

Abstract
Little information has been reported on the antitumor effects of the diterpenoid alkaloid constituents of Aconitum plants, used in the herbal drug “bushi”. This study was aimed at determining the antitumor activities of Aconitum C19-and C20-diterpenoid alkaloids and synthetic derivatives against lung (A549), prostate (DU145), nasopharyngeal (KB), and vincristine-resistant nasopharyngeal (KB-VIN) cancer cell lines. Newly synthesized C20-diterpenoid alkaloid derivatives showed substantial suppressive effects against all human tumor cell lines tested. In contrast, natural and derivatized C19-diterpenoid alkaloids showed only a slight or no effect. Most of the active compounds were hetisine-type C20-diterpenoid alkaloids, specifically kobusine and pseudokobusine analogs with two different substitution patterns, C-11 and C-11,15. Notably, several C20-diterpenoid alkaloids were more potent against multidrug-resistant KB subline KB-VIN cells. Pseudokobusine 11-3′-trifluoromethylbenzoate (94) is a possible promising new lead meriting additional evaluation against multidrug-resistant tumors.
Keywords: Diterpenoid alkaloids, Pseudokobusine, Antiproliferative agents
The genera Aconitum, Consolida and Delphinium (family Ranunculaceae) and the genus Spiraea (family Rosaceae) contain numerous diterpenoid alkaloids, which are classified structurally as C18-, C19-, and C20-diterpenoid alkaloids with the general structures and numbering systems shown in Figure 1.1,2 Aconitum plants are used in “bushi”, an herbal traditional Chinese medicine prescribed to treat hypometabolism, dysuria, cardiac weakness, chills, neuralgia, gout, and certain rheumatic diseases.3–5 Among the C19-diterpenoid alkaloids, aconitine (1), jesaconitine (3), mesaconitine (8), and hypaconitine (9) exhibit particularly high toxicity, while the C20-diterpenoid alkaloids lucidusculine (37), kobusine (51), pseudokobusine (71), and atisine are much less toxic. However, despite the extreme toxicities of some C19-diterpenoid alkaloids, only two studies appeared in the literature in 2005 and 2006.6,7 The first reported the antiproliferative activity of 8-O-azeloyl-14-benzoylaconine, an aconitine-type C19-diterpenoid alkaloid,6 and the second described the cytotoxic effects of various C19-diterpenoid alkaloids against tumor cell lines.7 Since 2007, many C19- and C20-diterpenoids as well as semisynthetic derivatives were evaluated for cytotoxicity by various assays, including cell growth, clonogenic, cell cycle distribution, and cell cycle-related, against four different human tumor cell lines, A172, A549, HeLa, and Raji.8–12
Figure 1.
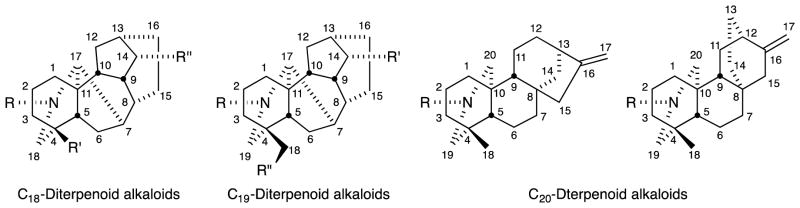
General structures and numbering systems for C18-, C19-, and C20-diterpenoid alkaloids.
In an initial survey of the pharmacological effects of natural diterpenoid alkaloids and their synthetic derivatives, we tested 108 compounds for antiproliferative activity against four tumor cell lines, lung (A549), prostate (DU145), nasopharyngeal (KB), and vincristine-resistant nasopharyngeal (KB-VIN) cancers. Here, we describe our results on the cytotoxic activities of these diterpenoid alkaloids and derivatives.
The C19-diterpenoid alkaloids may be divided into six types: aconitine, lycoctonine pyro, lactone, 7,17-seco, and rearranged.1,2 Most of the isolated C19-diterpenoid alkaloids are aconitine- and lycoctonine-types. The C20-diterpenoid alkaloids may be divided into ten types: atisine, denudatine, hetidine, hetisine, vakognavine, napelline, kusnezoline, racemulosine arcutine, and tricalysiamide.1,2 Most of the isolated C20-diterpenoid alkaloids are atisine-, hetisine-, and napelline-types. Table 1 lists the natural diterpenoid alkaloids and their source plant as well as the modified diterpenoid alkaloids generated for this study, and Figure 2 gives the structures of compounds 1–108.
Table 1.
Identities, types, and sources of diterpenoid alkaloids and derivatives
| Diterpenoid Alkaloid Type/Class | Natural Cmpds | Plant Source | References | Diterpenoid Alkaloid Type/Class | Modified Cmpds | References |
|---|---|---|---|---|---|---|
| Aconitine-type C19 | 1–6, 8–10, 12–14 | Aconitum japonicum Thunb. roots | 13–17 | Aconitine-type C19 | 7, 11 | 14, 18 |
| Lycoctonine-type C19 | 15, 17, 23–25 | A. yesoense var. macroyesoense (Nakai) Tamura roots | 19–22 | Lycoctonine-type C19 | 16, 18–22, 26, 27 | 5, 10, 23, 24 |
| Lycoctonine-type C19 | 28–32, 34, 36 | Delphinium elatum cv. Pacific Giant seeds | 25, 26 | Lycoctonine-type C19 | 33, 35 | 12 |
| Napelline- & hetisine-type C20 | 37–39, 41, 48–51, 71, 85 | A. yesoense var. macroyesoense roots | 19, 21, 22 | Napelline-type C20 | 40, 42–47 | 10, 19, 27 |
| Hetisine-type C20-analogs of 51 | 52–70 | 10, 11, 28, 29 | ||||
| Hetisine-type C20-analogs of 71 | 72–84, 86–108 | 10, 19, 28–31 |
Figure 2.




Structures of compounds 1–108
We tested 108 diterpenoid alkaloids for antiproliferative effects against four human tumor cell lines [lung carcinoma (A549), prostate carcinoma (DU145), nasopharyngeal (KB), and multi-drug resistant KB subline KB-VIN. Notably, we were interested in the antitumor activities against KB-VIN cells, because they overexpress drug transporter protein P-glycoprotein (P-gp), which effectively reduces intracellular drug concentration, especially of vinca and taxane alkaloids. The compounds included 24 natural (1–6, 8–10, 12–15, 17, 23–25, 28–32, 34, 36) and 12 synthesized (7, 11, 16, 18–22, 26, 27, 33, 35) C19-diterpenoid alkaloids, as well as 10 natural (37–39, 41, 48–51, 71, 85) and 62 synthesized (40, 42–47, 52–70, 72–84, 86–108) C20-diterpenoid alkaloids. Paclitaxel, a P-gp substrate anticancer agent, was used as an experimental control. The data are listed in Table 2. P-gp-overexpressing KB-VIN cells were over 100-fold resistant against paclitaxel, demonstrating that a lethal dose of paclitaxel may be required to be effective against MDR phenotype. The ratio of GI50 KB/GI50 KB-VIN demonstrated the efficacy of compound against KB-VIN. A549 (lung carcinoma), DU-145 (prostate cancer), and KB (epidermoid carcinoma) cell lines (ATCC) were supplied by Lineberger Comprehensive Cancer Center (UNC-CH). Professor Y.-C. Cheng, Yale University, CT generously provided KBvin (vincristine-resistant KB subline). Cells were cultured in RPMI 1640 medium containing 25 mM HEPES and 2 mM L-glutamine (Mediatech), supplemented with 10% heat-inactivated fetal bovine serum (Hyclone), 100 IU penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B (Mediatech). KB-VIN cells were grown in media containing 100 nM vincristine and were cultured for 7–10 days without vincristine before experiments were performed. Cells were maintained in a humidified 5% CO2 atmosphere at 37 °C, and passaged every 3–4 days. Cell viability was determined by the sulforhodamine B (SRB) colorimetric assay.32 In brief, cells (3–5 × 103 cells/well) were seeded in 96-well plates containing various concentrations of samples or paclitaxel (PXL) as an assay control, and incubated for 72 h. At the end of the exposure period, the attached cells were fixed with 50% trichloroacetic acid for 30 min followed by staining with 0.04% SRB (Sigma Chemical Co.) for 30 min. The bound SRB was solubilized in 10 mM Tris-base and the absorbance was measured at 515 nm on a Microplate Reader ELx800 (Bio-Tek Instruments, Winooski, VT) with Gen5 software. All results were representative of three or more experiments.
Table 2.
| Position/Substituent | GI50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Cmpd | 6 | 11 | 15 | A549 | DU145 | KB | KB-VIN | KB/KB-VIN ratio |
| 56 | H | OBz | OBz | 8.4 | 9.3 | 6.0 | 7.5 | 0.80 |
| 58 | H | OAs | OAs | 6.7 | 7.1 | 5.3 | 5.2 | 1.0 |
| 61 | H | ONB | ONB | 6.9 | 7.0 | 5.3 | 5.5 | 0.96 |
| 69 | H | OFB | OFB | 8.1 | 6.8 | 5.2 | 7.1 | 0.73 |
| 70 | H | OFMCM | OFMCM | 5.5 | 6.2 | 4.1 | 3.1 | 1.3 |
| 79 | OH | OBz | OBz | 8.8 | 7.6 | 5.2 | 6.3 | 0.82 |
| 89 | OH | ONB | OH | 5.8 | 7.2 | 6.4 | 6.4 | 1.0 |
| 93 | OH | OMNB | OMNB | 5.0 | 5.2 | 5.6 | 5.6 | 0.99 |
| 94 | OH | OMFMB | OH | 6.8 | 7.7 | 8.9 | 6.2 | 1.5 |
| 98 | OH | OCm | OH | 8.4 | 6.5 | 7.0 | 6.4 | 1.1 |
| 107 | OH | OTr | OH | 6.4 | 6.0 | 6.6 | 5.2 | 1.3 |
|
| ||||||||
| paclitaxel | 0.0071 | 0.0057 | 0.0064 | 0.95 | 0.0067 | |||
Compounds 33, 59, 64, 65, 78, 81, 84, 86, 87, 96 exhibited average GI50 values > 10 but < 20 μM. Compounds 1–32, 34–55, 57, 60, 62, 63, 66–68, 71–77, 80, 82, 83, 85, 88, 90–92, 95, 97, 99–106, 108 were inactive (GI50 > 20 μM) against all four cancer cell lines.
Cytotoxicity as GI50 values for each cell line, the concentration of compound that caused 50% reduction in absorbance at 515 nm relative to untreated cells using sulforhodamine B assay.
Lung carcinoma (A549), prostate carcinoma (DU145), nasopharyngeal (KB), and multidrug-resistant KB subline expressing P-glycoprotein (KB-VIN).
All tested aconitine-type C19-diterpenoid alkaloids, both the natural alkaloids (1–6, 8–10, 12–14) and synthetic analogs (7, 11), were inactive (GI50 > 20 μM). Among the lycoctonine-type C19-diterpenoid alkaloids, only compound 33, esterified with 3-trifluoromethylbenzoyl at C-6, showed any, although very weak, cytotoxic activity with GI50 values of 12.6, 14.9, and 11.9 μM against DU145, KB, and KB-VIN cell lines, respectively. The remaining natural alkaloids (15, 17, 23–25, 28–32, 34, 36) and synthetic analogs without a C-6 ester group (16, 18–22, 26, 27, 35) were inactive.
Among the C20-diterpenoid alkaloids, both natural (37–39, 41, 48–50) and synthetic (40, 42–47) napelline-type compounds, as well as hetidine-type synthetic analogs (72, 75, 76, 100), were inactive. Among hetisine-type C20-diterpenoid alkaloids, the natural parent alkaloids 51 and 71, as well as a related synthetic analog (108) of 71, were inactive against all four cancer cell lines. However, appropriate acylation, or in one case, etherification, of the two or three hydroxy groups in 51 and 71, respectively, led to cytotoxic synthetic analogs (see Table 1).
Nine different groups were present on hydroxyls of the 11 hetisine-type C20-diterpenoid alkaloids that exhibited average GI50 values of less than 10 μM: benzoyl (56, 79), anisoyl (58), 4-nitrobenzoyl (61, 89), 4-fluorobenzoyl (69), trans-3-trifluoromethylcinnamoyl (70), 3-nitrobenzoyl (93), 3-trifluoromethylbenzoyl (94), cinnamoyl (98), and trityl (107) groups. Nine additional compounds with six different ester groups exhibited average GI50 values between 10 and 20 μM: 78 (benzoyl), 81 (anisoyl), 59 (4-nitrobenzoyl), 64, 96 (4-trifluoromethylbenzoyl), 65 (4-trifluoromethoxybenzoyl), 84, 86, and 87 (veratroyl). However, all compounds esterified with acetyl (52, 53, 73, 74), 2-trifluoromethylbenzoate (62), propyl (105, 106), pivaloyl (101), and nicotinoyl (66, 102–104) groups, were inactive, regardless of the numbers or positions of the substitution. All six more potent (average GI50 < 10 μM) derivatives of 71 (79, 89, 93, 94, 98, 107) had a free hydroxy group at C-6. Comparison of corresponding analogs of 71 and 51 showed that some compounds with a C-6 OH rather than H exhibited higher potency (compare 89 to 59, 94 to 63), although this pattern was not universal (compare 56 to 79, 64 to 96).
Among analogs of 51, esterification of C-15 in addition to C-11 increased potency significantly (compare 59 to 61) or even converted an inactive to an active compound (compare 54 to 56, 57 to 58, 67 to 69). Consequently, all of the most analogs (56, 58, 61, 69, 70) of 51 were esterified at both C-11 and C-15. Among analogs of 71, four C-11 mono-substituted compounds (89, 94, 98, 107) and two C-11,15 diesterified compounds (79, 93) exhibited average GI50 values of less than 10 μM. Certain C-11 (81, 84, 96), C-6,11 (78, 86) and C-6,15 (87) esterified compounds were generally less potent, while all C-6 (77, 80, 88. 97) and C-15 (73, 85, 90, 99, 103, 105) mono-substituted compounds, as well as the tri-substituted analog (92), were inactive. Thus, all of the more active (GI50 < 10 μM) hetisine-type compounds had an ester or ether group on the C-11 hydroxyl and were either 11,15-diester analogs of 51 (H at C-6) or 11-monoester/11,15-diester analogs of 71 (OH at C-6).
Striking observations from the data in Table 2 were the degree and comparative ratio of KB/KB-VIN potency. Eleven compounds (56, 58, 61, 69, 70, 79, 89, 93, 94, 98, and 107) were quite potent (GI50 < 10 μM) against KB-VIN. Indeed, compound 70 exhibited a significantly low GI50 value of 3.1 μM. The ratios of KB to KB-VIN (GI50 KB/GI50 KB-VIN) were greater than 0.73 for all active compounds, with many compounds displaying comparable potency against the two cell lines, in contrast with paclitaxel (ratio of 0.0067). C20-Diterpenoid analogs 70, 94, and 107 showed over 1.3-fold selectivity with their greatest cytotoxic activity against KB-VIN (GI50 KB/GI50 KB-VIN: 1.3, 1.5 and 1.3, respectively).
In mechanism of action studies on selected diterpenoid alkaloids, the C20-diterpenoid alkaloid derivatives 81 and 96 showed important suppressive effects against Raji cells. Further study indicated that 96 inhibited extracellular signal-regulated kinase phosphorylation but induced enhanced phosphoinositide 3 kinase phosphorylation, leading to accumulation of Raji cells in the G1 or sub G1 phase.33 More investigation is certainly warranted.
In summary, we have synthesized acylated derivatives of various C19- and C20-diterpenoid alkaloids. Totally, 108 natural alkaloids and their derivatives were evaluated against four tumor cell lines. Eighty-seven compounds were non-toxic (GI50 > 20 μM), and ten compounds showed mild antiproliferative effects (GI50 = 10–20 μM). Except for 33, all of the most active compounds were hetisine-type C20-diterpenoid alkaloids with two different substitution patterns, C-11 and C-11,15. Compounds 56, 58, 61, 69, 70, 79, 89, 93, 94, 98 and 107, which are acylated or tritylated at the C-11 hydroxyl, exhibited the greatest potency over all four tested cell lines, including multidrug-resistant KB-VIN. These results demonstrate that modified hetisine-type C20-diterpenoid alkaloids are not substrates of P-gp and are effective against multi-drug resistant tumors. These promising new lead compounds merit continued studies to evaluate their potential as antitumor agents, particularly with enhanced resistant tumor selectivity. In addition, our results from modification-based antitumor activity studies can be used for further development of anticancer drugs overcoming a multidrug-resistant phenotype.
Supplementary Material
Acknowledgments
This work was supported by NIH grant CA177584 from the National Cancer Institute awarded to K. H. Lee.
Footnotes
Physical and spectroscopic data for compounds 19–22, 62, 65, 67–70, 79, 93, 95, 106, 107.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang FP, Chen QH. The C19-diterpenoid alkaloids. In: Cordell GA, editor. The Alkaloids. Vol. 69. New York: Elsevier Science; 2010. p. 1. [DOI] [PubMed] [Google Scholar]
- 2.Wang FP, Chen QH, Liu XY. Nat Prod Rep. 2010;27:529. doi: 10.1039/b916679c. [DOI] [PubMed] [Google Scholar]
- 3.Amiya T, Bando H. Aconitum alkaloids. In: Brossi A, editor. The Alkaloids. Vol. 34. San Diego: Academic Press; 1988. p. 95. [Google Scholar]
- 4.Pelletier SW, Page SW. Nat Prod Rep. 1984;1:375. doi: 10.1039/np9860300451. [DOI] [PubMed] [Google Scholar]
- 5.Pelletier SW, Page SW. Nat Prod Rep. 1986;3:451. doi: 10.1039/np9860300451. [DOI] [PubMed] [Google Scholar]
- 6.Chodoeva A, Bosc JJ, Guillon J, Decendit A, Petraud M, Absalon C, Vitry C, Jarry C, Robert J. Bioorg Med Chem. 2005;13:6493. doi: 10.1016/j.bmc.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 7.de Ines C, Reina M, Gavin JA, Gonzalez-Coloma A. Zeitschrift für Naturforschung C. 2006;61c:11. doi: 10.1515/znc-2006-1-203. [DOI] [PubMed] [Google Scholar]
- 8.Hazawa M, Wada K, Takahashi K, Mori T, Kawahara N, Kashiwakura I. Invest New Drugs. 2009;27:111. doi: 10.1007/s10637-008-9141-4. [DOI] [PubMed] [Google Scholar]
- 9.Hazawa M, Wada K, Takahashi K, Mori T, Kawahara N, Kashiwakura I. Invest New Drugs. 2011;29:1. doi: 10.1007/s10637-009-9327-4. [DOI] [PubMed] [Google Scholar]
- 10.Wada K, Hazawa M, Takahashi K, Mori T, Kawahara N, Kashiwakura I. J Nat Prod. 2007;70:1854. doi: 10.1021/np070270w. [DOI] [PubMed] [Google Scholar]
- 11.Wada K, Hazawa M, Takahashi K, Mori T, Kawahara N, Kashiwakura I. J Nat Med. 2011;65:43. doi: 10.1007/s11418-010-0452-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wada K, Ohkoshi E, Morris-Natschke SL, Bastow KF, Lee KH. Bioorg Med Chem Lett. 2012;22:249. doi: 10.1016/j.bmcl.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bando H, Kanaiwa Y, Wada K, Mori T, Amiya T. Heterocycles. 1981;16:1723. [Google Scholar]
- 14.Bando H, Wada K, Amiya T, Fujimoto Y, Kobayashi K. Chem Pharm Bull. 1988;36:1604. [Google Scholar]
- 15.Mori T, Bando H, Kanaiwa Y, Wada K, Amiya T. Chem Pharm Bull. 1983;31:2884. [Google Scholar]
- 16.Wada K, Bando H, Mori T, Wada R, Kanaiwa Y, Amiya T. Chem Pharm Bull. 1985;33:3658. [Google Scholar]
- 17.Wada K, Bando H, Watanabe M, Mori T, Amiya T. Chem Pharm Bull. 1985;33:4717. [Google Scholar]
- 18.Wada K, Bando H, Kawahara N, Mori T, Murayama M. Biol Mass Spect. 1994;23:97. [Google Scholar]
- 19.Bando H, Wada K, Amiya T, Kobayashi K, Fujimoto Y, Sakurai T. Heterocycles. 1987;26:2623. [Google Scholar]
- 20.Wada K, Kawahara N. Helv Chim Acta. 2009;92:629. [Google Scholar]
- 21.Wada K, Bando H, Amiya T. Heterocycles. 1985;23:2473. [Google Scholar]
- 22.Wada K, Bando H, Amiya T, Kawahara N. Heterocycles. 1989;29:2141. [Google Scholar]
- 23.Wada K, Ishizuki S, Mori T, Bando H, Murayama M, Kawahara N. Biol Pharm Bull. 1997;20:978. doi: 10.1248/bpb.20.978. [DOI] [PubMed] [Google Scholar]
- 24.Wada K, Mori T, Kawahara N. Chem Pharm Bull. 2000;48:660. doi: 10.1248/cpb.48.660. [DOI] [PubMed] [Google Scholar]
- 25.Bando H, Wada K, Tanaka J, Kimura S, Hasegawa E, Amiya T. Heterocycles. 1989;29:1293. [Google Scholar]
- 26.Wada K, Yamamoto T, Bando H, Kawahara N. Phytochemistry. 1992;31:2135. [Google Scholar]
- 27.Wada K, Mori T, Kawahara N. Chem Pharm Bull. 2000;48:1065. doi: 10.1248/cpb.48.1065. [DOI] [PubMed] [Google Scholar]
- 28.Wada K, Ishizuki S, Mori T, Fujihira E, Kawahara N. Biol Pharm Bull. 1998;21:140. doi: 10.1248/bpb.21.140. [DOI] [PubMed] [Google Scholar]
- 29.Wada K, Ishizuki S, Mori T, Fujihira E, Kawahara N. Biol Pharm Bull. 2000;23:607. doi: 10.1248/bpb.23.607. [DOI] [PubMed] [Google Scholar]
- 30.Wada K, Bando H, Wada R, Amiya T. Shoyakugaku Zasshi. 1989;43:50. [Google Scholar]
- 31.Wada K, Bando H, Kawahara N. Heterocycles. 1990;31:1081. [Google Scholar]
- 32.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. J Natl Cancer Inst. 1990;82:1107. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 33.Hazawa M, Takahashi K, Wada K, Mori T, Kawahara N, Kashiwakura I. Invest New Drugs. 2011;29:1. doi: 10.1007/s10637-009-9327-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.