or mutants of FLAG-tagged MLL and HA-tagged PU.1 (c). Anti-FLAG antibody immunoprecipitates (IP:FLAG) or cell lysates (Input) were subjected to immunoblotting with anti-HA or anti-FLAG antibodies. (d) PU.1-mediated Csf1r promoter-driven transcription. SaOS2 cells were transfected with the Csf1r–luciferase construct and PU.1, together with deletion mutants of MLL. Luciferase activity was analyzed 24 h after transfection. Error bars represent SD (n = 3).
Figure 4.
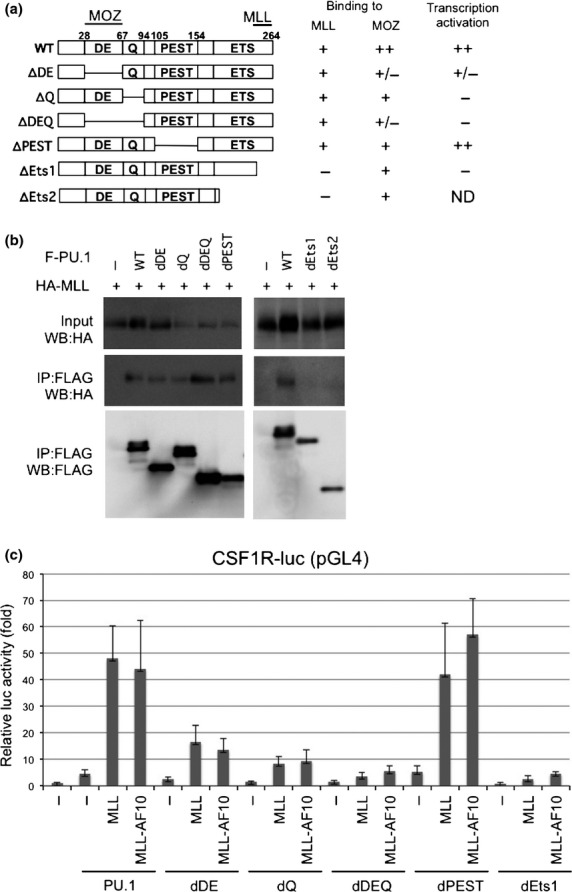
Functional domains of PU.1 (a) Deletion mutants of PU.1. The MOZ- and mixed lineage leukemia (MLL)-interacting domains and the results for interaction with PU.1 and PU.1-mediated transactivation of macrophage colony-stimulating factor receptor (Csf1r)-luc are indicated. ND, not determined. (b) Interaction of PU.1 mutants with MLL. 293T cells were co-transfected with HA-tagged MLL and WT or mutants of FLAG-tagged PU.1. Anti-FLAG antibody immunoprecipitates (IP:FLAG) or cell lysates (Input) were subjected to immunoblotting with anti-HA or anti-FLAG antibodies. (c) SaOS2 cells were co-transfected with the Csf1r–luciferase construct and the indicated effectors. Luciferase activity was analyzed 24 h after transfection. Error bars represent SD (n = 3).
To test whether MLL-AF10 stimulates PU.1-dependent induction of endogenous Csf-1r, Pu.1−/− myeloid progenitors expressing the PU.1-estrogen receptor fusion protein (PUER) were used. These cells can differentiate into macrophages after restoration of PU.1 activity by exposure to 4-hydroxytamoxifen (4-HT).32 PUER cells were infected with MSCV-MLL-AF10-ires-GFP or control retroviruses. The GFP+ cells were sorted and cultured in the presence of 4-HT. Five days after the addition of 4-HT, flow cytometry analysis indicated a strong increase in Csf-1r expression by cells expressing MLL-AF10, but only a slight increase in cells infected with the control vector (Fig.5a). Thus, MLL-AF10 induces expression of endogenous Csf-1r in a PU.1-dependent manner.
Figure 5.

PU.1 is critical for mixed lineage leukemia (MLL)-AF10-induced acute myeloid leukemia (AML). (a) PUER cells infected with MSCV-GFP or MSCV-FLAG-MLL-AF10-ires-GFP retroviruses were exposed to 100 nM 4-hydroxytamoxifen (4-HT) for 0, 2, or 5 days and analyzed by FACS for macrophage colony-stimulating factor (M-CSF) receptor (CSF-1R) expression. (b) Fetal liver cells of E12.5 PU.1+/+ and PU.1−/− mouse embryo littermates were infected with either MLL-AF10, and transplanted into irradiated mice. Leukemia-free survivals of the mice were analyzed (n = 6, P < 0.001). (c) Fetal liver cells of E14.5 PU.1flox/flox with ER-Cre were infected with MLL-AF10 and transplanted into irradiated mice. Bone marrow (BM) cells of the primary AML mice were transplanted into sublethally irradiated WT mice. Tamoxifen (tamo) (PU.1Δ/Δ) or solvent (PU.1f/f) was given to the secondary AML mice every 2 days by i.v. injection 17 days after transplantation, when GFP+ cells were detected in peripheral blood. Leukemia-free survivals of the secondary mice were investigated (n = 6, P < 0.001). (d,e) BM cells were prepared 0, 2, or 4 days after injection of tamoxifen and analyzed for expression of CSF-1R and c-Kit proteins (d) and for Csf1r, HoxA9, c-Kit, Meis1, and Gapdh mRNAs (e). (f) BM cells, untreated (f/f) or tamoxifen-treated for 4 days (Δ/Δ), were subjected to ChIP analysis using anti-Flag (MLL-AF10) antibodies. Semiquantitative real-time PCR was carried out on the co-precipitated DNAs.
To determine whether PU.1 is essential for initiation of MLL-AF10-induced AML, the WT and Pu.1−/− fetal liver cells of E12.5 litter mates were infected with MLL-AF10 retrovirus and transplanted into irradiated mice. Although the mice with WT cells expressing MLL-AF10 developed AML 2–3 months after transplantation, mice with Pu.1−/− cells were quite healthy for at least 6 months (Fig.5b).
To determine whether PU.1 is required for maintenance of MLL-AF10-induced AML, AML mice were generated using fetal liver cells of Pu.1 conditional KO mice (Pu.1flox/flox ERT2-Cre). The BM cells of the AML mice were transplanted into secondary recipient mice and deletion of the Pu.1 gene was induced 3 weeks after transplantation. All the control mice died within 1 month, whereas none of the mice with deletion of Pu.1 developed AML or died (Fig.5c). The population of Csf-1rhigh cells in BM decreased within 4 days after deletion of Pu.1 (Fig.5d). By contrast, c-Kit-positive cells still remained. These results indicate that PU.1 is required for both development and maintenance of MLL-AF10-induced AML. The RT-PCR analysis indicated that levels of Csf-1r mRNAs were decreased after Pu.1 deletion but levels of HoxA9, c-Kit, and Gapdh mRNAs were stable at least 4 days after tamoxifen treatment (Fig.5e). Chromatin immunoprecipitation analysis indicated that MLL-AF10 enrichment at the CSF-1R locus was reduced by deleting Pu.1 (Fig.5f).
CSF-1R is a promising target for AML therapy
To determine whether a high level of CSF-1R expression is an essential element of LICs, transgenic mice expressing drug-inducible FKBP-Fas suicide gene and EGFP under the control of the Csf-1r promoter were used (Fig.6a).17 In these mice, conditional ablation of Csf-1r-expressing cells can be induced by injection of the AP20187 dimerizer.17 c-Kit+ BM cells of transgenic mice were infected with MLL-AF10 retrovirus and transplanted into lethally irradiated WT mice. These mice developed AML approximately 2 months after transplantation, and their BM cells were transplanted into secondary recipient mice. Seven days after transplantation, the mice were injected with AP20187 as described previously.17 All untreated mice, and none of the AP20187-treated mice, developed AML 4–6 weeks after transplantation (Fig.6a), indicating that a high level of CSF-1R expression is a key LIC functional element in MLL-AF10-induced AML mice.
Figure 6.

Cure of mixed lineage leukemia (MLL)-AF10-induced acute myeloid leukemia (AML) by ablation of cells expressing high levels of macrophage colony-stimulating factor (M-CSF) receptor (CSF-1Rhigh). (a) Bone marrow (BM) cells from the transgenic (CSF-1R-EGFP-NGFR/FKBP1A/TNFRSF6) mice were infected with MSCV-MLL-AF10-ires-GFP and were transplanted into lethally irradiated C57BL/6 mice to induce AML. BM cells (1 × 104) of primary AML mice were transplanted into sublethally irradiated C57BL/6 mice. Administration of AP20187 or solvent (control) to the secondary AML mice was started by i.v. injection 3 weeks after transplantation. Leukemia-free survivals of the untreated (n = 8) and AP20187-treated (n = 8) secondary transplanted mice were investigated (P < 0.001). Right panel shows the structure of genes for the Csf1r promoter, EGFP, the NGFR–FKBP–Fas suicide construct, and activation of NGFR–FKBP–Fas. Note that in the transgenic mice, conditional ablation of cells expressing high levels of CSF-1R can be induced by exposure to the AP20187 dimerizer. (b) Fetal liver cells of E16.5 Csf1r+/+ and Csf1r−/− mice littermate embryos were infected with MLL-AF10-ires-GFP and transplanted into irradiated mice. The leukemia-free survivals of the mice were analyzed (n = 10, P < 0.001). (c,d). BM cells (105) from AML mice with MLL-AF10 were transplanted into non-irradiated mice. Mice were treated with vehicle, Ki20227 (Ki), Ara-C, or Ki plus Ara-C (AraC+Ki). Leukemia-free survivals were analyzed (c) (n = 10, P < 0.01 [control vs Ki; AraC vs AraC+Ki]). Peripheral blood cells were prepared 21 days after transplantation and analyzed for expression of GFP (d).
To determine if Csf-1r is essential for the development of MLL-AF10-induced AML, AML mice were generated using E16.5 fetal liver cells from Csf-1r−/−(18) and Csf-1r+/+ littermate embryos. The mice transplanted with the WT cells developed AML 6–9 weeks after transplantation whereas those transplanted with Csf-1r−/− cells developed AML 9–18 weeks after transplantation (Fig.6b). Thus, the CSF-1R is required for efficient induction of AML by MLL-AF10.
The present results suggest that signaling through CSF-1R may be a suitable therapeutic target for kinase inhibitors in MLL fusion-induced leukemogenesis. The effect of the CSF-1R-specific inhibitor Ki20227 was tested with or without AraC in MLL-AF10-induced AML in mice. Ki20227 and AraC slowed the onset of AML (Fig.6c) and inhibited the increase in GFP+ leukemic cells (Fig.6d). The combination of Ki20227 plus AraC was more effective than either agent alone.
Discussion
CSF-1R is a potential target for AML therapy
Acute myeloid leukemia is a highly malignant disease. Numerous genetic abnormalities are known in AML, among which chromosome translocations involving the MLL gene are associated with poor prognosis. Conventional chemotherapies are often effective in reducing the total number of leukemia cells, but are not curative in many cases of AML. Leukemic stem cells are capable of the limitless self-renewal necessary for cancer initiation and maintenance. As residual LSCs are a potential cause of AML relapse, eradication of LSCs is critical to cure the disease. The present results showed that LSCs are enriched in cells expressing high levels of CSF-1R. Relevant to our observations, a viral integration site of the Friend murine leukemia virus that is used in approximately 20% of virus-induced primary myeloid leukemias, was shown to be at the 5′-end of the Csf-1r gene and to result in high expression of a normal-sized Csf-1r mRNA.33 Using a mouse model expressing a drug-inducible suicide gene controlled by the Csf-1r promoter, ablation of Csf-1rhigh cells was shown to prevent AML mice from dying of the disease. Moreover, MLL-AF10-induced leukemia was suppressed by deletion of the Csf-1r gene. These results clearly show that CSF-1R is a promising target for novel AML therapy. CSF-1R is a receptor tyrosine kinase that regulates the survival, proliferation, and/or differentiation of macrophages, osteoclasts, and Paneth cells.34–36 A tyrosine kinase inhibitor specific for CSF-1R slowed the progress of MLL-AF10-induced leukemia. CSF-1R upregulation has been detected in LSCs in MOZ-TIF2-induced AML25 and human AML patients.26
CSF-1R expression is critical for AML initiation but not for immortalization in vitro
MLL leukemias are invariably associated with the expression of Hox genes. Upregulation of Hox genes in MLL leukemias is critical for LSC maintenance; however, Hox upregulation alone does not recapitulate all the biological and clinical features of MLL leukemias and is unlikely to support malignancy and the high LSC frequency observed in MLL leukemias. Forced expression of the HOXA9 gene can immortalize myeloid progenitors in vitro, but is not sufficient to initiate AML in vivo. By contrast, expression of MLL fusions such as MLL-AF10, MLL-AF9, and MLL-ENL is sufficient for both immortalization in vitro and initiation of AML in vivo. Our results indicate that cells expressing high levels of Csf-1r show strong AML initiation in vivo (Fig.1b,c) whereas cells expressing high and low levels of Csf-1r showed equivalent colony formation in vitro (Fig.1e). These findings suggest that CSF-1R expression is not important for immortalization in vitro but is critical for initiation of AML in vivo.
The LSC activity of MLL leukemia is known to be reduced after culture in vitro,37 suggesting that a certain in vivo microenvironment is required for LSC maintenance, as is also the case for hematopoietic stem cells. Our results show that the expression of Csf-1r is greatly reduced after culture in vitro (data not shown), suggesting that expression of CSF-1R is regulated by microenvironment-dependent epigenetics.
Differential regulation of CSF-1R and Hox
Both MLL and MLL fusion proteins form a complex with menin and LEDGF to regulate the expression of Hox genes.30,38 Our results show that PU.1 mediates the regulation of Csf1r transcription by MLL/MLL fusion proteins. The menin and LEGDF-interacting domains of MLL are not required for interaction with PU.1 or transactivation at the Csf1r promoter, suggesting that menin and LEGDF are unlikely to be involved in the regulation of CSF-1R expression. While expression of Csf-1r rapidly decreased after deletion of the Pu.1 gene (Fig.5d,e), HoxA9 mRNA levels were stable at least 4 days after Pu.1 deletion (Fig.5e). Moreover, HoxA9 mRNA levels were equivalent in AML cells expressing high and low levels of CSF-1R (Fig.1h). These results suggest that expression of the Csf1r and Hox genes is independently regulated by MLL fusion proteins.
The MLL-AF10-induced AML was shown to be cured by deletion of Pu.1 (Fig.5c). However, deletion of Csf-1r prolonged survival time but did not cure the AML completely. These facts suggest that CSF-1R is not the sole critical PU.1-dependent mediator of leukemogenesis, and that there are other PU.1-target genes critical for maintenance of MLL fusion-induced AML. Such genes may be involved in the leukemogenesis in collaboration with CSF-1R and Hox genes.
It has been suggested that second mutations, such as activating point mutations, in receptor tyrosine kinases (e.g., FLT3 and c-KIT) are required for fusion genes such as AML1-ETO, PML-RARa, or CBFb-MYH11 to induce acute leukemia.39 In contrast, MLL fusions alone can induce the rapid onset of AML.40 Our data suggest that MLL fusions induce the upregulation of the receptor tyrosine kinase Csf-1r and Hox genes, thereby inducing the rapid onset of AML by activating two classes of pathways (Fig.7). These pathways provide potential molecular targets for new approaches in the treatment of these forms of leukemia.
Figure 7.
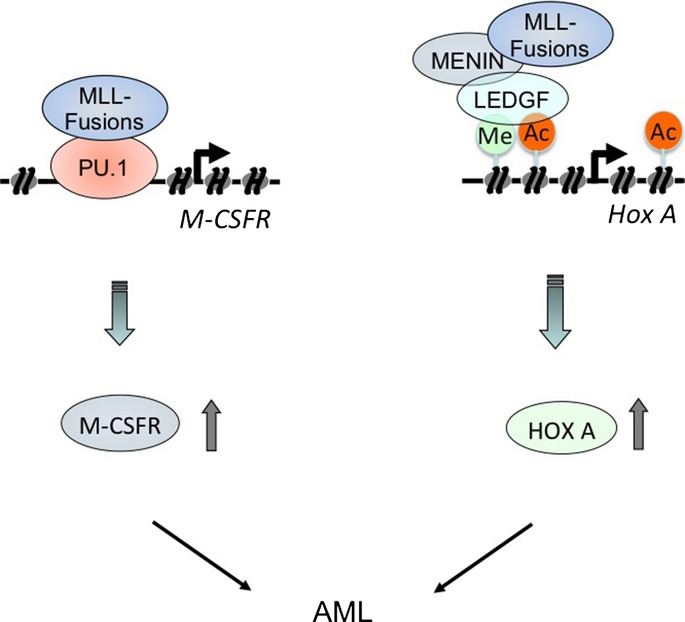
Model for transcriptional regulation of macrophage colony-stimulating factor receptor (Csf1r) and Hox genes by mixed lineage leukemia (MLL) fusion proteins. MLL fusions induce the rapid onset of acute myeloid leukemia by activating two classes of pathways, the CSF-1R and Hox pathways. Left, MLL fusions stimulate constitutive CSF-1R expression by binding to PU.1 to induce leukemia. Right, MLL fusion proteins form a complex with menin and LEDGF to regulate the expression of Hox genes.
Acknowledgments
We would like to thank Dr. D. E. Zhang for Csf-1r promoter mutant lacking PU.1-binding sites and Dr. Harinder Singh for PUER cells. This work was supported in part by Grants-in-Aid from the Ministry of Health, Labor and Welfare of Japan, the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Japanese National Cancer Center Research and Development Fund, and US National Institutes of Health grants HL112719, CA32551, and 5P30-CA13330.
Disclosure Statement
The authors have no conflicts of interest.
Supporting Information
Fig. S1. (A) Bone marrow (BM) cells from normal mice were analyzed by flow cytometry for expression of GFP and macrophage colony-stimulating factor receptor (Csf-1r). (B) Csf-1rhigh and Csf-1rlow/− cells from normal mixed lineage leukemia (MLL)-AF10–induced AML mice were sorted by flow cytometry. Levels of Csf1r (H) mRNAs were measured in Csf-1rhigh and Csf-1rlow/− cells prepared from normal and acute myeloid leukemia mouse BM. (C) Mll+/+ and Mll−/− Fetal liver cells were analyzed by flow cytometer for expression of Csf-1r.
References
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Warner JK, Wang JC, Hope KJ, Jin L, Dick JE. Concepts of human leukemic development. Oncogene. 2004;23:7164–77. doi: 10.1038/sj.onc.1207933. [DOI] [PubMed] [Google Scholar]
- Ayton PM, Cleary ML. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene. 2001;20:5695–707. doi: 10.1038/sj.onc.1204639. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- Dou Y, Hess JL. Mechanisms of transcriptional regulation by MLL and its disruption in acute leukemia. Int J Hematol. 2008;87:10–8. doi: 10.1007/s12185-007-0009-8. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Perales S, Cano F, Lobato MN, Rabbitts TH. MLL gene fusions in human leukaemias: in vivo modelling to recapitulate these primary tumourigenic events. Int J Hematol. 2008;87:3–9. doi: 10.1007/s12185-007-0001-3. [DOI] [PubMed] [Google Scholar]
- Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–35. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntly BJ, Shigematsu H, Deguchi K, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–96. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–7. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- Rozovskaia T, Feinstein E, Mor O, et al. Upregulation of Meis1 and HoxA9 in acute lymphocytic leukemias with the t(4: 11) abnormality. Oncogene. 2001;20:874–8. doi: 10.1038/sj.onc.1204174. [DOI] [PubMed] [Google Scholar]
- Yeoh EJ, Ross ME, Shurtleff SA, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–43. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003;17:2298–307. doi: 10.1101/gad.1111603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar AR, Hudson WA, Chen W, Nishiuchi R, Yao Q, Kersey JH. Hoxa9 influences the phenotype but not the incidence of Mll-AF9 fusion gene leukemia. Blood. 2004;103:1823–8. doi: 10.1182/blood-2003-07-2582. [DOI] [PubMed] [Google Scholar]
- Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007;21:2762–74. doi: 10.1101/gad.1602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar AR, Li Q, Hudson WA, et al. A role for MEIS1 in MLL-fusion gene leukemia. Blood. 2009;113:1756–8. doi: 10.1182/blood-2008-06-163287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10:257–68. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Burnett SH, Kershen EJ, Zhang J, et al. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J Leukoc Biol. 2004;75:612–23. doi: 10.1189/jlb.0903442. [DOI] [PubMed] [Google Scholar]
- Dai XM, Ryan GR, Hapel AJ, et al. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99:111–20. doi: 10.1182/blood.v99.1.111. [DOI] [PubMed] [Google Scholar]
- Iwasaki H, Somoza C, Shigematsu H, et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood. 2005;106:1590–600. doi: 10.1182/blood-2005-03-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibler J, Zevnik B, Kuter-Luks B, et al. Rapid generation of inducible mouse mutants. Nucleic Acids Res. 2003;31:e12. doi: 10.1093/nar/gng012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–6. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- Ohno H, Kubo K, Murooka H, et al. A c-fms tyrosine kinase inhibitor, Ki20227, suppresses osteoclast differentiation and osteolytic bone destruction in a bone metastasis model. Mol Cancer Ther. 2006;5:2634–43. doi: 10.1158/1535-7163.MCT-05-0313. [DOI] [PubMed] [Google Scholar]
- Zhang DE, Hetherington CJ, Chen HM, Tenen DG. The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol. 1994;14:373–81. doi: 10.1128/mcb.14.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Kitabayashi I, Ayton PM, Cleary ML, Ohki M. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood. 2002;100:3710–8. doi: 10.1182/blood-2002-04-1015. [DOI] [PubMed] [Google Scholar]
- Aikawa Y, Katsumoto T, Zhang P, et al. PU.1-mediated upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ-TIF2. Nat Med. 2010;16:580–5. doi: 10.1038/nm.2122. , 1p following 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikushige Y, Shima T, Takayanagi S, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7:708–17. doi: 10.1016/j.stem.2010.11.014. [DOI] [PubMed] [Google Scholar]
- DiMartino JF, Ayton PM, Chen EH, Naftzger CC, Young BD, Cleary ML. The AF10 leucine zipper is required for leukemic transformation of myeloid progenitors by MLL-AF10. Blood. 2002;99:3780–5. doi: 10.1182/blood.v99.10.3780. [DOI] [PubMed] [Google Scholar]
- Zhang DE, Hetherington CJ, Meyers S, et al. CCAAT enhancer-binding protein (C/EBP) and AML1 (CBF alpha2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol Cell Biol. 1996;16:1231–40. doi: 10.1128/mcb.16.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Zhao X, Wang L, et al. The ability of MLL to bind RUNX1 and methylate H3K4 at PU.1 regulatory regions is impaired by MDS/AML-associated RUNX1/AML1 mutations. Blood. 2011;118:6544–52. doi: 10.1182/blood-2010-11-317909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–17. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- Walsh JC, DeKoter RP, Lee HJ, et al. Cooperative and antagonistic interplay between PU.1 and GATA-2 in the specification of myeloid cell fates. Immunity. 2002;17:665–76. doi: 10.1016/s1074-7613(02)00452-1. [DOI] [PubMed] [Google Scholar]
- Gisselbrecht S, Fichelson S, Sola B, et al. Frequent c-fms activation by proviral insertion in mouse myeloblastic leukaemias. Nature. 1987;329:259–61. doi: 10.1038/329259a0. [DOI] [PubMed] [Google Scholar]
- Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18:39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Huynh D, Dai XM, Nandi S, et al. Colony stimulating factor-1 dependence of paneth cell development in the mouse small intestine. Gastroenterology. 2009;137:136–44. doi: 10.1053/j.gastro.2009.03.004. , 44 e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 2004;14:628–38. doi: 10.1016/j.tcb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–22. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–49. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–42. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 1997;16:4226–37. doi: 10.1093/emboj/16.14.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (A) Bone marrow (BM) cells from normal mice were analyzed by flow cytometry for expression of GFP and macrophage colony-stimulating factor receptor (Csf-1r). (B) Csf-1rhigh and Csf-1rlow/− cells from normal mixed lineage leukemia (MLL)-AF10–induced AML mice were sorted by flow cytometry. Levels of Csf1r (H) mRNAs were measured in Csf-1rhigh and Csf-1rlow/− cells prepared from normal and acute myeloid leukemia mouse BM. (C) Mll+/+ and Mll−/− Fetal liver cells were analyzed by flow cytometer for expression of Csf-1r.