

Abstract
Compounds that target isoprenoid biosynthesis in Plasmodium falciparum could be a welcome addition to malaria chemotherapy, since the methylerythritol phosphate (MEP) pathway used by the parasite is not present in humans. We previously reported that MMV008138 targets the apicoplast of P. falciparum and that its target in the MEP pathway differs from that of Fosmidomycin. In this article, we determine that the active stereoisomer of MMV008138 is 4a, which is (1R,3S)-configured. 2′,4′-Disubstitution of the D ring was also found to be crucial for inhibition of the parasite growth. Limited variation of the C3-carboxylic acid substitutent was carried out, and methylamide derivative 8a was found to be more potent than 4a; other amides, acylhydrazines, and esters were less potent. Finally, lead compounds 4a, 4e, 4f, 4h, 8a, and 8e did not inhibit growth of Escherichia coli, suggesting that protozoan-selective inhibition of the MEP pathway of P. falciparum can be achieved.
Keywords: malaria, Plasmodium, apicoplast, MEP pathway, Non-mevalonate
Malaria continues to be one of the most deadly diseases worldwide, with an estimated 584,000 deaths in 2013.1 The reductions seen in annual mortality over the last 10 years are greatly welcomed, but these advances are at risk due to several factors. Chief among these risk factors is emerging resistance to existing antimalarial drugs, such as chloroquine and artemether (Figure 1). Thus, there is a pressing need to develop antimalarial drugs that engage new targets. Since the discovery that the malaria parasite Plasmodium falciparum carried an apicoplast, there has been considerable interest in developing chemical agents that could specifically target this relict chloroplast, due to its unusual non-mammalian metabolism.2 Of particular interest is the fact that isoprenoid precursor biosynthesis in P. falciparum occurs in the apicoplast not via the mevalonate pathway, but rather through the methylerythritol phosphate (MEP) pathway.3 Moreover, supply of isoprenoid precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) is the sole essential function of this organelle during disease-causing asexual intraerythrocytic stages of the parasite4 and during gametocyte development, the malaria stages transmitted to the mosquito.5 Since the MEP pathway is absent in humans, agents targeting it could be both safe to the human host and effective. However, the MEP pathway is present in the human microbiome,6 therefore, compounds that can selectively inhibit the P. falciparum enzymes are highly desirable to reduce their impact on the host microbiome. Fosmidomycin (FOS) was shown to inhibit 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR or IspC), the second enzyme in the MEP pathway, and it cured mice infected with P. vinckei.3a In combination with clindamycin, FOS was evaluated in clinical trials.7 However, because of poor oral bioavailability, recent attention has focused on development of more hydrophobic FOS analogs.8
Figure 1.

Antimalarial agents used clinically (chloroquine, artemether) and MEP pathway-targeting drug candidates (FOS, MMV008138).
To identify chemotypes that might inhibit other enzymes in the MEP pathway, the compounds present in the publicly available Malaria Box9 were evaluated against asexual blood-stages of P. falciparum Dd2 strain. IPP supplementation4 was then employed to attempt chemical rescue of drug-treated parasites; in the case of FOS, 100% rescue was observed, consistent with its proposed mechanism of action.10 Of the 400 compounds in the Malaria Box that showed >95% growth inhibition at 5 μM, only MMV008138 exhibited >60% rescue following the addition of 200 μM IPP.10 Several antibiotics such as doxycycline kill malaria parasites by interfering with apicoplast genome replication, transcription, or protein translation.11 Parasites treated with these antibiotics initially present normal morphology and continue to grow and release daughter cells (merozoites) that are capable of invading new host cells. However, this progeny will not complete the subsequent intraerythrocytic cycle since the apicoplast will fail to replicate and segregate; this phenomenon is known as the delayed death phenotype.12 Like FOS, MMV008138 did not show a delayed death phenotype, and inhibition of apicoplast elongation was reversed by IPP supplementation; such rescue is not seen with antibiotics like doxycycline. However, the efficacy of MMV008138 observed against FOS-resistant parasites suggested that its target within the MEP pathway is not DXR.10
Since the stereochemical purity and identity of MMV008138 was not known, we synthesized all four possible stereoisomers by Pictet-Spengler reaction13 of (S)- and (R)-tryptophan methyl ester (Trp-OMe) with 2,4-dichlorobenzaldehyde 1a (Scheme 1).
Scheme 1.

Synthesis of the possible stereoisomers of MMV008138 (4a, 5a) and methyl ester precursors (2a, 3a). i) 4 Å molecular sieves, CH2Cl2, 20 h; TFA (2 equiv, 44 h). ii) Amberlyst hydroxide, THF/MeOH/H2O; AcOH, H2O.
Diastereomers 2a and 3a derive from (S)-Trp-OMe and were easily separated by column chromatography; the relative configuration of the C1 substituent (cis- or trans-) in each compound was determined by 13C NMR spectroscopy, using the empirical rule of Cook.14 Similarly (R)-Trp-OMe provided ent-2a and ent-3a. Hydrolysis of the esters using an Amberlyst resin catch and release protocol15 gave the desired stereoisomers of MMV008138, 4a, 5a, ent-4a, and ent-5a, free from inorganic salt contamination. 1H NMR spectroscopic analysis of commercial MMV008138 indicated that it was >95% trans-configured, like 4a or ent-4a (Supplementary Material, Figure S1). The four stereoisomers were then examined for growth inhibitory activity and reversal of growth inhibition by 200 μM IPP (Table 1), using MMV008138 and FOS as controls. As can be seen, 4a (IC50 = 250 ± 70 nM) was by far the most potent stereoisomer of the four (cf. entries 3, 4, 15 and 16). Compounds 5a and ent-4a showed no inhibition of growth at the highest concentration tested (10,000 nM), and ent-5a had an IC50 of 3,000 nM.
Table 1.
P. falciparum growth inhibitory activity and effect of IPP supplementation of MMV08138 stereoisomers and D-ring variants.
| ||||
|---|---|---|---|---|
| Entry | Compound | X | P. falciparum Dd2 straina IC50 (nM) | % Rescue (+ 200 μM IPP)b |
| 1 | FOS | 880 ± 70 | 100@ 10 μM | |
| 2 |
MMV 008138 |
2′,4′-Cl2 | 210 ± 90 | 100 @ 2.5 μM |
| 3 | 4a | 2′,4′-Cl2 | 250 ± 70 | 100 @ 2.5 μM |
| 4 | ent-4a | 2′,4′-Cl2 | >10,000 | ND |
| 5 | 4b | H | >10,000 | ND |
| 6 | 4c | 2′-Cl | 3,280 ± 990 | 60 @ 10 μM |
| 7 | 4d | 4′-Cl | 1,170 ± 60 | 50 @ 10 μM |
| 8 | 4e | 2′-Cl, 4′-Me | 410 ± 40 | 100 @ 2.5 μM |
| 9 | 4f | 2′-Me, 4′-Cl | 700 ± 90 | 100 @ 2.5 μM |
| 10 | 4g | 2′,4′-Me2 | 70% inhibition @ 10,000 | ND |
| 11 | 4h | 2′,4′-F2 | 780 ± 175 | 100 @ 5 μM |
| 12 | 4i | 2′,4′-(OMe)2 | >20,000 | ND |
| 13 | 4j | 2′,4′-(CF3)2 | >10,000 | ND |
| 14 | 4k | 3′,4′-(OMe)2 | >20,000 | ND |
| 15 | 5a | 2′,4′-Cl2 | >10,000 | ND |
| 16 | ent-5a | 2′,4′-Cl2 | 3,000 ± 200 | 60 @ 10 μM |
| 17 | 5b | H | >10,000 | ND |
| 18 | 5e | 2′-Cl, 4′-Me | >10,000 | ND |
| 19 | 5g | 2′,4′-Me2 | >10,000 | ND |
| 20 | 5h | 2′,4′-F2 | >20,000 | ND |
MRA-150, chloroquine-resistant (intermediate), pyrimethamine-resistant, mefloquine-resistant.
Drug at indicated concentration; nd signifies “not determined.”
Furthermore, the P. falciparum toxicity of 4a was reversed (100% at 2.5 μM) by supplementation with IPP, similar to MMV008138. These observations and the similar measured IC50 values for MMV008138 and 4a (210 ± 90 and 250 ± 70 nM, entries 2 and 3 respectively) confirm that MMV008138 is the pure (1R,3S)-stereoisomer and is thus properly depicted as 4a.
To ascertain the structural determinants of the apicoplast-targeting growth inhibitory activity of 4a, we explored the effect of variation of the D-ring substituents. With slight modification, the same synthetic methods were used to prepare (1R,3S)-configured acid derivatives 4b–k from benzaldehydes 1b–k, and a smaller number of (1S,3S)-configured acids 5b, e, g, h were also investigated (Table 2).
Table 2.
Synthesis and isolated yields for D-ring variants of 2a and 3a, and those of their acid derivatives (4 and 5).
| |||||||
|---|---|---|---|---|---|---|---|
| %Yield | %Yield | %Yield | %Yield | ||||
| 2b | 14 | 3b | 67 | 4b | 99 | 5b | 95 |
| 2c | 18 | 3c | 59 | 4c | 95 | ||
| 2d | 20 | 3d | 61 | 4d | 95 | ||
| 2e | 40 | 3e | 60 | 4e | 78 | 5e | 69 |
| 2f | 21 | 3f | 37 | 4f | 56 | ||
| 2g | 40 | 3g | 52 | 4g | 85 | 5g | 83 |
| 2h | 30 | 3h | 68 | 4h | 74 | 5h | 68 |
| 2i | 27 | 3i | 19 | 4i | 90 | ||
| 2j | 39 | 3j | 61 | 4j | 84 | ||
| 2k | 41 | 3k | 36 | 4k | 86 | ||
Bioassays of 4b–k revealed that potent growth inhibition requires 2′,4′-disubstitution of the D-ring. Compound 4b lacking D-ring substitution has no toxicity to P. falciparum at 10,000 nM (Table 1 entry 5). Growth inhibitory activity is regained with a 2′-Cl (4c) or a 4′-Cl (4d) substituent, though with substantially reduced potencies (Table 1 entries 6–7). Among the 2′,4′-disubstitution patterns explored, the relatively conservative 2′-chloro-4′-methyl (4e, Table 1 entry 8) and 2′-methyl-4′-chloro (4f, Table 1 entry 9) substitutions came closest to recapitulating the activity of 4a. Although 4e (IC50 = 410 ± 40 nM) is not as potent as 4a (cf. Table 1 entries 8 and 3), it demonstrated full rescue by 200 μM IPP supplementation. The size of the substituents at positions 2′ and 4′ of the D ring appears to be critical: the 2′,4′-difluoro analog (4h, Table 1 entry 11) is more potent than the 2′,4′-dimethyl analog (4g, Table 1 entry 10), and both the 2′,4′-dimethoxy and 2′,4′-bis(trifluoromethyl) analogs failed to show growth inhibitory activity at 10,000 or 20,000 nM (4i and 4j, Table 1 entries 12–13). The sole case of 3′,4′-disubstitution explored (4k, Table 1 entry 14) did not inhibit parasite growth at 20,000 nM. Thus the steric requirements for the D-ring substituents are very precise. It seems likely that the 2′- and 4′-substituents complement the shape of a rather tight binding pocket. In addition, the presence of the 2′-substituent may also constrain the D-ring to lie perpendicular to the C-ring. Finally, a limited number of (1S,3S)-configured analogs was explored (5b, 5e, 5g, 5h). As expected based on the low potency of 5a, none inhibited parasite growth at 10,000 or 20,000 nM (Table 1, entries 17–20).
Since the carboxylic acid group is often considered a liability in drug development, we investigated a limited series of analogs of 4a that varied in the C3 substituent (Table 3). Compound 2a, the methyl ester precursor of 4a had only weak growth inhibitory activity (IC50 = 6,800 ± 1,400 nM, Table 3 entry 1).
Table 3.
The effect of C3-substitution on the P. falciparum growth inhibitory activity of 4a and D-ring variants.
| ||||
|---|---|---|---|---|
| Entry | Compound | R | P. falciparum Dd2 straina IC50 (nM) | % Rescue (+ 200 μM IPP)b |
| 1 | 2a | CO2Me | 6,800 ± 1,400 | ~ 20 @ 20 μM |
| 2 | ent-2a | CO2Me | 7,700 ± 1,400 | 0 @ 20 μM |
| 3 | 2b | CO2Me | ~20% inhibition @ 20,000 | ND |
| 4 | (±)-6a | H | 10,000 ± 1,600 | 0 @ 20 μM |
| 5 | 7a | CONH2 | 1,200 ± 100 | 50 @ 10 μM |
| 6 | 8a | CONHMe | 190 ± 30 | 100 @ 2.5 μM |
| 7 | 8e | CONHMe | 340 ± 50 | 100 @ 2.5 μM |
| 8 | 8f | CONHMe | 1,850 ± 600 | 50 @ 10 μM |
| 9 | 8g | CONHMe | 70% inhibition @ 10,000 | ND |
| 10 | 8h | CONHMe | 1,200 ± 100 | 100 @ 2.5 μM |
| 11 | 8j | CONHMe | >10,000 | ND |
| 12 | 9a | CONHi-Pr | 50% inhibition @ 10,000 | ND |
| 13 | 10a | CONHBu | 80% inhibition @ 10,000 | ND |
| 14 | 11a | CONH c-C6H11 |
2,830 ± 500 | 0 @ 10 μM |
| 15 | 12a | CONHNHMe | 1,940 ± 200 | 80 @ 10 μM |
| 16 | tadalafil | NAc | >20,000 | ND |
MRA-150, chloroquine-resistant (intermediate), pyrimethamine-resistant, mefloquine-resistant.
Drug at indicated concentration; ND signifies “not determined.”
Not applicable.
Thus the methyl ester group cannot recapitulate the binding of the carboxylic acid/carboxylate of 4a, though it might serve well in a prodrug. A number of other esters were examined and found to be inactive; in particular ent-2a and 2b (the methyl ester precursors of ent-4a and 4b) had little growth inhibitory activity (Table 3, entries 2–3). Tryptamine was condensed with 1a to yield (±)-6a in 42 % yield, which lacks a C3-substitutent (Scheme 2). As can be seen in Table 3, (±)-6a is only a weak inhibitor of parasite growth (entry 4). More success was obtained with amide and hydrazide derivatives, that were simply prepared by treatment of the requisite methyl ester with the desired amine or hydrazine (Scheme 2).
Scheme 2.
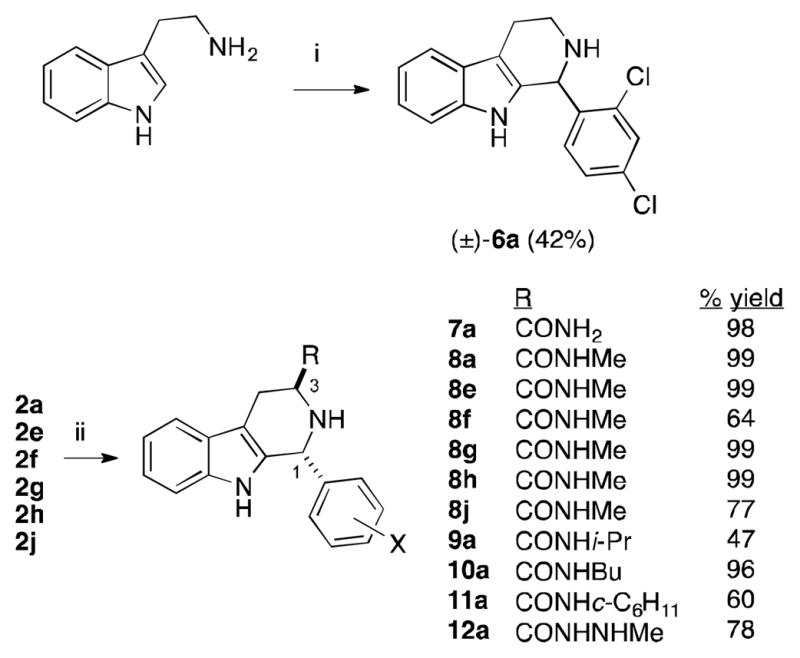
Synthesis of C3 variants of 4a/MMV008138. i) 2,4-dichlorobenzaldehyde (1a), 4 Å molecular sieves, CH2Cl2, 24 h. ii) Requisite amine or hydrazine, 25 °C or 50 ° C (9a, 11a), 24 h.
The primary amide 7a was weakly potent, but methylamide 8a proved slightly more potent than 4a (IC50 = 190 ± 30 nM, Table 3 entry 6, p < 0.04 ANOVA test) and exhibited 100% rescue upon IPP supplementation. X-ray crystallography of 8a confirmed (1R,3S)-configuration (Figure 2).
Figure 2.

Anisotropic displacement ellipsoid drawings (50%) of the X-ray structure of compound 8a. This structure was deposited at the Cambridge Structural Database (CCDC-1042325) and was visualized using OLEX2.16
Methylamide 8e featuring a 2′-chloro-4′-methylphenyl group in place of 2′,4′-dichlorophenyl also showed good potency (IC50 = 340 ± 50 nM, Table 3 entry 7); methylamides 8f–h proved to be less potent. Methylamide 8j had no activity, consistent with the low activity of the corresponding acid 4j. Amides featuring larger alkyl groups (9a–11a, Table 3 entries 12–14) were also less potent than methylamide 8a. One acylhydrazine 12a was explored, and proved to be less potent than 8a as well.
Finally, we noted that the amide derivatives bore a resemblance to the erectile dysfunction drug tadalafil. As can be seen in Table 3, this compound showed no growth inhibition at 20,000 nM. This low potency can be attributed to at least three factors: i) non-optimum (R,R)-configuration; ii) 3′,4′-disubstitution on the D-ring; and iii) the as yet unknown effect of rendering N2 non-basic.
To assess possible off-target effects of potent inhibitors 4a and 8a we explored their parasite growth inhibition activity in the presence of 200 μM IPP (Figure 3). As can be seen, the presence of 200 μM IPP raises the IC50 values of these compounds to roughly 10,000 nM. Similar results were seen for 4e, 4f, and 4h (Supplementary Material, Figure S2). Thus the off-target P. falciparum growth inhibition activity of these compounds occurs at concentrations roughly 40-fold higher than those that target the MEP pathway. In addition we note that when tested at 10–20 μM, many of the weakly potent (micromolar IC50 values) analogs in Tables 1 and 3 do not show full rescue upon IPP supplementation (e.g. 4c,d, ent-5a, 2a, ent-2a, (±)-6a, 7a, 8f, 11a, 12a). It is thus likely that the same off-target toxic mechanisms are also operative for these compounds. As mentioned above, the MEP pathway is present in the human microbiome and specificity of these inhibitors to the malaria parasite is highly desirable. Therefore, compounds 4a, 4e, 4f, 4h, 8a, 8e, and 8h, were tested for antibacterial activity against Escherichia coli. None of these seven compounds inhibited E. coli growth at 500 μM (Supplementary Material, Figure S3).
Figure 3.

Effects of 4a and 8a on P. falciparum Dd2 strain growth in the absence and presence of 200 μM IPP (72 h incubation). Data represent the average and standard error of five independent experiments.
In conclusion, we have demonstrated that the active stereoisomer of MMV008138 is structure 4a. In addition our survey of 33 analogs of 4a demonstrated that potent MEP pathway-targeting antiplasmodial activity within this scaffold requires a) (1R,3S)-configuration; b) a carboxylate/carboxylic acid or methylamide functionality at C3; and c) and sterically modest 2′,4′-disubstitution of the D-ring. Lead compounds 4a, 4e, 4f, 4h, 8a, and 8e have P. falciparum growth inhibition potencies ranging from 190 to 780 nM, and do not inhibit the growth of E. coli at 500 μM. Thus it is possible to selectively inhibit the MEP pathway of P. falciparum without affecting that of helpful gut bacteria; these tetrahydro-β-carboline derivatives therefore represent promising new antimalarial leads. Recently, other researchers have identified the molecular target of MMV008138 as IspD, the third enzyme of the MEP pathway; these authors also concluded that the active stereoisomer of MMV008138 was (1R,3S)-configured.17 Our characterization of the aforementioned lead compounds against that and related enzymes, and further structural optimization studies will be reported in due course.
Supplementary Material
Acknowledgments
We acknowledge the Fralin Life Science Institute, and the National Institutes of Health (AI082581 (PRC) and AI108819 (MBC)) for financial support. The following reagent was obtained through the MR4 as part of the BEI Resources Repository, NIAID, NIH: Plasmodium falciparum Dd2, MRA-150, deposited by D Walliker.
Footnotes
Supplementary data (38 pages) includes synthetic procedures, analytical tabulations, and copies of the 1H and 13C NMR spectra for all tested compounds; biological assay procedures and biological data can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.World Malaria Report. The World Health Organization; 2014. [last accessed 12/17/14]. available at http://apps.who.int/iris/bitstream/10665/144852/2/9789241564830_eng.pdf. [Google Scholar]
- 2.Ralph SA, D’Ombrain MC, McFadden GI. Drug Resist Updates. 2001;4:145. doi: 10.1054/drup.2001.0205. [DOI] [PubMed] [Google Scholar]
- 3.(a) Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Türbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Science. 1999;285:1573. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]; (b) Masini T, Hirsch AKH. J Med Chem. 2014;57:9740. doi: 10.1021/jm5010978. [DOI] [PubMed] [Google Scholar]
- 4.Yeh E, DeRisi JL. PLoS Biol. 2011;9:e1001138. doi: 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiley JD, Merino EF, Krai PM, McLean KJ, Tripathi AK, Vega-Rodriguez J, Jacobs-Lorena M, Klemba M, Cassera MB. Eukaryot Cell. 2014 doi: 10.1128/EC.00198-14.. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Science. 2006;312:1355. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oyakhirome S, Issifou S, Pongratz P, Barondi F, Ramharter M, Kun JF, Missinou MA, Lell B, Kremsner PG. Antimicrob Agents Chemother. 2007;51:1869. doi: 10.1128/AAC.01448-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Verbrugghen T, Vandurm P, Pouyez J, Maes L, Wouters J, Van Calenbergh S. J Med Chem. 2012;56:376. doi: 10.1021/jm301577q. [DOI] [PubMed] [Google Scholar]; (b) Xue J, Diao J, Cai G, Deng L, Zheng B, Yao Y, Song Y. ACS Med Chem Lett. 2013;4:278. doi: 10.1021/ml300419r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jansson AM, Wieckowska A, Bjorkelid C, Yahiaoui S, Sooriyaarachchi S, Lindh M, Bergfors T, Dharavath S, Desroses M, Suresh S, Andaloussi M, Nikhil R, Sreevalli S, Srinivasa BR, Larhed M, Jones TA, Karlen A, Mowbray SL. J Med Chem. 2013;56:6190. doi: 10.1021/jm4006498. [DOI] [PubMed] [Google Scholar]; (d) Konzuch S, Umeda T, Held J, Hahn S, Brucher K, Lienau C, Behrendt CT, Grawert T, Bacher A, Illarionov B, Fischer M, Mordmuller B, Tanaka N, Kurz T. J Med Chem. 2014;57:8827. doi: 10.1021/jm500850y. [DOI] [PubMed] [Google Scholar]
- 9.Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TNC, Willis P. PLOS One. 2013;8:e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowman JD, Merino EF, Brooks CF, Striepen B, Carlier PR, Cassera MB. Antimicrob Agents Chemother. 2014;58:811. doi: 10.1128/AAC.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodman CD, Su V, McFadden GI. Mol Biochem Parasitol. 2007;152:181. doi: 10.1016/j.molbiopara.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Dahl EL, Rosenthal PJ. Antimicrob Agents Chemother. 2007;51:3485. doi: 10.1128/AAC.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bailey PD, Hollinshead SP, McLay NR. Tetrahedron Lett. 1987;28:5177. [Google Scholar]
- 14.Ungemach F, Soerens D, Weber R, DiPierro M, Campos O, Mokry P, Cook JM, Silverton JV. J Am Chem Soc. 1980;102:6976. [Google Scholar]
- 15.Dandapani S, Lan P, Beeler AB, Beischel S, Abbas A, Roth BL, Porco JA, Panek JS. J Org Chem. 2006;71:8934. doi: 10.1021/jo061758p. [DOI] [PubMed] [Google Scholar]
- 16.Dolomanov OV, Bouris LJ, Gildea RJ, Howard JAK, Puschmann H. J Appl Cryst. 2009;42:339. doi: 10.1107/S0021889811041161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu W, Herrera Z, Ebert D, Baska K, Cho SH, DeRisi JL, Yeh E. Antimicrob Agents Chemother. 2015;59:356. doi: 10.1128/AAC.03342-14.(b) Reference 17a appeared online just as the present manuscript was in the final stages of preparation. No analytical data or synthetic procedures were provided for the stereoisomers of MMV008138. In the Supplemental material they disclosed antimalarial activity of less potent compounds, including (±)-6a and stereochemically undefined structures related to 4d, 4i, and 4k.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.