

Abstract
Patients with peripheral artery disease (PAD) develop a myopathy in their ischemic lower extremities, which is characterized by myofiber degeneration, mitochondrial dysfunction and impaired limb function. Desmin, a protein of the cytoskeleton, is central to maintenance of the structure, shape and function of the myofiber and its organelles, especially the mitochondria, and to translation of sarcomere contraction into muscle contraction. In this study, we investigated the hypothesis that disruption of the desmin network occurs in gastrocnemius myofibers of PAD patients and correlates with altered myofiber morphology, mitochondrial dysfunction, and impaired limb function. Using fluorescence microscopy, we evaluated desmin organization and quantified myofiber content in the gastrocnemius of PAD and control patients. Desmin was highly disorganized in PAD but not control muscles and myofiber content was increased significantly in PAD compared to control muscles. By qPCR, we found that desmin gene transcripts were increased in the gastrocnemius of PAD patients as compared with control patients. Increased desmin and desmin gene transcripts in PAD muscles correlated with altered myofiber morphology, decreased mitochondrial respiration, reduced calf muscle strength and decreased walking performance. In conclusion, our studies identified disruption of the desmin system in gastrocnemius myofibers as an index of the myopathy and limitation of muscle function in patients with PAD.
Keywords: Cytoskeleton, desmin, intermittent claudication, muscle disease, myofiber
Introduction
Peripheral artery disease (PAD) is a manifestation of atherosclerosis that produces progressive narrowing and occlusion of the arteries supplying the lower limb muscles. PAD affects approximately 8 million people in the USA, producing a considerable public health burden (Roger et al. 2012). The cardinal manifestation of PAD is claudication, a severe functional limitation identified as gait dysfunction and painful tightness or cramping in the muscles of the leg caused by walking and relieved by rest (Norgren et al. 2007).
Work from several laboratories including our own has demonstrated a myopathy in the affected lower extremities of patients with PAD, which is characterized by mitochondrial dysfunction, increased oxidative damage, myofiber degeneration, muscle fibrosis and decreased muscle force production (Brass and Hiatt 2000; Cluff et al. 2013; Hedberg et al. 1989; Hedberg et al. 1988b; Koutakis et al. 2010a; Makitie and Teravainen 1977; Makris et al. 2007; Marbini et al. 1986; McDermott et al. 2004; McDermott et al. 2012; Pipinos et al. 2007; Pipinos et al. 2003; Regensteiner et al. 1993; Weiss et al. 2013). This myopathy is closely related to leg function, daily activity, quality of life and even mortality of patients with PAD (Anderson et al. 2009; Brass 1996; Evans et al. 2011; Gardner et al. 2013; Garg et al. 2011; McDermott et al. 2009; McDermott et al. 2012; Thompson et al. 2014). Furthermore, worsening of the myopathy is associated with progression of PAD from claudication to rest pain and tissue loss (Cluff et al. 2013; Weiss et al. 2013). Investigations into the pathophysiology of PAD myopathy have focused largely on the myofibers and their contractile elements, with very little attention given to the myofiber cytoskeleton. The cytoskeleton is a key regulator of shape, size and function of the myofiber and its organelles, especially the mitochondria, and cytoskeletal abnormalities are closely associated with muscle pathology, including myofiber degeneration, muscle fibrosis, mitochondrial dysfunction and decreased muscle force production (Capetanaki 2002; Capetanaki et al. 2007; Capetanaki et al. 1997; Clemen et al. 2013; Goebel 1995; Goldfarb et al. 2004; Paulin et al. 2004). The cytoskeleton is an integrated network consisting of microfilaments (actins), microtubules (tubulins), and intermediate filaments (IFs). Desmin, a 53-kDa protein, is the main subunit of IFs in all types of muscle cells: skeletal, cardiac and smooth (Lazarides 1980). Desmin IFs invest myofibrils at the Z-disc forming a network that connects the myofibrils to proteins of the sarcolemma and acts as a 3D-scaffold that anchors and organizes organelles (Fig. 1; Capetanaki et al. 2007; Goldfarb et al. 2004). Desmin IFs bundle myofibrils such that they are aligned at the Z-discs (Capetanaki et al. 2007; Dalakas et al. 2000) and set neighboring myofibers into alignment at their Z-discs (Capetanaki et al. 1997; Carlsson and Thornell 2001; Lazarides 1980), thereby facilitating translation of sarcomeric contraction into muscle contraction (Bloch and Gonzalez-Serratos 2003; Capetanaki et al. 2007; Carlsson and Thornell 2001; Goldfarb et al. 2004; Paulin et al. 2004). This same system of IFs provides structure to the mitochondrial network (Capetanaki et al. 1997; Milner et al. 2000). Because of the functional significance of desmin and its potential association with many characteristics of PAD myopathy, we tested the hypothesis that cytoskeletal damage, which manifests as a disruption of the desmin network, occurs in gastrocnemius myofibers of patients with PAD and correlates with decreased mitochondrial respiration, altered myofiber morphology and impaired limb function.
Figure 1.
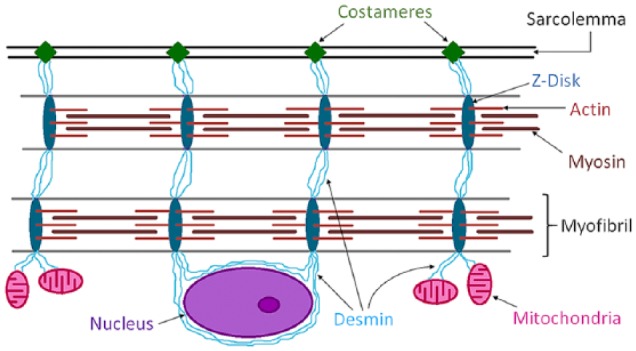
Schematic representation of the desmin intermediate filament network and its association with myofibrils, sarcolemma, mitochondria and the nucleus, in a skeletal myofiber. Desmin is a key protein of the intermediate filaments of the cytoskeleton in myofibers of skeletal muscle. Desmin’s main function is transmission of the forces produced by myofibrils. Desmin IFs link myofibrils into bundles through the Z-discs (dark blue) and connect them to specialized sites of adhesion (costameres) (green) in the sarcolemma. Myofibrillar forces are then transmitted from the costameres to the extracellular matrix and then to the tendons. In addition, Desmin provides a three-dimensional organization of the mitochondrial system (pink), connects the contractile apparatus to the myonuclei (purple), and may contribute to mechano-chemical signaling between the various compartments of the myofiber.
Materials & Methods
Subject Demographics
The experimental protocol was approved by the Institutional Review Board, of the VA Nebraska-Western Iowa and University of Nebraska Medical Centers, and all subjects gave informed consent.
PAD Group
We recruited 30 patients that were evaluated and diagnosed for PAD in the vascular surgery clinic (Table 1). For every patient, the diagnosis of PAD was established on the basis of medical history, physical examination, significantly decreased ankle-brachial index (ABI) and computerized or standard arteriography.
Table 1.
Demographics of Peripheral Artery Disease (PAD) and Control Subjects.
| Control | PAD | p-value | |
|---|---|---|---|
| Number of subjects | 30 | 30 | N/A |
| Mean Age (years) | 65.3 ± 7.6 | 62.2 ± 5.2 | 0.049 |
| Height (m) | 1.76 ± 0.06 | 1.75 ± 0.05 | 0.662 |
| Weight (kg) | 95.4 ± 15.4 | 88.9 ± 17.6 | 0.161 |
| Ankle brachial index (ABI) | 1.05 ± 0.08 | 0.55 ± 0.21 | <0.001 |
| Gender (%) (male/female) | 90.0/10.0 | 96.7/3.3 | 0.461 |
| Smoking (%) | |||
| Never | 23.3 | 6.7 | 0.017 |
| Current | 26.7 | 73.3 | <0.001 |
| Former | 50.0 | 20.0 | 0.001 |
| Coronary Artery Disease | 26.70% | 16.70% | 0.532 |
| Obesity | 43.30% | 36.70% | 0.598 |
| Diabetes | 20.00% | 36.70% | 0.252 |
| Dyslipidemia | 76.70% | 83.30% | 0.748 |
| Hypertension | 83.30% | 86.70% | 0.718 |
Continuous variables are presented as mean ± SD. Chi-squares were used for categorical variables and independent t-test for continuous variables.
Control Group
We recruited 30 patients with normal blood flow to their lower limbs, undergoing lower extremity operations for indications other than PAD (Table 1). These patients had no history of PAD symptoms, and all had normal lower extremity pulses at examination. All controls had normal ABIs at rest and after stress and all led sedentary lifestyles.
Muscle Biopsy
Gastrocnemius biopsies were obtained one day after limb function evaluation from the anteromedial aspect of the muscle belly, 10 cm distal to the tibial tuberosity. All biopsies were obtained with a 6-mm Bergstrom needle. The samples were placed immediately into cold methacarn. After 48 h in methacarn, the specimens were transferred to cold ethanol:H2O (50:50 v/v) and subsequently embedded in paraffin.
Western Blot Analysis
Total protein was extracted from muscle homogenates using MSM/EDTA/1% cholate buffer (220 mM Mannitol, 70 mM Sucrose, 5 mM Mops, 2 mM EDTA, pH 7.4) containing 1% protease inhibitor cocktail (Sigma-Aldrich; St. Louis, MO). For detection of desmin and β-actin (loading control), proteins were separated on a 12% Bis-Tris Mini gel (Life Technologies, Molecular Probes; Eugene, OR) using 10 μg of protein per well. Separated proteins were then transferred onto an Immuno-Blot PVDF membrane. The membrane was blocked for 2 h at room temperature and then incubated overnight in a cold room with a rabbit monoclonal anti-desmin antibody (ab32362; Abcam, Cambridge, MA), at a 1:500 dilution, and a mouse monoclonal anti-β-actin antibody (AM1829b-ev; Abgent, San Diego, CA) at a 1:1000 dilution. The next day the membrane was incubated for 1 h at room temperature with IRDye 680RD goat anti-rabbit IgG and IRDye 800cw goat anti-mouse IgG (LI-COR Biosciences; Lincoln, NE). Finally, the integrated fluorescence of each spot was determined with the LI-COR Odyssey System (LI-COR Biosciences), corrected for background, and the means of each spot were normalized against the loading control.
Quantification of Desmin Gene Transcripts in Muscle Homogenates by RT-qPCR
Total RNA was extracted from methacarn-fixed, paraffin-embedded tissues that had been mounted on slides. Briefly, 4 μm tissue sections from 4–5 slides were collected into a nuclease-free, 1.5-ml tube, and were deparaffinized in 200 μl of xylene. After 10-min incubation at room temperature, xylene was removed by centrifugation. For rehydration, the tissue sections were briefly suspended with 1 ml of absolute ethanol, and centrifuged. This step was repeated with 95%, 80% and 70% ethanol. Then, the pellets were washed twice with 400 μl of PBS. Finally, prior to RNA extraction, the tissue pellets were dissociated with 200 μl of proteinase K by incubating at 56°C for 1 h. Total tissue RNA was extracted with Trizol Reagent (Invitrogen; Austin, TX) per the manufacturer’s instructions. After DNase treatment, the purity and quantity of RNA isolated from the samples were determined using an Agilent Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA). Approximately 200 ng per sample was used for the synthesis of cDNA in a 20 μl reaction volume containing RNA, random hexamers (Promega; Madison, WI), 0.5 mM dNTPs, 1 U RNAsin Ribonulcease Inhibitor (Promega) and 1 U Improm-II Reverse Transcriptase (Promega) in a buffer containing 50 mM Tris-HCl, pH 8.3, 75 mM KCl, 3 mM MgCl2, and 2 mM DTT. RT reactions were incubated at 42°C for 1 h, followed by inactivation at 75°C for 15 min. After a brief chilling on ice, RNase H (Promega) was added to remove RNA strands from RT reactions, which subsequently were diluted 5-fold with nuclease-free water. The synthesized cDNA was aliquoted into small volumes and stored at -84°C, and subsequently amplified in a qPCR reaction (CFX Connect; Bio-Rad Laboratories, Hercules, CA). For qPCR, 10% of the cDNA was used with the desmin or myosin primer pairs (below) at 100 pmol each in DyNAmo SYBR Green qPCR reaction mix (Fisher Scientific; Pittsburgh PA) according to the manufacturer’s instructions. PCR was carried out using a CFX96 connect (Bio-Rad, Hercules, CA, USA). Cycling times were once at 95°C for 10 min followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. A melting curve was analyzed to ensure specificity. The myosin primers (199 bp; NCBI, NM_005963) were as follows: forward, 5’-TCCGAAAGTCTGAAAGGGAGCGAA-3’; reverse, 5’-GAGGGTTCATGGGGAAGACTTGGT-3’. The desmin primers (184 bp; NCBI, NM_001927) were as follows: forward, 5’-AGCCAGGCCTACTCGTCCAGCCA-3’; reverse, 5’-CCGCCCGACGTGCGCGACACCTG-3’.
Quantitative Fluorescence Microscopy
Paraffin-embedded biopsy specimens sectioned at 4 microns were labeled with fluorescent reagents for quantification of desmin in the myofibers. As a means of partitioning, individual myofiber sarcolemmas were labeled with wheat germ agglutinin (Kostrominova 2011). For measurement of desmin, specimens were treated with a rabbit monoclonal antibody (0.20 μg/ml; ab32362, Abcam) specific for the C-terminus of human desmin. For labeling of ATP Synthase (mitochondrial marker), specimens were treated with a mouse monoclonal antibody (10 μg/ml; ab14730 Abcam) specific for the human complex V beta subunit. Control slides were treated with IgG isotype control (0.20 μg/ml) from a non-immunized rabbit (08-6199; Life Technologies, Molecular Probes) and IgG isotype from a non-immunized mouse (5 μg/ml; 14-4714; eBioscience, Inc., San Diego, CA). After overnight incubation at 5C, the slides were labeled at room temperature with a mixture of Alexa Fluor® 488 goat anti-rabbit IgG (H+L) antibody, highly cross-adsorbed (10 μg/mL; A-11034; Life Technologies, Molecular Probes) and Alexa Fluor® 555 goat anti-mouse IgG (H+L) antibody, highly cross-adsorbed (10 μg/mL; A21424; Life Technologies, Molecular Probes). Myofiber sarcolemmas were labeled with Alexa Fluor® 647-conjugated wheat germ agglutinin (10 μg/ml; W32466; Life Technologies, Molecular Probes). The specimens were mounted in ProLong® Gold anti-fade medium with DAPI nuclear stain (P36931; Life Technologies, Molecular Probes).
Image Acquisition and Analysis
Desmin quantification within individual myofibers and myofiber morphometry were based on three-channel imaging (Huang et al. 2010; Huang et al. 2007; Weiss et al. 2013) of each microscopic field. The method has been previously described in detail (Cluff et al. 2013; Huang et al. 2007; Koutakis et al. 2014; Weiss et al. 2013). Briefly fluorescence images were captured with a 10× objective of a wide-field, epifluorescence microscope (Leica DMRXA2; North Central Instruments, Plymouth, MN) and a B/W CCD camera (Orca ER C4742-95; Hamamatsu Photonics, Bridgewater, NJ), with HCImage software (Hamamatsu, Sewickley, PA). All of the tissue that was mounted on each slide (150–300 microscopic fields per specimen, corresponding to 5000 to 10,000 myofibers) was captured in three fluorescence channels corresponding to 1) nuclei 2) desmin and 3) myofiber sarcolemma. The fluorescence signal produced by desmin within each myofiber was expressed as mean pixel intensity in grayscale units (gsu; on a 12-bit gray scale), representing concentration within the myofiber. The fluorescence signal was corrected for background (typically near the black level of the camera) and the mean of all myofibers in each specimen was determined.
Histology
Masson’s Trichrome staining was implemented with a kit from Thermo Fisher Scientific (#87019, Waltham, MA) according to the instructions provided with the kit. Briefly, paraffin-embedded biopsy specimens, sectioned at 4 μm, were deparaffinized and fixed in Bouin’s solution overnight. After incubation in Weigert’s Iron Hematoxylin Solution, the slides were stained with Biebrich Scarlet-Acid Fuchsin and Aniline Blue and dehydrated in ethanol and xylene. Extensive washes were done between each staining. Collagen was stained blue, the nuclei black and the myofibers red.
Muscle Morphology
Multiple morphometric parameters were determined for myofibers in each muscle specimen as described previously (Cluff et al. 2013). Briefly, we measured: 1) myofiber cross-sectional area (square microns), determined from the number of pixels enclosed within a segmented myofiber; 2) myofiber perimeter (microns), determined from the number of pixels on the boundary of the myofiber; 3) equivalent diameter (microns), defined as the diameter of a circle that has the same area as the segmented myofiber region; 4) roundness, defined as the equivalent diameter × π divided by the perimeter of the segmented region; and 5) solidity, defined as the myofiber cross-sectional area divided by the area of a fitted convex hull. Fiber density was defined as the area occupied by the myofibers divided by the area occupied by the myofibers plus interstitial tissue.
Evaluation of Mitochondrial Electron Transport Chain Complexes
Respiration of skinned (saponin-permeabilized) gastrocnemius muscle fibers was measured with a Clark electrode (YSI) in four separate assays corresponding to the following sets of conditions: 1) 5 mM glutamate, 5 mM malate, and 1 mM ADP (complex I-dependent respiration); 2) 3 μM rotenone (to inhibit complex I), 1 mM ADP, and 10 mM succinate (complex II-dependent respiration); 3) 3 μM rotenone, 1 mM ADP, and 1 mM duroquinol (complex III-dependent respiration); and 4) 3 μM rotenone, 1 mM ADP, 10 mM ascorbate, and 0.2 mM N,N,N′,N′-tetramethyl-p-phenylenediamine (complex IV-dependent respiration). Respiratory rates (nanoatoms of oxygen per min) were normalized to Citrate Synthase (CS) activity (a measure of mitochondrial content) after completion of the respiratory measurements. CS activity was measured as the increase in absorbance (412 nm) from the reduction of 5,5′-dithiobis-2-nitrobenzoic acid by newly formed CoA-SH.
Muscle Strength Measurement
Muscle strength production from the ankle plantarflexors (the muscles of the posterior compartments of the calf) was measured using a Biodex dynamometer (System 4.0; Biodex Medical Systems, Shirley, NY). Thirty PAD and 15 control subjects were secured onto the Biodex, with the foot positioned in the standard ankle plantar flexion/dorsiflexion position of 90° between the foot and the shank. In order to isolate the plantarflexor muscles, the ipsilateral thigh, waist and chest of all subjects were secured. The leg was positioned so that the ipsilateral thigh was braced and supported with a knee angle of 30°. The parameter of the muscle strength we measured was the peak force produced by the ankle plantarflexors during a maximum isometric contraction of 10 s. Data are represented in ft*lbs.
Graded Treadmill Test and 6-Minute Walking Distance
Claudication onset distance (COD) and peak walking distance (PWD) were measured for all PAD subjects using the Gardner graded treadmill test. The patients walked at a constant speed of 3.2 km·h−1 on a 0% grade, with a 2% grade increase every 2 min. The COD was recorded at the onset of claudication pain and PWD was recorded as the maximum distance walked. In addition, all PAD subjects were evaluated with the 6-minute walk test. The test was performed in an indoor 20-m hallway for 6 min. The procedure was performed under technical supervision and patients were instructed to cover as much distance as possible.
Statistics
Baseline characteristics of PAD and controls subjects were compared using independent t-tests for continuous variables and chi-square tests for categorical variables. Categorical variables that were different between the two groups were used as covariates in subsequent analyses. Differences between parameters for PAD and control subjects were evaluated by analysis of covariance, with adjustments for age and smoking status. The relationships of desmin with myofiber morphology, mitochondrial respiration, muscle strength and walking distances were evaluated by a Pearson correlation. All analyses were implemented with SAS statistical software version 9.3 (SAS Institute Inc., Cary, NC). Data are presented as the mean and standard deviation (SD), unless stated otherwise, and significance was set at p<0.05.
Results
Subject Demographics
The demographic information for both the PAD and control subjects is presented in Table 1. Only smoking status (χ2=17.9, p<0.001), age (t=3.14, p=0.002) and ABI (t=15.4, p<0.001) were significantly different between the PAD and control subjects.
Abnormal Accumulation of Desmin in Gastrocnemius Myofibers of PAD Subjects
Previous studies identified abnormal accumulations of desmin in myofibers of patients with desminopathies (Carlsson and Thornell 2001; Claeys and Fardeau 2013; Clemen et al. 2013; Goldfarb et al. 2008; Goldfarb et al. 2004). Accumulated desmin was seen as cytoplasmic aggregates and was associated with myofibril fragility that produces severe muscle dysfunction (Capetanaki et al. 2007; Dalakas et al. 2000; Li and Dalakas 2001). Using wide-field fluorescence microscopy, we examined the distribution of desmin in longitudinal sections and cross-sections of myofibers in the gastrocnemii from PAD and control subjects and present representative images of these sections (Fig. 2). As seen in longitudinal sections, labeling of Z-disk desmin in control muscle revealed a regular pattern of striation representing the Z-disks of aligned myofibrils (Fig. 2A–2C). In contrast, longitudinal sections of PAD myofibers (Fig. 2D–2F) revealed an irregular pattern with intense labeling in some areas and desmin cross-links that appeared to connect multiple Z-disks. Desmin cross-links between adjacent Z-disks occurred frequently throughout these myofibers and were associated with desmin aggregates. In addition, large cavities lined with intense desmin labeling were observed. Desmin aggregates and desmin-lined cavities were not seen in control myofibers. In cross-sections, control myofibers (Fig. 2G–2I) exhibited a highly regular honeycomb pattern with even labeling, representing numerous myofibril bundles. In contrast, PAD myofibers (Fig. 2J–2L) exhibited an irregular honeycomb pattern with uneven, intense labeling. Using quantitative fluorescence microscopy, we found that myofibers of PAD (n=30) muscle contained, on average, 21.5% more desmin as compared with the control (n=30) muscle (mean: 1306 ± 41 gsu vs 1075 ± 40 gsu at p<0.001; median: 1242 (1009–2049) gsu vs 1023 (599–1453) gsu, at p<0.001). These results were supported by western blot analyses (Fig. 3A), which also identified in the PAD specimens a second desmin band at approximately 42 kDa, suggesting post-translational modification of the protein and/or protein degradation (Winter et al. 2014). The increased accumulation of desmin in PAD myofibers was associated with a mean 126-fold increase (SD, ±36) and median 124.5-fold increase (range, 8 to 288; p<0.001) in desmin gene transcripts as compared with the control (Fig. 3B). The disparity between the abundances of desmin protein and gene transcripts may reflect inefficient translation and/or a high rate of desmin degradation and proteolysis in the damaged myofibers.
Figure 2.

Slide-mounted gastrocnemius sections from a control subject (A–C, G–I) and a Peripheral Artery Disease (PAD) subject (D–F, J–L) were labeled for fluorescence microscopy and captured with 20×, 40× and 100× objectives. In longitudinally sectioned control myofibers (A–C), desmin (green) exhibited a normal distribution along the Z-disks, seen as regular, horizontal striations of bright green labeling and representing a well-organized cytoskeletal network. In PAD myofibers (D–F), desmin was disorganized and seen as a more dense deposition along the Z-disks in some areas (arrowheads), where abnormal cross-links between adjacent Z-disks are evident, and dense accumulation along the walls of large vacuoles (filled arrows). In cross-sections of control myofibers (G–I), desmin exhibited a normal honeycomb appearance throughout the myofiber (100×), representing numerous myofibrils in cross-section, and a high concentration in the subsarcolemma region (20×, 40× and 100×) (filled arrows). In cross-sections of PAD myofibers (J–L), desmin exhibited an irregular honeycomb appearance seen as intense and uneven labeling along the boundaries of the myofibrils (100×) (line arrows) and extensive, irregular cytoplasmic aggregates (arrowheads).
Figure 3.

(A) Representative western blot showing increased desmin content in gastrocnemius tissue from Peripheral Artery Disease (PAD) subjects as compared with that of control subjects. A desmin band of higher mobility in PAD muscle suggests post-translational modification, possibly oxidative damage, and/or protein degradation. (B) Desmin mRNA content normalized to myosin mRNA was higher in PAD gastrocnemius specimens. (C) Mean desmin mRNA normalized to myosin mRNA was 126 times higher in PAD (n=30) as compared with control (n=30) gastrocnemius, suggesting inefficient translation of the gene transcript and/or high protein degradation.
Myofiber Morphology and Density Change in association with Desmin Accumulation in PAD Myofibers
Previous studies have demonstrated that myofiber cross-sectional area is reduced in PAD myofibers as compared with controls (Regensteiner et al. 1993; Weiss et al. 2013). To evaluate the relationship between desmin accumulation and myofiber morphology, we compared multiple parameters of myofiber morphology in cross-sections of gastrocnemius biopsies from the same PAD (n=30) and control (n=30) subjects whose myofibers exhibited significant differences in quantities of desmin protein and gene transcripts. In PAD gastrocnemius as compared with controls, myofiber cross-sectional area, major axis, minor axis, equivalent diameter and perimeter, and measures of myofiber size were all significantly reduced (Table 2). Moreover, myofiber roundness was increased and solidity was decreased in PAD muscle, indicating an abnormal morphology (Table 2). Finally, we observed a decrease in myofiber density per unit area of cross-sectioned specimens associated with increased fibrosis in PAD muscle (Fig. 4), and this was consistent with reduced myofiber size. These changes are characteristic of desminopathies (Abraham et al. 1998; Clemen et al. 2013; Selcen et al. 2004).
Table 2.
Myofiber Morphometry in the Gastrocnemius of Controls and Patients with Peripheral Artery Disease (PAD).
| Control (n=30) | PAD (n=30) | p-value | |
|---|---|---|---|
| Cross-Sectional Area (μ2) | 4999 ± 1151 | 4058 ± 988 | 0.004 |
| Major Axis (μ) | 99.27 ± 11 | 93.09 ± 9 | 0.037 |
| Minor Axis (μ) | 61.27 ± 7.4 | 56.55 ± 5.4 | 0.009 |
| Equivalent Diameter (μ) | 75.21 ± 9.7 | 71.25 ± 5.5 | 0.047 |
| Perimeter (μ) | 293.25 ± 35 | 270.84 ± 26 | 0.008 |
| Roundness | 0.804 ± 0.02 | 0.825 ± 0.03 | 0.008 |
| Solidity | 0.951 ± 0.01 | 0.943 ± 0.01 | 0.014 |
| Fiber Density | 0.822 ± 0.07 | 0.741 ± 0.05 | < 0.001 |
Data are adjusted for age and smoking status. Values are presented as the mean ± SD.
Figure 4.
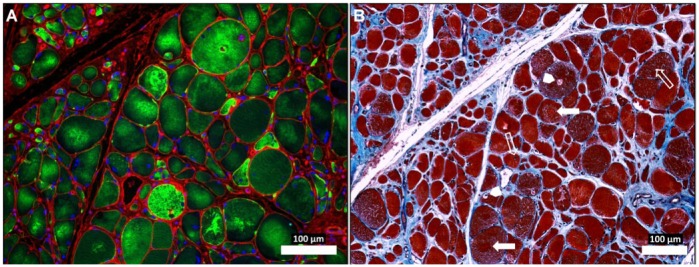
Abnormal desmin accumu-lation in the myofibers of Peripheral Artery Disease (PAD) gastrocnemius (A) and associated changes in myofiber morphology and extracellular matrix (B). Sections from the gastrocnemius of a PAD subject with moderate to severe myopathy were captured under fluorescence (A) and bright-field (B) conditions with a 10× objective. The specimen (A) was labeled with desmin antibody (green), wheat germ agglutinin (membranes and connective tissue; red) and DAPI nuclear stain (blue). The neighboring section (B) was stained with Masson’s Trichrome (sarcoplasm, red; connective tissue, blue; nuclei, black). The PAD myofibers display dense, irregular accumulations of desmin in the sarcoplasm (A). The myofibers are irregular in shape and size and include many large, rounded and swollen fibers and many small, irregularly shaped fibers (A and B). Several of the large myofibers show evidence of target lesions (filled arrows), vacuoles (arrow heads) and internal nuclei (line arrows) (B), findings consistent with myofiber degeneration. There is obvious expan-sion of the extracellular matrix. Scale, 100 µm.
The observed differences between PAD and control gastrocnemii specimens suggested that the abnormal accumulation of desmin in PAD myofibers may correlate with myofiber morphometrics. We evaluated correlations between desmin protein, as quantified by quantitative fluorescence microscopy, and parameters of myofiber morphology in the PAD subjects (n=30). Cross-sectional area (r = -0.405; p=0.027), perimeter (r = -0.480; p=0.007), solidity (r = -0.594; p=0.001) and fiber density (r = -0.549; p=0.002) decreased with increasing myofiber content of desmin. Roundness, representing abnormal myofiber morphology, increased (r = 0.372; p=0.049) with increasing desmin content.
Mitochondrial Respiration Decreases in association with Desmin Accumulation in PAD Myofibers
In skeletal muscle fibers, mitochondria typically are organized around the Z-disc of the sarcomere throughout the length of the myofibril (Milner et al. 2000). Ultrastructural studies have established a connection between mitochondria topology and function and intermediate filaments (Capetanaki et al. 2007; Milner et al. 2000). In this study, we found that abnormal desmin accumulation in PAD myofibers was associated with irregular and patchy distribution of the mitochondria and a characteristic absence of mitochondria in the areas of dense desmin accumulation (Fig. 5). Mitochondrial function, determined as respiratory activity, which is dependent on electron transport chain complexes I and IV normalized to mitochondrial content, was reduced in the myofibers of PAD subjects (n=30) as compared with control subjects (n=30) (Table 3). These observations suggest that the abnormal accumulation of desmin in PAD myofibers may correlate with mitochondrial function in PAD gastrocnemius. We evaluated correlations between desmin protein, as quantified by quantitative fluorescence microscopy, and respiratory activity dependent on electron transport chain complexes I and IV in the PAD subjects (n=30). We found that activity dependent on both complex I and complex IV decreased with increasing desmin accumulation (r = -0.422, p=0.005; r = -0.414, p=0.006, respectively).
Figure 5.

Representative fluorescence images of desmin (green) and ATP-Synthase (red) in slide-mounted sections of the gastrocnemius of control (row A) and Peripheral Artery Disease (PAD) subjects (row B). There is an irregular and patchy distribution of mitochondria in PAD myofibers, with an absence of mitochondrial labeling in the areas where desmin is abundant and highly aggregated. The mitochondria exhibit an uneven distribution throughout the remainder of the sarcoplasm and in the sub-sarcolemma region.
Table 3.
Complex-Dependent Mitochondrial Respiration Normalized to Citrate Synthase Activity.
| Control (n=30) | PAD (n=30) | p-value | |
|---|---|---|---|
| CI/CS | 74.72 ± 26 | 58.36 ± 16 | 0.009 |
| CII/CS | 41.00 ± 21 | 35.93 ± 13 | 0.312 |
| CIII/CS | 159.11 ± 40 | 154.75 ± 45 | 0.504 |
| CIV/CS | 184.89 ± 56 | 158.80 ± 32 | 0.043 |
Data are adjusted for age and smoking status. Values are presented as the mean ± SD of nanoatom O2 per min per unit of CS activity in controls and patients with peripheral artery disease (PAD). CI–IV, electron transport chain complexes; CS, citrate synthase.
Plantarflexor Strength Declines with Increasing Desmin Accumulation in PAD Myofibers
Desmin IFs combine the forces produced by individual myofibrils and transmit these to costameres (Ervasti 2003; Grounds et al. 2005), specialized linkage sites in the sarcolemma (Fig. 1). These forces are transmitted via the costameres to the extracellular matrix (ECM) and towards the tendons (Bloch and Gonzalez-Serratos 2003; Capetanaki et al. 2007; Paulin et al. 2004). Electron microscopy studies of PAD muscles have identified extensive abnormalities of the myofibrils (Makitie and Teravainen 1977) and studies by our group have established impairment of plantarflexor power of PAD patients (Koutakis et al. 2010a; Koutakis et al. 2010b; Wurdeman et al. 2012). We tested the hypothesis that plantarflexor strength decreases with increasing desmin accumulation in gastrocnemius myofibers in patients with PAD. First, we compared plantarflexor strength of PAD (n=30) and control (n=30) subjects. Plantarflexor strength was decreased in PAD subjects as compared with control subjects (58.89 ± 3.8 vs. 81.25± 2.8 ft*lbs; p<0.001). This difference suggested that the abnormal accumulation of desmin in myofibers may correlate with plantarflexor strength. We evaluated the association of desmin protein, as quantified by quantitative fluorescence microscopy, and plantarflexor strength in PAD subjects (n=30). We found that isometric plantarflexor strength decreased with increasing desmin accumulation (r = -0.612, p<0.001) (Fig. 6). There was no association between desmin accumulation and isometric plantarflexor strength in the control subjects.
Figure 6.
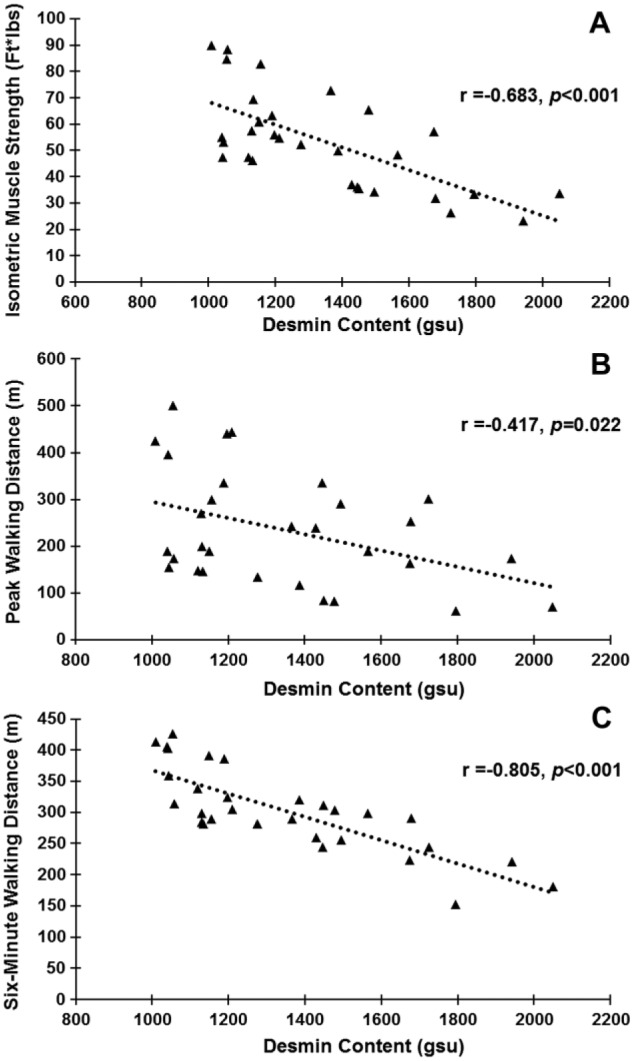
Desmin content in Peripheral Artery Disease (PAD; n=30) gastrocnemius myofibers is inversely correlated with limb function. (A) Muscle strength exerted from the ankle plantarflexors during maximum isometric contraction. (B) Peak walking distance results. (C) Six-minute walking distance results.
Six-Minute and Peak Walking Distances Decrease with Increasing Desmin Accumulation in PAD Myofibers
Our finding of a negative correlation between plantarflexor strength and desmin accumulation in PAD myofibers suggested that the distances PAD patients can walk may be affected by a disruption in the desmin system in myofibers. The average distances our PAD subjects were able to walk were: 303 ± 67 m for 6-minute walking distance, 76.9 ± 70 m for COD and 235.2 ± 120 m for PWD. We evaluated correlations of walking distances with desmin accumulation in the PAD subjects (n=30). PWD and 6-minute walking distances decreased with increasing desmin accumulation (r = -0.417, p=0.022; and r = -0.805, p<0.001, respectively) (Fig. 6). COD exhibited no correlation.
Discussion
Our study has established, as part of the myopathy of PAD gastrocnemius, an abnormal accumulation of the cytoskeletal protein desmin and extensive disruption of its normal registration along Z-discs of the myofibrils, as compared with that observed in healthy myofibers (Fig. 2). This pathological change in PAD myofibers correlated with altered myofiber morphology, reduced mitochondrial respiration and walking impairment. These findings, for the first time, link disruption of the cytoskeleton to the myopathy of PAD. Using quantitative fluorescence microscopy, we demonstrated an increase in the mean content of desmin in PAD as compared with control myofibers and found that, in cross-sections of PAD myofibers with increased desmin content, desmin was present as an irregular honeycomb and/or amorphous aggregates of varying size. Longitudinal sections revealed uneven labeling of desmin and numerous deviations of labeled desmin from Z-disc registration in many PAD myofibers. This was in contrast to normal gastrocnemius myofibers that exhibited a highly organized registration of evenly labeled desmin along the Z-discs of myofibrils. The observed abnormalities of desmin abundance and distribution in PAD myofibers are similar to those seen in the myofibers of patients with myofibrillar myopathies (Janue et al. 2007a; Janue et al. 2007b; Nakano et al. 1996; Olive et al. 2008; Selcen et al. 2004). Myofibrillar myopathies are a clinically and genetically heterogeneous group of muscle disorders characterized by degradation of the myofibrils in association with disintegration of the Z-disc, accumulation of myofibrillar degradation products and an abnormal expression, disorganization and sarcoplasmic aggregation of desmin and other proteins of the cytoskeleton, including myotilin, α,β-crystallin and dystrophin (Claeys and Fardeau 2013; Winter et al. 2014). The primary clinical feature of myofibrillar myopathies is progressive skeletal muscle weakness and, in some patients, associated cardiomyopathy and peripheral neuropathy. Desminopathies are a subgroup of myofibrillar myopathies, caused by mutations in the desmin gene and are characterized by increased content of desmin in the myofibers, disorganization of the desmin filament network, accumulation of insoluble desmin-containing aggregates and disruption of the sarcomere of striated muscle (Dalakas et al. 2000; Goldfarb et al. 2004; Selcen et al. 2004). Current information concerning the pathophysiology of PAD and myofibrillar myopathies suggests that desmin may be directly damaged by reactive oxygen species (ROS). Several groups, including our own, have shown increased oxidative damage in PAD muscle (Pipinos et al. 2008). More detailed studies established that oxidative damage to myofibers in PAD gastrocnemius increased as the hemodynamics of the blood flow to the leg worsened and as the disease advanced from Fontaine stage II to stage III and stage IV (Weiss et al. 2013). Studies of myofibrillar myopathies have shown that ROS and reactive nitrogen species (RNS) damage cytoskeletal and other proteins and impair their processing through the ubiquitin-proteasome pathway, favoring accumulation of non-degraded damaged proteins as protein aggregates (Clemen et al. 2013; Janue et al. 2007a; Janue et al. 2007b). Desmin is a key target for oxidative and nitrosative damage in myofibrillar myopathies (Janue et al. 2007a; Janue et al. 2007b), leading to inactivation and misfolding of the protein and exposing hydrophobic surfaces for protein-protein interactions, which promote aggregation (Grune et al. 1997; Janue et al. 2007a; Janue et al. 2007b). Furthermore, desmin is a preferred site for the development of advanced glycation end products (AGEs) in cardiomyopathies, both in mice and in humans (Diguet et al. 2011). Diguet et al. (2011) demonstrated that, in dilated cardiomyopathy, oxidatively damaged desmin progressively loses its striated pattern and accumulates in the sarcoplasm. Desmin has been found to be particularly susceptible to the formation of AGEs and to damage by lipid peroxidation end-products in animal models of tissue ischemia and reperfusion (Canton et al. 2004; Janue et al. 2007a; Janue et al. 2007b). AGEs and lipid peroxidation end-products accumulate in PAD muscle and, particularly, in PAD myofibers (Norgren et al. 2007; Pipinos et al. 2008, 2007).
Desmin links neighboring myofibrils into bundles through their Z-discs (Capetanaki et al. 2007; Dalakas et al. 2000), aligns the Z-discs of neighboring myofibers (Capetanaki et al. 1997; Carlsson and Thornell 2001; Lazarides 1980), organizes the mitochondria into a well-defined functional network around the myofibrils (Capetanaki et al. 1997; Milner et al. 2000) and facilitates transmission of the force of sarcomere contraction to the ECM (Bloch and Gonzalez-Serratos 2003; Capetanaki et al. 2007; Carlsson and Thornell 2001; Goldfarb et al. 2004; Paulin et al. 2004). Mitochondrial dysfunction in PAD skeletal muscle is well documented (Makris et al. 2007; Pipinos et al. 2006; Pipinos et al. 2003; Pipinos et al. 2000). In the present study, we found that mitochondrial Complex I- and Complex IV-dependent respiration correlated inversely with desmin accumulation in PAD myofibers. In addition, our fluorescence microscopy work revealed an irregular and patchy distribution of mitochondria in the myofibers, with a characteristic absence of mitochondria in areas where desmin accumulation was high. The distribution of mitochondria was uneven throughout the remainder of the sarcoplasm. These findings are consistent with previous reports concerning desminopathies (Capetanaki et al. 1997; Milner et al. 2000).
In the gastrocnemius of PAD subjects, myofibers have reduced size (decrease in cross-sectional area, major and minor axes, equivalent diameter and perimeter) and altered shape (less polygonal and more rounded) and the endomysium and perimysium are expanded, consistent with decreased myofiber density. Studies of muscle in patients with desminopathies have demonstrated a similar myopathy characterized by rounding of muscle fibers, fiber splitting, internalization of myonuclei, and increased connective and fat tissue (Abraham et al. 1998; Clemen et al. 2013; Selcen et al. 2004). Loss of normal size and shape of myofibers is indicative of significant damage to the myofibrillar, cytoskeletal and membranous components of the myofiber (Bloch and Gonzalez-Serratos 2003; Sanger et al. 2004). The present work suggests that the cytoskeleton of myofibers in PAD gastrocnemius is also damaged and that this damage produces changes in myofiber morphology.
A finding of the present work, which has clinical and translational relevance, is that of a negative correlation between desmin accumulation in the myofibers of PAD gastrocnemius and the strength of the ankle plantarflexors and walking performance (6-minute walk distance and PWD) of PAD patients. The strength of the ankle plantarflexors of PAD patients was significantly reduced as compared with controls, consistent with myofiber degeneration in the muscles of PAD legs (Hedberg et al. 1989; Hedberg et al. 1988a; Koutakis et al. 2010a; Koutakis et al. 2010b; McDermott et al. 2004; McDermott et al. 2007; McDermott et al. 2001; Regensteiner et al. 1993). Dalakas et al. (2000) proposed that accumulation of damaged desmin in association with disruption of the filamentous network weakens the sarcomere and causes increased mechanical stress within and between myofibrils, myofiber fragility, Z-disc disintegration and myofibrillar damage. Similarly, the accumulation and abnormal distribution of desmin in PAD myofibers, which signal damage to the cytoskeleton, may contribute to decreased strength of leg muscles, impaired limb function and decreased activity in patients with PAD (Hedberg et al. 1988b; Koutakis et al. 2010a; Koutakis et al. 2010b; McDermott et al. 2004; McDermott et al. 2007; McDermott et al. 2001; Regensteiner et al. 1993). Myofibers, in long skeletal muscles, are much shorter than the muscle length and, as a result, most of them start and finish without attaching themselves to either tendon (Gaunt and Gans 1990; Schejter and Baylies 2010; Trotter 2002). The forces generated by myofibrils (the contractile units of myofibers composed mainly of myosin, actin, titin, tropomyosin and troponin) are transferred through the cytoskeletal proteins to sarcolemma (membrane of the myofiber) proteins and then to the ECM (endomysium, perimysium and epimysium) proteins through which they reach the tendons (Monti et al. 1999; Patel and Lieber 1997; Turrina et al. 2013). It is apparent that the changes we are describing in the non-contractile components (mainly desmin and the cytoskeleton, but also the ECM) of PAD muscle can be key factors contributing to the functional deterioration of patients with PAD.
This is a correlational study that identifies damage to desmin as an objective measure of the extent of muscle damage and, therefore, a potential biomarker for the efficacy of therapeutic intervention and responsiveness of patients with PAD to treatment options. The study does not identify cause/effect relationships; however, the high-quality human data that we have produced provides a direction for future mechanistic studies, which are essential for the development of improved therapy and prognosis of PAD. With mechanistic information concerning desmin damage in PAD muscle, it will be possible to prioritize, for evaluation, therapies proposed for the management of myofibrillar myopathies. These include exercise, nutrition, as well as pharmacological, stem cell and gene therapies (Clemen et al. 2013; Goldfarb et al. 2008; Goldfarb et al. 2004; Segard et al. 2013).
In summary, our work demonstrated the abnormal accumulation and distribution of desmin in the gastrocnemius myofibers of subjects with PAD, indicating damage to the cytoskeleton, as compared with control subjects. These changes correlated with decreased mitochondrial function, abnormal myofiber morphology and impaired limb function. Our findings provide insight into the chronic and progressive nature of PAD, where damage to desmin in myofibers may contribute to the myopathy of PAD and deterioration of performance of the ischemic lower limb. Finally, our use of muscle biopsy specimens for the quantitative microscopic analysis of cell structure and biomolecules, such as desmin, which serve high-level cellular functions, is an effective translational approach that can identify potential mechanisms of disease and targets for therapeutic intervention as well as biomarkers for enhanced diagnosis, treatment and prognosis of PAD.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was primarily supported by NIH grant R01AG034995, by the Charles and Mary Heider Fund for Excellence in Vascular Surgery, by the Alexander S. Onassis Public Benefit Foundation and by the American Heart Association Pre-Doctoral Fellowship 13PRE13860010. Secondary funding was provided by the Department of Veterans’ Affairs, Veterans Health Administration, Rehabilitation Research and Development Service 1I01RX000604. Furthermore, this material is the result of work supported with resources and the use of facilities at the VA Nebraska-Western Iowa Health Care System.
References
- Abraham SC, DeNofrio D, Loh E, Minda JM, Tomaszewski JE, Pietra GG, Reynolds C. (1998). Desmin myopathy involving cardiac, skeletal, and vascular smooth muscle: report of a case with immunoelectron microscopy. Hum Pathol 29:876-882. [DOI] [PubMed] [Google Scholar]
- Anderson JD, Epstein FH, Meyer CH, Hagspiel KD, Wang H, Berr SS, Harthun NL, Weltman A, Dimaria JM, West AM, Kramer CM. (2009). Multifactorial determinants of functional capacity in peripheral arterial disease: uncoupling of calf muscle perfusion and metabolism. J Am Coll Cardiol 54:628-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch RJ, Gonzalez-Serratos H. (2003). Lateral force transmission across costameres in skeletal muscle. Exerc Sport Sci Rev 31:73-78. [DOI] [PubMed] [Google Scholar]
- Brass EP. (1996). Skeletal muscle metabolism as a target for drug therapy in peripheral arterial disease. Vasc Med 1:55-59. [DOI] [PubMed] [Google Scholar]
- Brass EP, Hiatt WR. (2000). Acquired skeletal muscle metabolic myopathy in atherosclerotic peripheral arterial disease. Vasc Med 5:55-59. [DOI] [PubMed] [Google Scholar]
- Canton M, Neverova I, Menabo R, Van Eyk J, Di Lisa F. (2004). Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am J Physiol Heart Circ Physiol 286:H870-877. [DOI] [PubMed] [Google Scholar]
- Capetanaki Y. (2002). Desmin cytoskeleton: A potential regulator of muscle mitochondrial behavior and function. In Trends in Cardiovascular Medicine. 339-348 [DOI] [PubMed] [Google Scholar]
- Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S. (2007). Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res 313:2063-2076. [DOI] [PubMed] [Google Scholar]
- Capetanaki Y, Milner DJ, Weitzer G. (1997). Desmin in muscle formation and maintenance: knockouts and consequences. Cell Struct Funct 22:103-116. [DOI] [PubMed] [Google Scholar]
- Carlsson L, Thornell LE. (2001). Desmin-related myopathies in mice and man. Acta Physiol Scand 171:341-348. [DOI] [PubMed] [Google Scholar]
- Claeys KG, Fardeau M. (2013). Myofibrillar myopathies. Handb Clin Neurol 113:1337-1342. [DOI] [PubMed] [Google Scholar]
- Clemen CS, Herrmann H, Strelkov SV, Schroder R. (2013). Desminopathies: pathology and mechanisms. Acta Neuropathol 125:47-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluff K, Miserlis D, Naganathan GK, Pipinos, Koutakis P, Samal A, McComb RD, Subbiah J, Casale GP. (2013). Morphometric analysis of gastrocnemius muscle biopsies from patients with peripheral arterial disease: objective grading of muscle degeneration. Am J Physiol Regul Integr Comp Physiol 305:R291-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. (2000). Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 342:770-780. [DOI] [PubMed] [Google Scholar]
- Diguet N, Mallat Y, Ladouce R, Clodic G, Prola A, Tritsch E, Blanc J, Larcher JC, Delcayre C, Samuel JL, Friguet B, Bolbach G, Li Z, Mericskay M. (2011). Muscle creatine kinase deficiency triggers both actin depolymerization and desmin disorganization by advanced glycation end products in dilated cardiomyopathy. J Biol Chem 286:35007-35019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM. (2003). Costameres: the Achilles’ heel of Herculean muscle. J Biol Chem 278:13591-13594. [DOI] [PubMed] [Google Scholar]
- Evans NS, Liu K, Criqui MH, Ferrucci L, Guralnik JM, Tian L, Liao Y, McDermott MM. (2011). Associations of calf skeletal muscle characteristics and peripheral nerve function with self-perceived physical functioning and walking ability in persons with peripheral artery disease. Vasc Med 16:3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner AW, Parker DE, Montgomery PS, Blevins SM, Teague AM, Casanegra AI. (2013). Monitored Daily Ambulatory Activity, Inflammation, and Oxidative Stress in Patients With Claudication. Angiology 65:491-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg PK, Liu K, Ferrucci L, Guralnik JM, Criqui MH, Tian L, Sufit R, Nishida T, Tao H, Liao Y, McDermott MM. (2011). Lower extremity nerve function, calf skeletal muscle characteristics, and functional performance in peripheral arterial disease. J Am Geriatr Soc 59:1855-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaunt AS, Gans C. (1990). Architecture of chicken muscles: short-fibre patterns and their ontogeny. Proc R Soc Lond B Biol Sci 240:351-362. [DOI] [PubMed] [Google Scholar]
- Goebel HH. (1995). Desmin-related neuromuscular disorders. Muscle Nerve 18:1306-1320. [DOI] [PubMed] [Google Scholar]
- Goldfarb LG, Olive M, Vicart P, Goebel HH. (2008). Intermediate filament diseases: desminopathy. Adv Exp Med Biol 642:131-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb LG, Vicart P, Goebel HH, Dalakas MC. (2004). Desmin myopathy. Brain 127:723-734. [DOI] [PubMed] [Google Scholar]
- Grounds MD, Sorokin L, White J. (2005). Strength at the extracellular matrix-muscle interface. Scand J Med Sci Sports 15:381-391. [DOI] [PubMed] [Google Scholar]
- Grune T, Reinheckel T, Davies KJ. (1997). Degradation of oxidized proteins in mammalian cells. FASEB J 11:526-534. [PubMed] [Google Scholar]
- Hedberg B, Angquist KA, Henriksson-Larsen K, Sjostrom M. (1989). Fibre loss and distribution in skeletal muscle from patients with severe peripheral arterial insufficiency. Eur J Vasc Surg 3:315-322. [DOI] [PubMed] [Google Scholar]
- Hedberg B, Angquist KA, Sjostrom M. (1988a). Peripheral arterial insufficiency and the fine structure of the gastrocnemius muscle. Int Angiol 7:50-59. [PubMed] [Google Scholar]
- Hedberg B, Langstrom M, Angquist KA, Fugl-Meyer AR. (1988b). Isokinetic plantar flexor performance and fatiguability in peripheral arterial insufficiency. Effects of training vs. vascular surgery. Acta Chir Scand 154:363-369. [PubMed] [Google Scholar]
- Huang D, Casale GP, Tian J, Lele SM, Pisarev VM, Simpson MA, Hemstreet GP., 3rd (2010). Udp-glucose dehydrogenase as a novel field-specific candidate biomarker of prostate cancer. Int J Cancer 126:315-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Casale GP, Tian J, Wehbi NK, Abrahams NA, Kaleem Z, Smith LM, Johansson SL, Elkahwaji JE, Hemstreet GP., 3rd (2007). Quantitative fluorescence imaging analysis for cancer biomarker discovery: application to beta-catenin in archived prostate specimens. Cancer Epidemiol Biomarkers Prev 16:1371-1381. [DOI] [PubMed] [Google Scholar]
- Janue A, Odena MA, Oliveira E, Olive M, Ferrer I. (2007a). Desmin is oxidized and nitrated in affected muscles in myotilinopathies and desminopathies. J Neuropathol Exp Neurol 66:711-723. [DOI] [PubMed] [Google Scholar]
- Janue A, Olive M, Ferrer I. (2007b). Oxidative stress in desminopathies and myotilinopathies: a link between oxidative damage and abnormal protein aggregation. Brain Pathol 17:377-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrominova TY. (2011). Application of WGA lectin staining for visualization of the connective tissue in skeletal muscle, bone, and ligament/tendon studies. Microsc Res Tech 74:18-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutakis P, Johanning JM, Haynatzki GR, Myers SA, Stergiou N, Longo GM, Pipinos (2010a). Abnormal joint powers before and after the onset of claudication symptoms. J Vasc Surg 52:340-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutakis P, Pipinos, Myers SA, Stergiou N, Lynch TG, Johanning JM. (2010b). Joint torques and powers are reduced during ambulation for both limbs in patients with unilateral claudication. J Vasc Surg 51:80-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutakis P, Weiss DJ, Miserlis D, Shostrom VK, Papoutsi E, Ha DM, Carpenter LA, McComb RD, Casale GP, Pipinos (2014). Oxidative damage in the gastrocnemius of patients with peripheral artery disease is myofiber type selective. Redox Biol 2:921-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarides E. (1980). Desmin and intermediate filaments in muscle cells. Results Probl Cell Differ 11:124-131. [DOI] [PubMed] [Google Scholar]
- Li M, Dalakas MC. (2001). Abnormal desmin protein in myofibrillar myopathies caused by desmin gene mutations. Ann Neurol 49:532-536. [PubMed] [Google Scholar]
- Makitie J, Teravainen H. (1977). Histochemical changes in striated muscle in patients with intermittent claudication. Arch Pathol Lab Med 101:658-663. [PubMed] [Google Scholar]
- Makris KI, Nella AA, Zhu Z, Swanson SA, Casale GP, Gutti TL, Judge AR, II P. (2007). Mitochondriopathy of peripheral arterial disease. Vascular 15:336-343. [DOI] [PubMed] [Google Scholar]
- Marbini A, Gemignani F, Scoditti U, Rustichelli P, Bragaglia MM, Govoni E. (1986). Abnormal muscle mitochondria in ischemic claudication. Acta Neurol Belg 86:304-310. [PubMed] [Google Scholar]
- McDermott MM, Criqui MH, Greenland P, Guralnik JM, Liu K, Pearce WH, Taylor L, Chan C, Celic L, Woolley C, O’Brien MP, Schneider JR. (2004). Leg strength in peripheral arterial disease: associations with disease severity and lower-extremity performance. J Vasc Surg 39:523-530. [DOI] [PubMed] [Google Scholar]
- McDermott MM, Ferrucci L, Guralnik J, Tian L, Liu K, Hoff F, Liao Y, Criqui MH. (2009). Pathophysiological changes in calf muscle predict mobility loss at 2-year follow-up in men and women with peripheral arterial disease. Circulation 120:1048-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott MM, Guralnik JM, Ferrucci L, Tian L, Pearce WH, Hoff F, Liu K, Liao Y, Criqui MH. (2007). Physical activity, walking exercise, and calf skeletal muscle characteristics in patients with peripheral arterial disease. J Vasc Surg 46:87-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott MM, Liu K, Tian L, Guralnik JM, Criqui MH, Liao Y, Ferrucci L. (2012). Calf muscle characteristics, strength measures, and mortality in peripheral arterial disease: a longitudinal study. J Am Coll Cardiol 59:1159-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott MM, Ohlmiller SM, Liu K, Guralnik JM, Martin GJ, Pearce WH, Greenland P. (2001). Gait alterations associated with walking impairment in people with peripheral arterial disease with and without intermittent claudication. J Am Geriatr Soc 49:747-754. [DOI] [PubMed] [Google Scholar]
- Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y. (2000). Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol 150:1283-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti RJ, Roy RR, Hodgson JA, Edgerton VR. (1999). Transmission of forces within mammalian skeletal muscles. J Biomech 32:371-380. [DOI] [PubMed] [Google Scholar]
- Nakano S, Engel AG, Waclawik AJ, Emslie-Smith AM, Busis NA. (1996). Myofibrillar myopathy with abnormal foci of desmin positivity. I. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol 55:549-562. [DOI] [PubMed] [Google Scholar]
- Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG, Group TIW. (2007). Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J Vasc Surg 45 Suppl S:S5-67. [DOI] [PubMed] [Google Scholar]
- Olive M, van Leeuwen FW, Janue A, Moreno D, Torrejon-Escribano B, Ferrer I. (2008). Expression of mutant ubiquitin (UBB+1) and p62 in myotilinopathies and desminopathies. Neuropathol Appl Neurobiol 34:76-87. [DOI] [PubMed] [Google Scholar]
- Patel TJ, Lieber RL. (1997). Force transmission in skeletal muscle: from actomyosin to external tendons. Exerc Sport Sci Rev 25:321-363. [PubMed] [Google Scholar]
- Paulin D, Huet A, Khanamyrian L, Xue Z. (2004). Desminopathies in muscle disease. J Pathol 204:418-427. [DOI] [PubMed] [Google Scholar]
- Pipinos, Judge AR, Selsby JT, Zhu Z, Swanson SA, Nella AA, Dodd SL. (2008). The myopathy of peripheral arterial occlusive disease: Part 2. Oxidative stress, neuropathy, and shift in muscle fiber type. Vasc Endovascular Surg 42:101-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipinos, Judge AR, Selsby JT, Zhu Z, Swanson SA, Nella AA, Dodd SL. (2007). The myopathy of peripheral arterial occlusive disease: part 1. Functional and histomorphological changes and evidence for mitochondrial dysfunction. Vasc Endovascular Surg 41:481-489. [DOI] [PubMed] [Google Scholar]
- Pipinos, Judge AR, Zhu Z, Selsby JT, Swanson SA, Johanning JM, Baxter BT, Lynch TG, Dodd SL. (2006). Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic Biol Med 41:262-269. [DOI] [PubMed] [Google Scholar]
- Pipinos, Sharov VG, Shepard AD, Anagnostopoulos PV, Katsamouris A, Todor A, Filis KA, Sabbah HN. (2003). Abnormal mitochondrial respiration in skeletal muscle in patients with peripheral arterial disease. J Vasc Surg 38:827-832. [DOI] [PubMed] [Google Scholar]
- Pipinos, Shepard AD, Anagnostopoulos PV, Katsamouris A, Boska MD. (2000). Phosphorus 31 nuclear magnetic resonance spectroscopy suggests a mitochondrial defect in claudicating skeletal muscle. J Vasc Surg 31:944-952. [DOI] [PubMed] [Google Scholar]
- Regensteiner JG, Wolfel EE, Brass EP, Carry MR, Ringel SP, Hargarten ME, Stamm ER, Hiatt WR. (1993). Chronic changes in skeletal muscle histology and function in peripheral arterial disease. Circulation 87:413-421. [DOI] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, American Heart Association Statistics C, Stroke Statistics S (2012). Executive summary: heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation 125:188-197. [DOI] [PubMed] [Google Scholar]
- Sanger JW, Sanger JM, Franzini-Armstrong C. (2004) Assembly of the skeletal muscle cell. In Engel AG, Franzini-Armstrong C, eds. Myology. New York, NY, USA, McGraw-Hill, 45-65. [Google Scholar]
- Schejter ED, Baylies MK. (2010). Born to run: creating the muscle fiber. Curr Opin Cell Biol 22:566-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segard BD, Delort F, Bailleux V, Simon S, Leccia E, Gausseres B, Briki F, Vicart P, Batonnet-Pichon S. (2013). N-acetyl-L-cysteine prevents stress-induced desmin aggregation in cellular models of desminopathy. PLoS One 8:e76361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcen D, Ohno K, Engel AG. (2004). Myofibrillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain 127:439-451. [DOI] [PubMed] [Google Scholar]
- Thompson JR, Swanson SA, Haynatzki G, Koutakis P, Johanning JM, Reppert PR, Papoutsi E, Miserlis D, Zhu Z, Casale GP, Pipinos (2014). Protein Concentration and Mitochondrial Content in the Gastrocnemius Predicts Mortality Rates in Patients With Peripheral Arterial Disease. Ann Surg. March 25 [Epub]. 10.1097/SLA.0000000000000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter JA. (2002). Structure-function considerations of muscle-tendon junctions. Comp Biochem Physiol A Mol Integr Physiol 133:1127-1133. [DOI] [PubMed] [Google Scholar]
- Turrina A, Martinez-Gonzalez MA, Stecco C. (2013). The muscular force transmission system: role of the intramuscular connective tissue. J Bodyw Mov Ther 17:95-102. [DOI] [PubMed] [Google Scholar]
- Weiss DJ, Casale GP, Koutakis P, Nella AA, Swanson SA, Zhu Z, Miserlis D, Johanning JM, Pipinos (2013). Oxidative damage and myofiber degeneration in the gastrocnemius of patients with peripheral arterial disease. J Transl Med 11:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter DL, Paulin D, Mericskay M, Li Z. (2014). Posttranslational modifications of desmin and their implication in biological processes and pathologies. Histochem Cell Biol 141:1-16. [DOI] [PubMed] [Google Scholar]
- Wurdeman SR, Koutakis P, Myers SA, Johanning JM, Pipinos, Stergiou N. (2012). Patients with peripheral arterial disease exhibit reduced joint powers compared to velocity-matched controls. Gait Posture 36:506-509. [DOI] [PMC free article] [PubMed] [Google Scholar]