

Abstract
The pathogenesis of DLBCL is strongly linked to perturbation of epigenetic mechanisms. The germinal center (GC) B-cells from which DLBCLs arise are prone to instability in their cytosine methylation patterns. DLBCLs inherit this epigenetic instability and display variable degrees of epigenetic heterogeneity. Greater epigenetic heterogeneity is linked with poor clinical outcome. Somatic mutations of histone modifying proteins have also emerged as a hallmark of DLBCL. The effect of these somatic mutations may be to disrupt epigenetic switches that control the GC phenotype and “lock in” certain oncogenic features of GC B-cells resulting in malignant transformation. DNA methyltransferase and histone methyltransferase inhibitors are emerging as viable therapeutic approaches to erase aberrant epigenetic programming, suppress DLBCL growth and overcome chemotherapy resistance. This review will discuss these recent advances and their therapeutic implications.
Features of the epigenome relevant to normal B-cell development and DLBCL pathogenesis
The phenotype of different cell types is determined by epigenetic instructions. These instructions are encoded by chemical languages that collectively control transcriptional regulation, RNA splicing, DNA replication, response to environmental stimuli, DNA damage responses and other functions [1]. Major components of the epigenome include cytosine modifications, histone modifications and non-coding RNA molecules [1]. Features such as histone isoform exchange and other DNA associated proteins such as Hp1A and HMG proteins are emerging as additional epigenetic control mechanisms[2]. Precisely controlled epigenetic programming is required for normal B-cell development, and DLBCLs universally feature profound disruption of their epigenomes. This review will focus in particular on epigenetic changes that occur when resting B-cells are activated to form germinal centers (GCs) as well as epigenetic switches that terminate the GC reaction and induce memory or plasma cell differentiation. Perturbation of GC epigenetic control mechanisms appears to play a fundamental role in DLBCL pathogenesis.
Several basic considerations must be taken into account when considering the role of the epigenome in normal B-cells and DLBCL. First, the significance of epigenetic modifications is strongly linked to the “geography” and topology of the genome[3]. The meaning of epigenetic modifications such as DNA methylation is profoundly different depending on where they are located. DNA methylation of CpG rich gene promoters is linked to transcriptional silencing, whereas cytosine methylation of intragenic regions is linked to gene activation. In GC B-cells, loss of DNA methylation often occurs at promoters of functionally relevant genes and transcription factor binding sites [4, 5]. Aberrant DNA methylation patterning in DLBCL involves specific chromosomal regional patterns as well as at focal sites proximal to gene promoters [6], suggesting the effect of altered DNA methylation on DLBCL pathogenesis is location-dependent. DNA methylation of transcriptional factor binding sites can result in either transcriptional activation or repression. For example, cytosine methylation of specific residues within the first intron of the BCL6 locus disrupts binding of CTCF, resulting in transcriptional activation of BCL6 in lymphoma cells due to loss of the repressor effect of CTCF[7].
Second, the epigenome is endowed with significant plasticity, and different epigenetic marks have different degrees of plasticity[1]. On the one hand, plasticity enables cells to rapidly switch from one phenotype state to another as occurs when resting B-cells are activated to form GCs and when GC B-cells undergo selection after immunoglobulin affinity maturation to become memory or plasma cells. In this case, epigenetic marks are actively reprogrammed due to signals from the microenvironment (as described below) and result in specific changes in gene expression that determine cell phenotype shifts. On the other hand, plasticity may occur in a more stochastic manner during cell proliferation or exposure to stress, features that are characteristic of GC B-cells and DLBCLs [4]. Stochastic redistribution of marks such as cytosine methylation can result in epigenetic heterogeneity among populations of cells such as GC B-cells[4]. Random switching of epigenetic marks may confer advantages to particular cells and contribute to their clonal outgrowth, independent of the presence of somatic mutations. Both directed and stochastic epigenetic reprogramming are implicated in DLBCL pathogenesis.
Third, epigenetic marks are combinatorial[1, 2]. It is tempting to focus on single epigenetic mark to keep things simple. However the reality of these biochemical instructions is that they form highly complex and textured regions throughout the genome. The functionality of these regions depends on the sum of epigenetic marks present at a given location. It may be difficult to link any particular cytosine or histone modification to specific effects on gene expression when taken out of context. Indeed comprehensive epigenome mapping studies have illustrated that combinatorial epigenomic patterning more accurately reveals the functional significance of epigenetic marks[2]. For example, the combination of repressive mark H3K27me3 and activation mark H3K4me3 at gene promoters define “bivalent” chromatin domains that are linked to transcriptional poising (e.g. [8, 9]). Bivalent chromatin places promoters in a transiently repressed state whereupon genes can be definitively repressed if they subsequently lose the H3K4me3 mark or definitively activated if they lose the H3K27me3 mark[9]. During development genes that determine cell lineage start out as bivalent poised genes in stem cells and gradually switch to monovalent active or repressed as cells choose their fate[9]. Bivalent chromatin marks play a key role in establishing the GC B-cell phenotype and preventing their premature differentiation to plasma cells[10]. Alteration of bivalency at poised promoters affects normal GC B-cell plasma cell differentiation and results in potential malignant transformation[10]. Another example of combinatorial effect of histone modifications resides in the control of enhancer elements. Enhancers are DNA elements that can loop across long distances to come into physical contact with gene promoters and induce their transcriptional activation. From the epigenetic standpoint enhancers feature H3K4 mono or dimethylation and absence of H3K4 trimethylation [11, 12]. Enhancers are active if they contain histone H3 lysine 27 (H3K27) acetylation but are poised and inactive if they lack this mark[13]. Knowledge of the combination of H3K4 methylation and H3K27 acetylation along with other marks is required to understand enhancer function and how they may become perturbed in lymphomas.
These features of the epigenome form the backbone of this review, which will focus specifically on cytosine methylation and several key histone modifications in DLBCL. However, additional epigenetic layers such as the three dimensional topology of the genome, non-coding RNAs, and a much broader diversity of cytosine and histone modifications are also likely to be critically important. New types of modifications, such as histone lysine crotonylation, have been discovered[14]. The role of these new modifications have yet to be defined in normal GC B-cells and malignant DLBCLs. One final caveat that must be taken into account when interpreting epigenetic studies is that many histone modifying proteins can also modify non-histone proteins. Hence it is important to consider that mutation or altered expression of these enzymes may have many effects independent of those linked to epigenetic regulation of the genome.
Epigenetic switches controlling the GC reaction
The next section of this review will focus on two epigenetic switching mechanisms that enable the transient establishment of the GC centroblastic phenotype, both of which are implicated in DLBCL pathogenesis (Figure 1). The pathogenesis of DLBCL is intimately linked to the biology of the GC reaction during the humoral immune response. Upon T-cell dependent activation, resting follicular B-cells follow at least two distinct maturation pathways. A subset of B-cells immediately undergo immunoglobulin class switch recombination and differentiate into low affinity plasma cells[15]. Another subset of B-cells migrate within lymphoid follicles wherein they become centroblasts and form the GC dark zone. Centroblasts are physically larger than resting B-cells and exhibit irregular nuclei with lax chromatin. They undergo rapid proliferation, somatic hypermutation of their immunoglobulin loci and tolerate replicative stress, genomic instability, metabolic reprogramming, and other stresses[15]. The centroblast phenotype facilitates acquisition of somatic mutations induced in part through the actions of activation induced cytosine deaminase (AICDA). In addition to targeting the immunoglobulin loci, AICDA introduces point mutations at cytosine residues throughout the genome at regions with accessible chromatin and specific sequence features[15]. Acquisition of advantageous somatic mutations enables malignant transformation of centroblasts. As centroblasts undergo clonal expansion they encounter and interact with T-cells and follicular dendritic cells in the GC light zone. Interactions with these cells stops proliferation and induces a second phenotype shift in B-cells, which become centrocytes[15]. These signaling events select centrocytes that express high affinity immunoglobulin for survival and differentiation to memory or plasma cells. Those that have not generated high affinity immunoglobulin undergo apoptosis or convert back to proliferating centroblasts for additional rounds of somatic mutation.
Figure 1. Epigenetic switches controlling the GC phenotype.
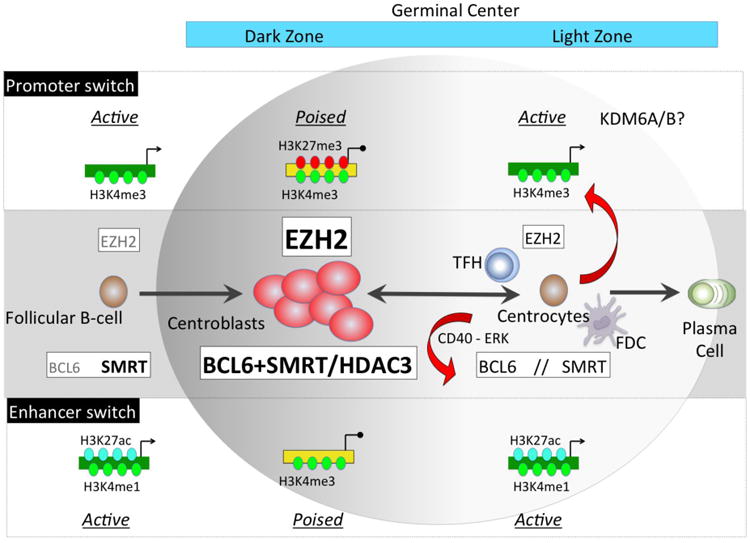
Upon T-cell dependent antigen stimulation a subset of B-cells migrate within lymphoid follicles and upregulate EZH2 and BCL6 proteins. EZH2 catalyzes addition of H3k27me3 to a set of activated promoters causing their transient transcriptional poising. BCL6 forms a complex with SMRT at active enhancers (marked with H3K4me1) causing their H3K27 deacetylation through HDAC3 and placing them in a repressed/poised configuration. Both of these switches result in transcriptional repression of genes involved in proliferation checkpoint, differentiation and apoptosis, thus defining the centroblast phenotype. When GC B-cells migrate to the GC light zone a second switch occurs via signaling from T follicular helper (TFH) and follicular dendritic cells (FDC). These result in downregulation of EZH2 and cytoplasmatic shuttling of SMRT complexes due to ERK mediated phosphorylation (red arrows). As a consequence there is histone acetylation and derepression of BCL6 targets, and H3K27 demethylation of EZH2 target bivalent promoters possibly by KDM6A/B. These events result in activation of proliferation checkpoints and induction of GC exit genes. The effects are reversible and can be switched back off in B-cells that undergo additional rounds of somatic hypermutation.
a) An epigenetic switch for gene promoters in GC B-cells
As introduced earlier, the establishment of bivalency at key gene promoters is a critical step for resting B-cells to switch on GC B-cell phenotype, and EZH2 plays an essential role in this process. Centroblasts feature prominent upregulation of EZH2[16, 17], a histone methyltransferase that mediates H3K27 mono and di methylation and to a lesser extent H3K27 trimethylation (H3K27me3). EZH2 is the enzymatic component of Polycomb Repressor Complex 2 (PRC2), and requires PRC2 subunits EED and SUZ12 for its enzymatic activity (reviewed in [18]). These other PRC2 subunits are also upregulated in GC B-cells[16, 17]. Deposition of H3K27me3 at gene promoters induces epigenetic silencing of transcription at least in part through recruitment of the Polycomb Repressive Complex 1 (PRC1), which can induce chromatin compaction and suppress RNA polymerase II activity among other functions[18]. EZH2 is essential and required for B-cells to form GCs. Mice engineered to specifically delete EZH2 in their GC B-cells fail to form GCs and undergo immunoglobulin affinity maturation[10, 19]. The GC function of EZH2 is dependent on its enzymatic activity since administration of specific EZH2 inhibitors to mice phenocopies EZH2 knockout[10].
It is well established that EZH2 plays essential roles in embryonic stem (ES) cells. However, the direct target genes of EZH2 in primary human GC B-cells are only partially overlapping with those that it binds in ES cells. Strikingly, only the GC specific subset of EZH2 targets that are preferentially repressed in GC B-cells, indicating that EZH2 functions in a cell-context specific manner[16]. The mechanism of action of EZH2 in GC B-cells has been inferred from studies mapping the epigenome of human B-cells. Specifically, it was observed that EZH2 adds H3K27me3 mark to ∼1000 active promoters that are marked by H3K4me3 in resting B-cells, resulting in the de novo formation of bivalent chromatin domains and transcriptional silencing[10]. EZH2 induced bivalent chromatin is enriched at genes required for memory and plasma cell differentiation such as IRF4 and PRDM1[10]. Others are proliferation checkpoint regulators such as CDKN1A (p21) and CDKN1B. EZH2 knockdown or enzymatic inhibition in GC derived DLBCL cells induces re-expression of these genes with consequent proliferation arrest and induction of plasma cell differentiation[10]. EZH2 levels decline in centrocytes through mechanisms that remain to be defined[10]. It is possible that H3K27 demethylases such as KDM6A or KDM6B might become active at this stage and help to erase bivalent domains. At the same time CD40 and BCR signaling by T-cells and FDCs strongly induce activation of differentiation and checkpoint genes[15], thus terminating the transient EZH2 poising effect, presumably switching bivalent promoters to active state, allowing differentiation to occur.
EZH2 thus functions as a gene promoter epigenetic switching mechanism that transiently places the B-cell differentiation and proliferation control program into a poised bivalent state so that B-cells can proliferate and undergo somatic hypermutation(Figure 1). Bivalent promoter poising is dynamic and apparently rapidly reversed when B-cells encounter T-cells and FDCs in the light zone. Notably, it generally believed that bivalent domains become gradually depleted as cells undergo terminal differentiation[9]. If this is true then GC B-cells represent an exception to this rule. There are many unanswered questions regarding the mechanism of action of EZH2 in GC B-cells. For example, EZH2 does not directly bind to chromatin and it is not known how it is that EZH2 is recruited to a GC-specific set of target genes. In various cell types EZH2 was shown to be recruited by specific long-noncoding RNA species[20]. It is possible that GC B-cell lncRNAs might be involved in recruitment. Another puzzling finding is that core PRC1 components such as BMI-1 are downregulated in GC B-cells raising the question of how bivalent genes are repressed in these cells[17]. One clue to this question is that the transcriptional repressor BCL6 which is also required for GC formation binds to many of the EZH2 induced bivalent chromatin domains[21]. Hence EZH2 may cooperate in some way with BCL6 to mediate repression of bivalent chromatin. Recent data suggest the joint recruitment of a non-canonical PRC1-like complex by EZH2 and BCL6[22].
b) An epigenetic switch for gene enhancers in GC B-cells
BCL6 protein is absent from resting B-cells and strongly upregulated in GC B-cells[15]. Genomic localization studies revealed that BCL6 is more extensively bound to enhancers rather than promoters in GC B-cells[21]. In resting B-cells the enhancers that BCL6 binds when it is expressed in GCs tend to be active, bound by EP300 and enriched for H3K27 acetylation[21]. However upon BCL6 upregulation these enhancers become H3K27 deacetylated, lose EP300 binding and their neighboring genes become repressed. These enhancers do not lose their H3K4me1 or H3K4me2 marks however, which means they remain epigenetically marked even though “toggled” into a poised configuration by BCL6[21]. BCL6 mediated enhancer poising involves recruitment of the SMRT and NCOR corepressor proteins through the BCL6 BTB domain[21]. SMRT and NCOR form a complex with HDAC3, which in turn mediates H3K27 deacetylation. Accordingly SMRT-HDAC3 complexes deacetylate H3K27 in a BCL6 dependent manner in GC B-cells. BCL6 binding is also associated with reduction in EP300 enrichment[21]. This may be due to steric competition but could also be linked to BCL6 mediated direct repression of EP300 expression[23].
BCL6 enhancer poising is rapidly reversible[21]. In GC light zone, CD40 signaling to GC derived B-cells induces SMRT phosphorylation through ERK, resulting SMRT export to the cytoplasm, followed by histone acetylation and derepression of BCL6 target genes such as ATR[24]. Derepression of BCL6 enhancer targets presumably restores DNA damage sensing checkpoints and enables activation of genes required for B-cells to exit the germinal center reaction[24]. However, cessation of CD40 signaling results in relocation of SMRT to the nucleus and restoration of BCL6 repressive activity[24]. It is notable that the set of genes regulated through the enhancer toggling mechanism are largely different than those regulated through the promoter epigenetic switch mechanism[21]. Hence these two highly dynamic epigenetic switching mechanisms, one based on EZH2 formation of bivalent chromatin at promoters and the other based on BCL6 mediated H3K27 deacetylation with maintenance of H3K4me1 and H3K4me2 at enhancers, appear to cooperate in transiently impose the GC phenotype on B-cells until they are switched off in the light zone(Figure 1).
Somatic mutation of epigenetic regulators in DLBCL pathogenesis
Somatic mutation of epigenetic modifier proteins is a hallmark of DLBCL [25, 26]. DLBCLs tend to have heavy mutation burdens and exhibit considerable intratumoral heterogeneity. Yet mutation of epigenetic modifiers tend to be widely distributed among lymphoma cells in individual patients suggesting that they occur at early stages of lymphomagenesis [27-29]. We present views on the most frequently mutated epigenetic modifiers in DLBCL which are EZH2, EP300, CREBBP and KMT2D (also known as MLL4 or MLL2, Figure 2).
Figure 2. Effect of somatic mutation of histone modifying enzymes.
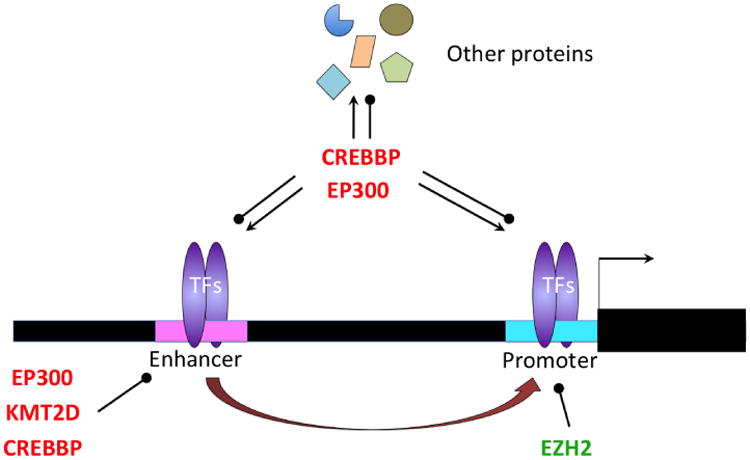
Somatic mutation in EZH2 results in an enzymatic gain of function that increased H3K27me3 at target gene promoters that causes their transcriptional repression. Somatic mutation of EP300 and CREBBP results in either loss of function or dominant negative effects that may result in failure to induce acetylation of BCL6 target enhancers which could cause them to remain in a poised/repressed conformation. By the same token mutation of EP300 and CREBBP enzymes could lead to failure to acetylate histones located at gene promoters, dysfunction of transcription factors that are controlled through lysine acetylation such as TP53, BCL6 and others, as well as the altered acetylation of many other proteins. Lymphoma somatic mutation of KMT2D, and enzyme that mediates H3K4 monomethylation of gene enhancers is expected to result in failure to maintain the activity of enhancers that could limit the propensity of centroblasts to transform into DLBCL cells.
a) EZH2
Missense mutations of EZH2 occur in 21.7% of patients with GCB type DLBCL but are absent from ABC-DLBCL[30]. These mutations are always heterozygous and the vast majority of them affect tyrosine residue 641[30], with only a small percentage affecting alanine 682 or 692[25], all of which are located within the EZH2 catalytic SET domain. Enzymatic assays revealed that Y641 mutant EZH2 proteins exhibit a subtle alteration of their catalytic activity. Whereas wild type EZH2 most efficiently mediates mono and di methylation of H3K27, the Y641 mutant EZH2 is more efficient at adding a third methyl group to H3K27[31, 32]. This results in massive accumulation of H3K27me3 in Y641 mutant DLBCL cells. This effect likely requires biochemical cooperation between the wild type and Y641 mutant EZH2 proteins [10, 31, 32], which may explain why EZH2 Y641 (and alanine 682 or 692) mutations are always heterozygous.
Since EZH2 involves dynamic switching of promoter chromatin bivalency in GC B-cells [10], it would be logical to predict that a stoichiometric shift in H3K27 methylation activity of mutant EZH2 would disrupt this delicate balance. Indeed conditional knockin of Y641 mutant EZH2 into GC B-cells induces massive GC hyperplasia presumably through the dual effects of maintaining centroblast proliferation and blocking terminal differentiation[10]. This is accompanied by massive accumulation of H3K27me3 and concordant profound silencing of bivalent chromatin at promoters of EZH2 target genes[10]. Furthermore, expression of Y641 mutant EZH2 cooperates with oncogenes such as BCL2 and MYC to accelerate lymphomagenesis[10, 33]. Likewise, DLBCL patients with Y641 mutant EZH2 display a GC bivalent gene repression signature. These data establish a new paradigm in malignant transformation whereby subtle stoichiometric disruption of a bivalent chromatin epigenetic switch results in development of a malignancy – in this case B-cell lymphomas (Figure 2). A recent report suggests an additional role of Y641 mutation in disrupting the interaction of EZH2 with the FBXW1 ubiquitin ligase, which may prevent protein turnover of EZH2 and further reinforce its actions on H3K27 trimethylation [34].
EZH2 mutation also occurs in up to 12% of patients with follicular lymphoma (FL), which like DLBCL, originates from GC B-cells[27]. The same study revealed that 29% of the transformed FLs carried EZH2 mutations. However EZH2 is never mutated in ABC-DLBCLs. The reason for this may be that EZH2 represses the same germinal center exit CD40 and NFkB target genes that drive the unique phenotype of ABC-DLBCLs. Indeed EZH2 shRNA or inhibition with small molecules does not seem to suppress the growth of most ABC-DLBCL cell lines[10]. On the other hand, EZH2 is expressed in most GCB type DLBCLs regardless of somatic mutation. Given the role of EZH2 in normal GC B-cells it might be expected that GCB-DLBCLs could be addicted to this protein as a lineage factor. Indeed wild type EZH2 GCB-DLBCLs are also biologically dependent on EZH2 for their proliferation and differentiation state[10]. Patients with GCB-DLBCL and expressing high levels of WT EZH2 also displayed a GC bivalent gene repression signature similar to EZH2 Y641 mutant patients[10]. EZH2 thus plays a central role in imposing the centroblast transcriptional program and biological features characteristic of GCB-DLBCL.
b) EP300 and CREBBP
EP300 and CREBBP are members of the KAT3 family of histone acetyl transferases (HATs). Their major potential substrates are H3K18 and H3K27 given that the global acetylation levels of these two lysine residues are substantially reduced in Ep300 and Crebbp double null mouse fibroblasts [35]. Mutations in both genes are common in DLBCLs. Since H3K27ac is strongly associated with active enhancers, it is conceivable that these mutations may disrupt normal GC B-cell enhancer network.
Somatic mutations of EP300 were first identified in DLBCL cell lines [36] and shown to occur in up to 5-10% of DLBCL cases [23, 25, 26, 37]. Mutations are usually heterozygous and generally result in truncations or disruption of the HAT domain giving rise to a catalytically dead enzyme that may or may not have dominant negative activity. In addition, 5% of DLBCLs contained monoallelic deletions spanning the EP300 locus[37]. Truncated EP300 facilitates the growth of lymphoma cell lines and disrupts the transcriptional activity of REL[36]. EP300 is a direct target gene of BCL6 and expression of dominant negative EP300 or an EP300 inhibitor could rescue DLBCL cells from withdrawal of BCL6 [23]. Hence EP300 may function as a tumor suppressor protein in DLBCL. It is interesting to speculate that one mechanism through which EP300 loss of function could contribute to lymphomagenesis is by reinforcing enhancer repression via BCL6-SMRT complexes. Since EP300 mediates enhancer H3K27 acetylation [13] and seems to compete with BCL6-SMRT complexes [21], it is plausible that its loss of function would leave BCL6-SMRT-HDAC3 complexes unopposed and tilt the stoichiometric balance of histone modification towards deacetylation. Thus EP300 mutation might represent a perturbation in epigenetic switching of enhancers in GC B-cells preventing termination of the BCL6 transcriptional program.
Somatic mutations of CREBBP, a HAT protein with structural similarities to EP300, are even more frequent, occurring in 18-23% of DLBCL cases with a preference to GCB-DLBCL [25, 37]. About half of these mutations are inactivating mutations, including nonsense mutations, frameshift insertions and deletions of base pairs, and splicing variants. Similar to the case of EP300, these mutations tend to be heterozygous and either eliminate or perturb the HAT domain. Up to 11% DLBCL cases also exhibit monoallelic deletion of the CREBBP locus[37]. It is not yet clear if somatic mutation of CREBBP and EP300 constitute the same lesion, since these proteins have both overlapping and non-overlapping functions. However, in a large DLBCL cohort, only two cases harbored genetic alterations of both genes[37], arguing the mechanisms of which CREBBP or EP300 lesions lead to lymphomagenesis may be distinct to each other. Moreover, it is not yet clear whether CREBBP is relevant to the BCL6 enhancer mechanism described above. In additional to histone acetylation, it is important to note that CREBBP and EP300 may acetylate more than 1000 non-histone proteins. For example, in vitro experiments suggest that loss of CREBBP and EP300 HAT activity might impair acetylation of TP53 and BCL6[37]. Reduced TP53 acetylation would be predicted to attenuate its activity, whereas in contrast reduced BCL6 acetylation might enhance its functionality. Thus mutant CREBBP might be difficult to ascribe to any single function in DLBCL pathogenesis (Figure 2).
c) KMT2D
KMT2D is a histone methyltransferase that primarily mediates H3K4 monomethylation to regulate gene enhancers and as such mediates epigenetic activation of gene expression[38, 39]. KMT2D forms a complex with KMT2C and the H3K27 demethylase UTX[40]. Germline somatic mutations of KMT2D and UTX are the cause of congenital Kabuki syndrome, which features intellectual disability, short stature and various other manifestations[41]. Somatic missense and nonsense mutations of KMT2D occur in approximately 23-27% of DLBCL patients and in most cases are heterozygous[25, 26, 42]. Zhang et al reported that KMT2C, which forms complex with KMT2D, was the most frequently mutated gene in their cohort of DLBCL cases (of note is that KMT2D was not included in the exome capture library used in this study), further confirming the important role of this complex in DLBCL pathogenesis[43]. Many of KMT2D lesions result in mutation or truncations predicted to affect the catalytic SET domain. These mutations may result in loss of function or dominant negative effect in B-cells. Although KMT2D is expressed in follicular and GC B-cells, its role in B-cells is not yet described. Given KMT2D and UTX were linked to cell proliferation and invasiveness in solid tumors[44, 45], it seems plausible that KMT2D may form part of an enhancer switching mechanism governing critical functions during B-cell development, including B-cell proliferation, migration and trafficking. Disruption of KMT2D early in the pathogenesis of DLBCL would be expected to cause loss of function of enhancers linked these processes (Figure 2).
The contribution of aberrant cytosine methylation to DLBCL pathogenesis
GC B-cells display massive redistribution of cytosine methylation as compared to resting follicular B-cells consisting mostly of promoter hypomethylation[4, 5]. There is a trend for these changes to be inversely correlated with gene expression in these cell types and among these are genes with potentially significant functions in cell proliferation and survival, such as UHRF2 and IKBKE[4]. DNA methylation is catalyzed by the DNMT1, DNMT3A and DNMT3b enzymes. Among these the DNMT3A protein is strongly downregulated in GC B-cells[4], which could potentially explain some of the focal hypomethylation observed in GC B-cells. In contrast, DNMT1 is strongly upregulated in GC B-cells and is required for the development of GCs in mice[4]. Given that DNMT1 plays crucial roles during replication both in maintaining DNA methylation and genomic integrity it is possible that both of these roles are relevant in the GC context. Indeed, DNMT1 hypomorph mice displayed an increase in DNA damage in their remnant GC B-cells[4].
DNA demethylation in GC B-cells could also occur through induction of AICDA. Cytosine deaminases including AICDA directly demethylate cytosine residues through base excision repair replacing methylated cytosines with unmethylated nucleotides. This activity of AICDA plays crucial roles in germ cell and induces pluripotent cell epigenetic reprogramming [46-49]. In GC B-cells, sites of hypomethylation are enriched for AICDA binding motifs. In vitro experiments in B-cells failed to confirm an effect for AICDA in demethylation[50]. However, examination of DNA methylation profiles in Aicda knockout mice revealed striking loss of the expected cytosine methylation redistribution by Aicda in GC B-cells[51]. Hence it seems that in addition to its role in somatic hypermutation, AICDA also plays a significant role in epigenetic changes in the GC.
Perturbation of cytosine methylation profiles is a universal event in DLBCL[52, 53]. Hypermethylation and silencing of specific genes has been widely reported in DLBCL, and some of these (e.g. such as CDKN2A) may have prognostic significance (e.g. as reviewed recently in[54]). The GCB and ABC DLBCL subtypes exhibit distinct DNA methylation patterns[52] and can be classified according to cell of origin based on a 16 gene DNA methylation classifier.
One of the most striking features of DLBCLs is their epigenetic heterogeneity (Figure 3). Individual DLBCL patients exhibit markedly heterogeneous DNA methylation patterning among their lymphoma cells[6]. DLBCL was the first disease where epigenetic heterogeneity was shown to be clinically relevant[6]. DNA methylation diversity might increase the likelihood of a given patient containing clones epigenetically programmed to better tolerate exposure to chemotherapy. Indeed, those patients with higher epigenetic heterogeneity had inferior clinical outcomes after RCHOP therapy[6]. The putative significance of epigenetic clonality was further reinforced by the finding that relapsed DLBCLs display reduced epigenetic heterogeneity, consistent with selection for chemotherapy resistant clones[55]. Importantly, epigenetic heterogeneity was not associated with genetic clonality at relapse suggesting that genetic and epigenetic clonality are not necessarily linked and may have independent biological significance[55]. Finally, DLBCL patients can be classified based on their degree of DNA methylation variability. Cohorts of patients with highest variability indices also display inferior survival after RCHOP therapy[56].
Figure 3. DNA methylation heterogeneity in the pathogenesis of DLBCL.
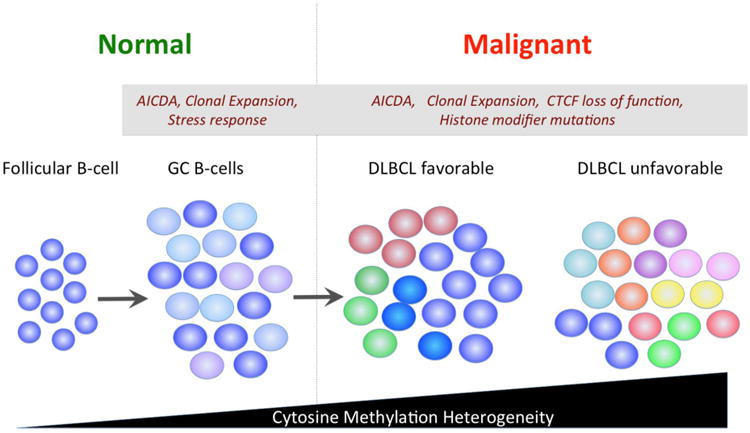
The DNA methylation pattern of follicular B cells is well-defined. However when these cells become centroblasts they acquire more heterogeneity regarding the methylation status of cytosine residues. These effects are strongly linked to the action of AICDA, which causes both diffuse and focal heterogeneity most likely because of its removal of cytosine residues. However stress responses and proliferation may also contribute. DLBCLs feature much more exaggerated diffuse and focal regions of cytosine methylation heterogeneity. Greater heterogeneity is linked to worse outcome. Increasing heterogeneity may be linked to loss of CTCF function and mutations in histone modifying proteins, in addition to factors driving heterogeneity in GC B cells.
These observations raise the question of what might be the source of epigenetic heterogeneity in DLBCLs. Examination of methylomes of primary human GC B-cells revealed a significant increase in DNA methylation heterogeneity as compared to resting B-cells[4, 6]. Hence epigenetic variability is a property inherent to the cell of origin of DLBCL and simply becomes more exaggerated in the tumors (Figure 3). Sites of epigenetic variability in normal GC B-cells and DLBCL are enriched for putative AICDA binding sites[4, 6]. Aicda knockout GC B-cells display less variability than their wild type counterparts[51]. Hence the continuous presence of AICDA induced cytosine turnover may be an important contributing factor to this phenomenon. Sites of epigenetic variability are also enriched in binding sites for the epigenetic regulatory factor CTCF[6]. Notably, Ctcf heterozygous mice are prone to develop B-cell lymphomas[57]. Pre-malignant tissues of Ctcf heterozygous mice exhibited hotspots of focal cytosine methylation variability[57]. Finally, sites of cytosine methylation and H3K27me3 are mutually exclusive in normal GC B-cells[16]. In contrast cytosine methylation and H3K27me3 tend to overlap in DLBCLs[16] and hypermethylated genes in DLBCL partially overlap with EZH2 targets[53]. The genomic distribution of H3K27me3 is highly variable among DLBCL cell lines [10, 58]. It is therefore possible that variation in cytosine methylation could be linked to deregulated functions of EZH2. Other potential influences on epigenetic heterogeneity could be linked to age and proliferative rate of B-cells. However the magnitude of such effects was shown to not be concordant with the degree of DNA methylation heterogeneity in DLBCLs[6].
Epigenetic targeted therapy for DLBCL
Translating knowledge of epigenetic mechanism into effective therapy is a major challenge. Epigenetic therapy is by definition geared towards reprogramming tumor cells as opposed to inducing cytotoxic effects. Hence traditional pre-clinical and clinical parameters developed to evaluate chemotherapy drugs are not necessarily appropriate for guiding the deployment of drugs targeting epigenetic mechanisms. Dosing, timing and sequencing of epigenetic drugs must be guided by relevant pharmacodynamic markers. Many of the drugs described as “epigenetic targeted therapy” have numerous non-epigenetic off target effects. DLBCL cell lines are epigenetically distinct from primary human DLBCLs and may have limited suitability to predict the activity of these drugs against primary human tumors. Finally, epigenetic programming is combinatorial and targeting only one modification may not be sufficient to reverse tumor associated epigenetic abnormalities. Nonetheless the increasing availability of small molecules targeting epigenetic modifiers and improved methods for pre-clinical assessment and biomarker discovery will increasingly facilitate rational translation of such therapies (Table 1).
Table 1. List of epigenetic therapy approaches for DLBCL.
| Drugs | Site of Action | Goals | DLBCLS to Treat* |
|---|---|---|---|
| DNMTi (demethylating dose only) | Sites of focal hypermethylation Site of focal or diffuse epigenetic heterogeneity |
Reverse methylation of tumor suppressors Reverse methylation of genes linked to chemo-resistance Erase heterogeneity, reduce clonality |
Patients with SMAD1 or CDKN2A methylation Relapsed or newly diagnosed high risk patients |
| HDACi | Enhancer and promoter H3K9, H3K18, H3K27 acetylation >1000 non-histone proteins |
Reverse centroblast enhancer poising Alter the functions of TP53, BCL6, NFkB, etc. |
Mutant EP300 and CREBBP: marker for sensitivity or resistance? |
| EZH2i | “Locked-in” bivalent chromatin domains | Restore expression of proliferation checkpoint and differentiation genes | GCB type DLBCLs with or without EZH2 mutations Possible biomarkers: H3K27me3, EZH2 target gene signature |
| BCL6i | BCL6 target promoters and enhancers, including bivalent domains and poised enhancers | Induce cell death and proliferation arrest | GCB and ABC-DLBCLs, possibly those with BCL6 target gene signature |
No biomarkers for these drugs are currently validated. This list is speculative based on publications discussed in the text.
There is a compelling rationale for testing DNA methyltransferase inhibitors (DNMTi) as targeted therapy for DLBCL: i) DLBCLs are highly proliferative and DNMTi require cell replication to hit their target; ii) relatively short exposure to drug should allow its excellent incorporation into the DNA of tumor cells, iii) DNMT1 is required for GC B-cell and DLBCLs may inherit this dependency; iv) epigenetic clonal complexity may contribute to pathogenesis; v) aberrant hypermethylation of tumor suppressors documented as being clinically significant and vi) low demethylating dose of DNMTi below the levels that induce DNA damage should not have cross-toxicity with chemotherapy. Only one study so far has tested DNMTis at strictly non-DNA damaging doses in pre-clinical assays. After several days of demethylating dose, DNMTi induced partial senescence with incomplete growth arrest in DLBCL cells, and sensitized tumor cells from patients with chemotherapy refractory disease to cytotoxic chemotherapy agents [59]. DNA methylation profiling studies identified hypermethylation of SMAD1 as a biomarker for predicting chemotherapy resistance in DLBCL[59, 60]. Functional assays confirmed that expression of SMAD1 induced chemotherapy responsiveness in chemotherapy resistant DLBCL cells[59]. Based on these findings, a phase I study administered 5′azacytidine to newly diagnosed high risk DLBCL patients for five days followed by RCHOP. This regimen was well tolerated. Serial biopsies were performed on a subset of patients before and after the first cycle of 5′azacytidine and confirmed reversal of SMAD1 methylation and derepression of SMAD1 expression[59]. The initial diagnostic patient specimens tested were resistant to clinically relevant concentrations of doxorubicin ex vivo, whereas the follow-up biopsy specimens became chemotherapy responsive[59]. In contrast, there was considerable toxicity in a different phase I study using decitabine alone for longer exposure times in various lymphoid neoplasms [61]. Overall, there seems to be a rational to evaluate low dose and limited duration of DNMTi to enhance response to chemotherapy in high risk or relapsed DLBCLs in larger scale studies.
HDAC inhibitors suppress many DLBCL cell lines in pre-clinical studies in vitro and in vivo, but exhibited limited activity in clinical trials. Many of these results have been reviewed in detail elsewhere (e.g. [62-64]). Most of the currently available HDAC inhibitors are pleiotropic in their actions and inhibit multiple HDAC enzymes. Thousands of proteins are regulated through lysine acetylation and are affected by inhibition of HDACs. Hence it is difficult to interpret available pre-clinical and clinical data. Developing biomarkers to guide deployment of these relatively non-targeted drugs is challenging. Nonetheless the responsiveness of DLBCL cell lines and the subset of patients who respond to these drugs justify continued efforts to refine mechanistic studies to target HDACs in DLBCL. Inhibitors for specific HDAC family members are needed to better understand these targets and improve rational therapeutic targeting of lymphomas. It is intriguing to postulate that patients with CREBBP and EP300 mutations might be more responsive to these drugs since suppression of HDACs could ostensibly partially restore the stoichiometry of protein and/or histone lysine acetylation. On the other hand published data suggested that DLBCL cells with disruption of EP300 function were less sensitive to the HDAC inhibitor SAHA[23]. Therefore, it is possible that without EP300 HAT activity the HDAC effect might be attenuated. An intriguing recent report suggested that exposure to HDAC inhibitor could rescue histone methylation defects in KMT2D heterozygous murine embryo fibroblasts and partially rescue some of the neurologic phenotypes in mouse neurogenesis[65]. Perhaps inhibition of HDACs could increase the histone acetylation and activity of enhancers that are aberrantly repressed by KMT2D loss of function.
The recent development of specific EZH2 inhibitors portends the development of more specific forms of epigenetic therapy for DLBCL[58, 66-69]. EZH2 inhibitors can be considered as a form of “bivalent chromatin targeted therapy” based on the epigenetic switch mechanism described above. In mice bearing DLBCL xenografts, EZH2 inhibitors exerted a potent anti-lymphoma effect without evidence of significant toxicity [58, 70]. The primary effect of EZH2 inhibitors on lymphoma cells is primarily growth arrest and induction of differentiation, which is only evident after several days of exposure [10, 58, 67, 69]. Whereas initial reports suggested that these drugs would be active specifically in those DLBCLs with mutant EZH2, subsequent studies suggested that GCB type DLBCLs may also respond albeit with slower kinetics[10]. EZH2 inhibitors synergize with glucocorticoids in suppressing DLBCL cells suggesting a possible approach for combinatorial therapy[71]. Moreover, BCL6-SMRT induced enhancer poising can be reversed through the use of specific BCL6 inhibitors that block the binding of SMRT to BCL6 and restore H3K27 to BCL6 repressed enhancers in DLBCL cells[21, 72, 73]. Hence both epigenetic switching mechanisms can be overcomed by novel targeted therapies. Finally, enhancers that drive expression of key oncogenes in DLBCL may be targetable using bromodomain inhibitors that cause a partial attenuation of their transcriptional activation effects[74]. Following these observations, it appears that targeting epigenetic switches could represent a focus point for future targeted epigenetic therapies.
Concluding remarks
The past several years have seen a dramatic expansion of our mechanistic understanding of DLBCL pathogenesis with epigenetic mechanisms coming into focus as a driving force in these tumors. Somatic mutations in epigenetic modifiers are clearly initiating events and occur early in disease. In many cases several epigenetic mechanisms may be simultaneously at play, including switching mechanisms that control promoter and enhancer poising. Epigenetic heterogeneity and clonality represents a novel dimension pointing towards evolutionary fitness of more chemotherapy resistant DLBCLs. The current panoply of novel epigenetic targeted therapies offers the opportunity to therapeutically reprogram the epigenome but requires deeper mechanistic knowledge and improved biomarkers to guide their deployment. Future studies combining drugs that target various components of the epigenetic code in a rigorous and rational manner may profoundly impact and change the way we treat patients with DLBCL
Acknowledgments
YJ was supported by an American Society of Hematology Scholar Award. AM is supported by NIH R01 CA187109, Leukemia and Lymphoma Society Translational Research grant 6141-14, a Burroughs Wellcome Translational Scientist Award and the Chemotherapy Foundation.
Footnotes
Conflict of Interest: AM: Consultant: Epizyme, Roche, Celgene.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nature reviews Cancer. 2010;10:457–69. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maze I, Noh KM, Soshnev AA, Allis CD. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nature reviews Genetics. 2014;15:259–71. doi: 10.1038/nrg3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME, et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood. 2011;118:3559–69. doi: 10.1182/blood-2011-06-357996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai AY, Mav D, Shah R, Grimm SA, Phadke D, Hatzi K, et al. DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome research. 2013;23:2030–41. doi: 10.1101/gr.155473.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De S, Shaknovich R, Riester M, Elemento O, Geng H, Kormaksson M, et al. Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLoS genetics. 2013;9:e1003137. doi: 10.1371/journal.pgen.1003137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai AY, Fatemi M, Dhasarathy A, Malone C, Sobol SE, Geigerman C, et al. DNA methylation prevents CTCF-mediated silencing of the oncogene BCL6 in B cell lymphomas. The Journal of experimental medicine. 2010;207:1939–50. doi: 10.1084/jem.20100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 9.Gan Q, Yoshida T, McDonald OG, Owens GK. Concise review: epigenetic mechanisms contribute to pluripotency and cell lineage determination of embryonic stem cells. Stem cells. 2007;25:2–9. doi: 10.1634/stemcells.2006-0383. [DOI] [PubMed] [Google Scholar]
- 10.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer cell. 2013;23:677–92. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–12. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nature reviews Genetics. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 13.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21931–6. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–28. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatzi K, Melnick A. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends in molecular medicine. 2014;20:343–52. doi: 10.1016/j.molmed.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010;116:5247–55. doi: 10.1182/blood-2010-04-280149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raaphorst FM, van Kemenade FJ, Blokzijl T, Fieret E, Hamer KM, Satijn DP, et al. Coexpression of BMI-1 and EZH2 polycomb group genes in Reed-Sternberg cells of Hodgkin's disease. The American journal of pathology. 2000;157:709–15. doi: 10.1016/S0002-9440(10)64583-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chase A, Cross NC. Aberrations of EZH2 in cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:2613–8. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 19.Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. The Journal of clinical investigation. 2013;123:5009–22. doi: 10.1172/JCI70626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benetatos L, Voulgaris E, Vartholomatos G, Hatzimichael E. Non-coding RNAs and EZH2 interactions in cancer: long and short tales from the transcriptome. International journal of cancer Journal international du cancer. 2013;133:267–74. doi: 10.1002/ijc.27859. [DOI] [PubMed] [Google Scholar]
- 21.Hatzi K, Jiang Y, Huang C, Garrett-Bakelman F, Gearhart MD, Giannopoulou EG, et al. A hybrid mechanism of action for BCL6 in B cells defined by formation of functionally distinct complexes at enhancers and promoters. Cell reports. 2013;4:578–88. doi: 10.1016/j.celrep.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beguelin W, Teater M, Hatzi K, Popovic R, Jiang Y, Bunting KL, et al. EZH2 and BCL6 Cooperate To Create The Germinal Center B-Cell Phenotype and Induce Lymphomas Through Formation and Repression Of Bivalent Chromatin Domains. Blood. 2013;122 [Google Scholar]
- 23.Cerchietti LC, Hatzi K, Caldas-Lopes E, Yang SN, Figueroa ME, Morin RD, et al. BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. The Journal of clinical investigation. 2010 doi: 10.1172/JCI42869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polo JM, Ci W, Licht JD, Melnick A. Reversible disruption of BCL6 repression complexes by CD40 signaling in normal and malignant B cells. Blood. 2008;112:644–51. doi: 10.1182/blood-2008-01-131813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nature genetics. 2011;43:830–7. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bodor C, Grossmann V, Popov N, Okosun J, O'Riain C, Tan K, et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood. 2013;122:3165–8. doi: 10.1182/blood-2013-04-496893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nature genetics. 2014;46:176–81. doi: 10.1038/ng.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Y, Redmond D, Nie K, Eng KW, Clozel T, Martin P, et al. Deep sequencing reveals clonal evolution patterns and mutation events associated with relapse in B-cell lymphomas. Genome biology. 2014;15:432. doi: 10.1186/s13059-014-0432-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nature genetics. 2010;42:181–5. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20980–5. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–9. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berg T, Thoene S, Yap D, Wee T, Schoeler N, Rosten P, et al. A transgenic mouse model demonstrating the oncogenic role of mutations in the polycomb-group gene EZH2 in lymphomagenesis. Blood. 2014;123:3914–24. doi: 10.1182/blood-2012-12-473439. [DOI] [PubMed] [Google Scholar]
- 34.Sahasrabuddhe AA, Chen X, Chung F, Velusamy T, Lim MS, Elenitoba-Johnson KS. Oncogenic Y641 mutations in EZH2 prevent Jak2/beta-TrCP-mediated degradation. Oncogene. 2014 doi: 10.1038/onc.2013.571. [DOI] [PubMed] [Google Scholar]
- 35.Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. The EMBO journal. 2011;30:249–62. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garbati MR, Alco G, Gilmore TD. Histone acetyltransferase p300 is a coactivator for transcription factor REL and is C-terminally truncated in the human diffuse large B-cell lymphoma cell line RC-K8. Cancer letters. 2010;291:237–45. doi: 10.1016/j.canlet.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, et al. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes & development. 2012;26:2604–20. doi: 10.1101/gad.201327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife. 2013;2:e01503. doi: 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, et al. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Molecular and cellular biology. 2007;27:1889–903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bogershausen N, Wollnik B. Unmasking Kabuki syndrome. Clinical genetics. 2013;83:201–11. doi: 10.1111/cge.12051. [DOI] [PubMed] [Google Scholar]
- 42.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3879–84. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1398–403. doi: 10.1073/pnas.1205299110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo C, Chen LH, Huang Y, Chang CC, Wang P, Pirozzi CJ, et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget. 2013;4:2144–53. doi: 10.18632/oncotarget.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JH, Sharma A, Dhar SS, Lee SH, Gu B, Chan CH, et al. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer research. 2014;74:1705–17. doi: 10.1158/0008-5472.CAN-13-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar R, DiMenna L, Schrode N, Liu TC, Franck P, Munoz-Descalzo S, et al. AID stabilizes stem-cell phenotype by removing epigenetic memory of pluripotency genes. Nature. 2013;500:89–92. doi: 10.1038/nature12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–5. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–7. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. The Journal of biological chemistry. 2004;279:52353–60. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 50.Fritz EL, Rosenberg BR, Lay K, Mihailovic A, Tuschl T, Papavasiliou FN. A comprehensive analysis of the effects of the deaminase AID on the transcriptome and methylome of activated B cells. Nature immunology. 2013;14:749–55. doi: 10.1038/ni.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dominguez M, Teater M, Chambwe N, Redmond D, Vuong B, Chaudhuri J, et al. Demethylase Activity of Aid during Germinal Center B Cell Maturation Could Contribute to Lymphomagenesis. Blood. 2014;124 [Google Scholar]
- 52.Shaknovich R, Geng H, Johnson NA, Tsikitas L, Cerchietti L, Greally JM, et al. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood. 2010;116:e81–9. doi: 10.1182/blood-2010-05-285320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin-Subero JI, Kreuz M, Bibikova M, Bentink S, Ammerpohl O, Wickham-Garcia E, et al. New insights into the biology and origin of mature aggressive B-cell lymphomas by combined epigenomic, genomic, and transcriptional profiling. Blood. 2009;113:2488–97. doi: 10.1182/blood-2008-04-152900. [DOI] [PubMed] [Google Scholar]
- 54.Jiang Y, Hatzi K, Shaknovich R. Mechanisms of epigenetic deregulation in lymphoid neoplasms. Blood. 2013;121:4271–9. doi: 10.1182/blood-2012-12-451799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pan H, Jiang Y, Redmond D, Nie K, Cerchietti L, Shaknovich R, et al. Epigenomic Evolution In Diffuse Large B-Cell Lymphomas. Blood. 2013;122 doi: 10.1038/ncomms7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chambwe N, Kormaksson M, Geng H, De S, Michor F, Johnson NA, et al. Variability in DNA methylation defines novel epigenetic subgroups of DLBCL associated with different clinical outcomes. Blood. 2014;123:1699–708. doi: 10.1182/blood-2013-07-509885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kemp CJ, Moore JM, Moser R, Bernard B, Teater M, Smith LE, et al. CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell reports. 2014;7:1020–9. doi: 10.1016/j.celrep.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 59.Clozel T, Yang S, Elstrom RL, Tam W, Martin P, Kormaksson M, et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer discovery. 2013;3:1002–19. doi: 10.1158/2159-8290.CD-13-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leshchenko VV, Kuo PY, Jiang Z, Thirukonda VK, Parekh S. Integrative genomic analysis of temozolomide resistance in diffuse large B-cell lymphoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:382–92. doi: 10.1158/1078-0432.CCR-13-0669. [DOI] [PubMed] [Google Scholar]
- 61.Blum KA, Liu Z, Lucas DM, Chen P, Xie Z, Baiocchi R, et al. Phase I trial of low dose decitabine targeting DNA hypermethylation in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: dose-limiting myelosuppression without evidence of DNA hypomethylation. British journal of haematology. 2010;150:189–95. doi: 10.1111/j.1365-2141.2010.08213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunology and cell biology. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 63.Guo SQ, Zhang YZ. Histone deacetylase inhibition: an important mechanism in the treatment of lymphoma. Cancer biology & medicine. 2012;9:85–9. doi: 10.3969/j.issn.2095-3941.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cerchietti L, Leonard JP. Targeting the epigenome and other new strategies in diffuse large B-cell lymphoma: beyond R-CHOP. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2013;2013:591–5. doi: 10.1182/asheducation-2013.1.591. [DOI] [PubMed] [Google Scholar]
- 65.Bjornsson HT, Benjamin JS, Zhang L, Weissman J, Gerber EE, Chen YC, et al. Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Science translational medicine. 2014;6:256ra135. doi: 10.1126/scitranslmed.3009278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garapaty-Rao S, Nasveschuk C, Gagnon A, Chan EY, Sandy P, Busby J, et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chemistry & biology. 2013;20:1329–39. doi: 10.1016/j.chembiol.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 67.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nature chemical biology. 2012;8:890–6. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 68.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS chemical biology. 2013;8:1324–34. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21360–5. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Molecular cancer therapeutics. 2014;13:842–54. doi: 10.1158/1535-7163.MCT-13-0773. [DOI] [PubMed] [Google Scholar]
- 71.Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, et al. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PloS one. 2014;9:e111840. doi: 10.1371/journal.pone.0111840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cerchietti LC, Ghetu AF, Zhu X, Da Silva GF, Zhong S, Matthews M, et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer cell. 2010;17:400–11. doi: 10.1016/j.ccr.2009.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cerchietti LC, Yang SN, Shaknovich R, Hatzi K, Polo JM, Chadburn A, et al. A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood. 2009;113:3397–405. doi: 10.1182/blood-2008-07-168773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer cell. 2013;24:777–90. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]