

Colorectal tumors become more heterogeneous after progression on EGFR-inhibitors by developing multiple independent mutations. Upon discontinuation of treatment, the mutant allele burden declines over several months, which suggests that the changes may be dynamic. Monitoring genetic changes during treatment using ctDNA may provide opportunities to optimize therapeutic strategies.
Keywords: ctDNA, KRAS acquired mutation, EGFR acquired mutation, anti-EGFR mAbs
Abstract
Introduction
KRAS and EGFR ectodomain-acquired mutations in patients with metastatic colorectal cancer (mCRC) have been correlated with acquired resistance to anti-EGFR monoclonal antibodies (mAbs). We investigated the frequency, co-occurrence, and distribution of acquired KRAS and EGFR mutations in patients with mCRC refractory to anti-EGFR mAbs using circulating tumor DNA (ctDNA).
Patients and methods
Sixty-two post-treatment plasma and 20 matching pretreatment archival tissue samples from KRAS wt mCRC patients refractory to anti-EGFR mAbs were evaluated by high-sensitivity emulsion polymerase chain reaction for KRAS codon 12, 13, 61, and 146 and EGFR 492 mutations.
Results
Plasma analyses showed newly detectable EGFR and KRAS mutations in 5/62 [8%; 95% confidence interval (CI) 0.02–0.18] and 27/62 (44%; 95% CI 0.3–0.56) samples, respectively. KRAS codon 61 and 146 mutations were predominant (33% and 11%, respectively), and multiple EGFR and/or KRAS mutations were detected in 11/27 (41%) cases. The percentage of mutant allele reads was inversely correlated with time since last treatment with EGFR mAbs (P = 0.038). In the matching archival tissue, these mutations were detectable as low-allele-frequency clones in 35% of patients with plasma mutations after treatment with anti-EGFR mAbs and correlated with shorter progression-free survival (PFS) compared with the cases with no new mutations (3.0 versus 8.0 months, P = 0.0004).
Conclusion
Newly detected KRAS and/or EGFR mutations in plasma ctDNA from patients refractory to anti-EGFR treatment appear to derive from rare, pre-existing clones in the primary tumors. These rare clones were associated with shorter PFS in patients receiving anti-EGFR treatment. Multiple simultaneous mutations in KRAS and EGFR in the ctDNA and the decline in allele frequency after discontinuation of anti-EGFR therapy in a subset of patients suggest that several resistance mechanisms can co-exist and that relative clonal burdens may change over time. Monitoring treatment-induced genetic alterations by sequencing ctDNA could identify biomarkers for treatment screening in anti-EGFR-refractory patients.
introduction
The anti-epidermal growth factor receptor (anti-EGFR) monoclonal antibodies (mAbs) cetuximab and panitumumab have improved clinical outcomes for patients with metastatic colorectal cancer (mCRC) who have tumors expressing wild-type KRAS [1].
Researchers have found a relationship between the occurrence of novel genetic events in key downstream effectors of the EGFR pathway and treatment-acquired resistance. In one study, an EGFR extracellular domain (S492R) mutation was newly detected in 2/20 patients whose disease initially responded but later progressed on cetuximab [2]. Other studies have reported the presence of acquired KRAS mutations in patients in whom resistance to anti-EGFR mAbs developed after an initial clinical benefit [3–5]. Retrospective studies had identified pre-existing low-frequency KRAS mutations using high-sensitivity sequencing techniques, despite the fact that standard-of-care KRAS testing had failed to detect these mutations. These low-frequency KRAS mutations seemed to correlate with a poor response to anti-EGFR treatment [6, 7].
In this present retrospective study, we evaluated the frequency and distribution of newly detectable KRAS and EGFR mutations after progression on anti-EGFR mAb by sequencing cell-free circulating tumor DNA (ctDNA), and correlated these findings with high-sensitivity testing of the primary tumor to characterize the presence of pre-existing rare KRAS and EGFR mutant clones.
Additional goals were to understand whether multiple genetic changes may develop under treatment pressure and to track the temporal dynamics of these mutations upon completion of anti-EGFR therapy.
materials and methods
study population
We retrospectively studied 70 patients with mCRC or unresectable locally advanced colorectal cancer who had been treated with systemic chemotherapy and anti-EGFR mAbs. Patients had been enrolled in the Assessment of Targeted Therapies in Colorectal Cancer (ATTACC) biomarker screening program, undergoing to evaluation for biomarker-matched clinical trials at The University of Texas MD Anderson Cancer Center. All patients had KRAS and BRAF sequenced as part of standard-of-care testing in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory. Tumor measurements by RECIST1.1 were calculated from computed tomography (CT) scans obtained in proximity to plasma sample collection. Tumor burden assessment (Figure 2B) was calculated from CT scans using the commercially available volumetric-dedicated software MyrianXL-Onco (Intrasense, Montpellier, France) [8]. This study was carried out under an Institutional Review Board-approved protocol, and informed consent was collected from all patients enrolled.
Figure 2.
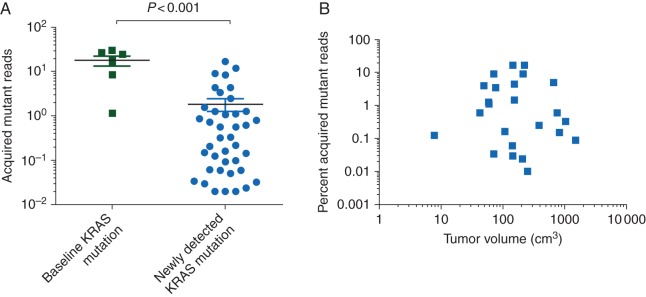
Concentration of ctDNA-detectable alleles by timing of mutation and tumor size. (A) Patients with tumors carrying de novo KRAS mutations had a significantly higher percentage of KRAS mutant alleles (defined as the number of mutant KRAS allele reads divided by the total number of wild-type or mutant KRAS allele reads) detected in plasma ctDNA than patients characterized as having wild-type KRAS initially but who had newly detected KRAS mutations after progression on EGFR mAb treatment (P < 0.001, Mann–Whitney U-test). (B) Tumor volumes were calculated by three independent readers by manually tracing the outer edge of the lesions on each image, and the whole-tumor burden was calculated by summing each lesion volumes. No correlation was found between percentage of acquired mutant alleles and tumor burden.
sample collection, ctDNA isolation, and KRAS and EGFR mutation analysis
Baseline KRAS mutation analysis carried out in a CLIA-compliant laboratory on patient formalin-fixed paraffin-embedded (FFPE) tissue samples using either DNA sequencing (IonTorrent PGM, Life Technologies, Gray Island, NY) or allele-specific polymerase chain reaction (PCR) combined with matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (MassARRAY, Sequenom, San Diego, CA) for mutations in the four most common hot spots (codons 12, 13, 61, and 146) of KRAS. After resistance on EGFR-inhibitors blood samples for ctDNA were collected and processed within 2 h of collection in cell preparation tubes (BD, Franklin Lakes, NJ). Plasma was separated after a single centrifugation step and stored in 2-ml aliquots at −80°C until analysis. ctDNA was purified from the plasma aliquots at Sysmex Inostics Laboratory (Hamburg, Germany). A subsequent KRAS and EGFR analysis was carried out using high-sensitivity emulsion PCR on DNA extracted from FFPE samples of primary tumor tissues from patients who had newly detected KRAS and EGFR mutations identified in post-progression plasma samples. DNA was extracted from five 10-μm-thick slides made from the FFPE sample [9]. Sequencing was carried out using Bead Emulsion Amplification Magnetic (BEAMing) technology, and samples with a detectable mutation rate above 0.02% were considered positive [9]. Specific KRAS and EGFR mutations were analyzed (supplementary Table S1).
statistical analysis
Differences in progression-free survival (PFS) for initial treatment with EGFR inhibitors were evaluated using the log-rank test (Mantel–Cox test). The relationship between pre- and post-treatment KRAS mutation reads was assessed by a Mann–Whitney non-parametric test. The correlation between KRAS mutation reads as a percentage of total reads and months from last anti-EGFR treatment was analyzed by the non-parametric Kruskal–Wallis test. Comparisons were considered significant if the P value was ≤0.05. Statistical analyses were carried out using GraphPad Prism software, version 6.1 (GraphPad Software, Inc., La Jolla, CA).
results
Of the 70 patients studied, 62 had acquired resistance to anti-EGFR mAbs after an initial response, 8 had tumors known to carry KRASmut and were used as positive controls for KRAS mutation detection in the plasma. Among the 62 patients with anti-EGFR mAb resistance, 55 were treated with a cetuximab-based regimen, 4 with panitumumab-based regimen, and 3 received both. Median patient age was 60 years, and 74% of the patients had primary colon cancer, whereas 26% had tumors that arose in the rectum. Anti-EGFR mAbs were administered as first-line treatment in 4/62 (6%) patients, as second-line treatment in 30/62 (49%), as third-line treatment in 24/62 (39%), and as fourth-line treatment in 4/62 (6%). Analysis of specific EGFR and KRAS mutations was possible in all samples. KRAS mutational analysis carried out on ctDNA extracted from post-treatment plasma samples from the eight control patients known to harbor a mutation in KRAS confirmed the presence of such mutations detected at the time of diagnosis. No EGFR mutations were detected in any of these samples.
For the 62 patients initially characterized as having KRASwt tumors, acquired mutations in KRAS and EGFR following resistance to anti-EGFR therapies were seen in 27/62 cases [44%, 95% confidence interval (CI) of 30% to 56%] and 5/62 cases (8%, 95% CI of 2% to 19%), respectively. Although KRAS codon 12 and 13 mutations represented, as expected, the majority of the acquired mutations, KRAS mutations in codons 61 and 146 occurred more frequently in patients with acquired KRAS mutations after anti-EGFR mAb treatment than in treatment-naïve patients with KRAS mutations. Codon 61 and 146 mutations were identified in 9/27 (33%) and 3/27 (11%) samples compared with 9/240 (4%) and 5/240 (2%) samples, respectively, of untreated colorectal tumors classified as KRAS-mutant at baseline and analyzed by standard-of-care sequencing (P < 0.01 for comparison with acquired mutation frequencies, Figure 1). Newly detected EGFR S492R mutations were seen in five of the plasma samples from patients treated with cetuximab but in none of those treated with panitumumab. Interestingly, 4/5 patients with acquired EGFR mutations also had newly detected KRAS mutations suggesting that these mutations are not mutually exclusive. Multiple acquired KRAS codon mutations were simultaneously present in the ctDNA of 11/27 (41%) patients (supplementary Table S3).
Figure 1.
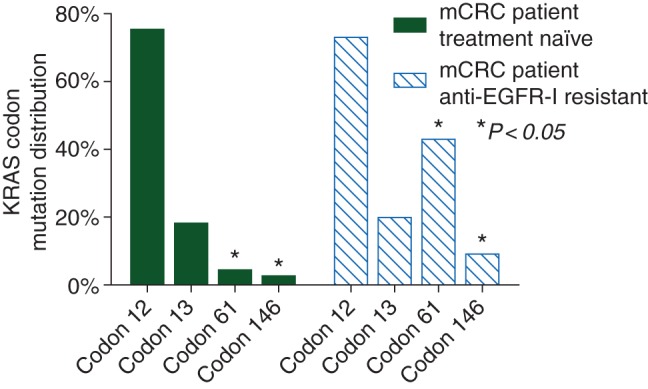
KRAS codon mutation distribution in treatment-naïve patients versus patients with acquired EGFR-inhibitor resistance (EGFR-I resistant). Atypical KRAS codon 61 and codon 146 mutations were more frequent in metastatic colorectal cancer patients with acquired KRAS mutations after anti-EGFR mAb treatment than in treatment-naïve patients with KRAS mutations (*P < 0.05) .
The presence of multiple clones carrying different genetic alterations and the dynamic clonal selection induced by anti-EGFR treatment pressure was confirmed by an analysis of serial samples from one of the patients (supplementary Figure S1). Analysis of KRAS and EGFR mutations in the patient's primary tumor archival sample using BEAMing confirmed KRASwt and EGFRwt status, which correlated with the patient's response to the initial anti-EGFR treatment. The blood samples collected after progression and before the time of anti-EGFR re-challenge within a clinical trial showed multiple newly detected KRAS and EGFR mutations. After retreatment with cetuximab, a sample demonstrated an increased percentage of KRAS mutant allele reads and continued acquisition of new KRAS and EGFR mutations.
The quantitative analysis of the plasma sample data showed that the acquired KRAS mutations occurring after progression on anti-EGFR therapy had a lower concentration of detectable mutant alleles than the KRAS mutants identified at diagnosis (Figure 2A, P < 0.001). These differences remained after controlling for metastatic tumor volume (Figure 2B).
Because it is possible that clones with acquired KRAS and EGFR mutations no longer have a growth advantage relative to other clones after discontinuation of the selective pressure, we evaluated the impact of time since the last cycle of anti-EGFR mAb treatment on the mutant/wild-type allele ratio for the most prevalent acquired allele in each patient. A significant inverse relationship between mutant allele levels and time since last treatment was observed (P = 0.038, Figure 3).
Figure 3.
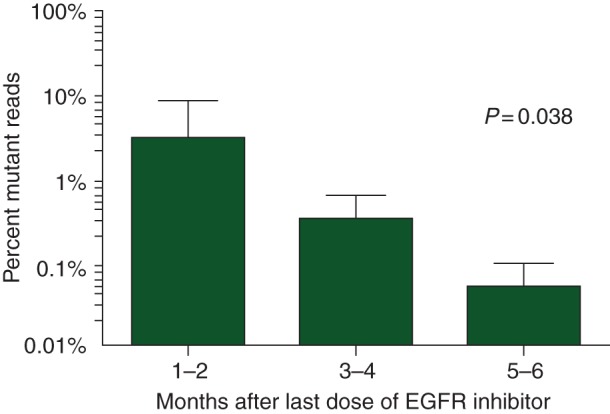
Time from last treatment with EGFR mAbs correlates with quantification of KRAS mutations detected in ctDNA. Time from last dose of EGFR mAbs correlated with the percentage of KRAS mutation reads detected in ctDNA from the plasma of metastatic colorectal cancer patients who developed resistance to treatment (P = 0.038).
To determine whether the newly detected KRAS and EGFR mutations were already present in the treatment-naïve primary tumors as undetectable low-frequency clones or were actual acquired mutations, we used BEAMing to re-analyze the archival samples from 20 of the 27 patients positive for newly detected KRAS mutations. KRAS mutations were detectable in 7 (35%) of the 20 primary tumors analyzed (95% CI of 0.15–0.69), whereas EGFR extracellular domain mutations were not detected in any samples [10] (supplementary Table S2). Although the sample size was small, patients with low-frequency mutations in their primary tumor samples had significantly worse PFS than patients with KRASwt primary tumors after BEAMing reanalysis of their archival tissue (median PFS: 3.0 versus 8.0 months, P = 0.0004, Figure 4).
Figure 4.

Low-frequency KRAS mutation expression in patient archival tissue and patient plasma samples correlate with shorter PFS with anti-EGFR treatment. (A) In 7of 20 patients with newly detected KRAS mutations in the plasma and wild-type KRAS by standard-of-care testing in the primary tumor, there were detectable low-frequency mutations present in microdissected tumor analyzed by BEAMing assay. (B) Patients with low-frequency KRAS mutations detected in archival primary tumor tissues had shorter PFS than patients with no detectable mutations by BEAMing (P = 0.0004).
discussion
Our findings demonstrate that KRAS mutations newly detected in the plasma of patients with colorectal cancer who had disease progression following treatment with anti-EGFR mAbs were already present as minor clones in more than a third (7/20) of the archival tissue collected at the time of diagnosis and that the pattern of genetic alterations in plasma and tissue samples was consistent.
In agreement with our findings, Diaz et al. [3] presented modeling data suggesting that acquired KRAS mutations were pre-existent in minor clones and expanded under selective pressure. Our data confirm this in a subset of patients by demonstrating the presence of clones missed by standard-of-care sequencing.
The pattern of genetic alterations in cancer patients is a dynamic model affected by several intrinsic and extrinsic factors including tumor genetic instability and treatment pressure. The presence of low-frequency KRAS mutations in the primary tumor correlated with a worse response to anti-EGFR treatment. Our findings confirm the data presented by Tougeron et al. [6] who demonstrated that low-frequency KRAS mutations, detected by pyrosequencing, were present in ∼20% of tumors previously characterized as KRASwt and that these mutations were associated with a shorter PFS. Our data extend this finding by demonstrating that these low-frequency mutations are indeed the dominant clone by allele frequency in ctDNA at the time of progression on anti-EGFR therapy.
Overall, these data support the hypothesis that utilizing a high-sensitivity mutation detection technique that provides quantitative information about the presence of mutant alleles in the gene of interest would allow for the selection of patients most likely to benefit from anti-EGFR treatment. However, it remains to be seen whether low-frequency KRAS-mutant clones in the primary tumor provide relative or absolute resistance to anti-EGFR mAb therapy; this question will require evaluation in randomized clinical trials. An additional limitation is that standard-of-care sequencing rarely utilizes microdissection, and it is possible that higher prevalence KRAS-mutant clones may not be detected in a stromal-rich primary tumor. This was seen in 4 of our 14 primary tumors, where microdissected tumor resulted in allele frequencies >10% that were missed by routine standard-of-care testing. Our study did not include other mutations that have subsequently been shown to be associated with EGFR resistance, including NRAS mutations and EGFR codon 491 [11]. Further efforts should utilize a larger panel of mutations to more fully characterize the spectrum molecular changes with acquired resistance.
In our study, newly detected KRAS mutations were present in multiple concomitant KRAS codons within the same patient in several cases, whereas EGFR extracellular domain mutations were confirmed in only a few patients after acquired resistance to cetuximab treatment. Multiple KRAS codon mutations are not usually seen in primary tumors because KRAS is considered an early mutation in tumor formation and is associated with a low degree of mutational heterogeneity [12]. KRAS mutations occurring in codons 61 and 146 are considered rare events in untreated colorectal cancers, and multiple studies have suggested that these biomarkers predict a lack of response to anti-EGFR therapies [13]. In contrast, tumors with codon 13 mutations have been suggested to retain sensitivity to EGFR inhibition in some, but not all, studies [14]. The acquisition of codon 13, 61, and 146 mutations in our study provides strong evidence that these mutations provide a selective growth advantage in the presence of EGFR inhibition; thus, our study provides orthogonal data suggesting that, similar to codon 12 mutations, these mutations are predictive biomarkers of anti-EGFR mAb resistance. It is also of interest that ‘atypical’ codon 61 and 146 KRAS mutations were expressed more frequently in acquired resistance mutations than in the general colorectal cancer population [15]. Although the mechanism for this finding is unknown, prior studies have demonstrated that codon 61 and 146 mutations have weaker RAS-GTPase activity in transforming assay, and therefore may have lower growth advantage compared with exon 2 mutations that may only expand once the tumor is placed under treatment pressure with an EGFR inhibitor [16].
The presence of an EGFR S492R mutation has been reported in one study to be a biomarker predictive of cetuximab resistance but panitumumab sensitivity, owing to the different binding sites for each of the drugs [2]. In our study, the EGFR S492R mutation was detected in 5/62 (8%) of the EGFR mAb-resistant plasma samples analyzed but in none of the archival tissues [10], and it should be noted that 4/5 patients with newly detectable EGFR mutations had concomitant KRAS mutations. Although the number of patient treated with panitumumab was very limited, in our study the EGFR S492R mutation seemed a more common feature of patients progressing on cetuximab, consistently with the ASPECCT trial data where acquired EGFR S492R mutation where detected in 16% of the patient treated with cetuximab and 1% of those treated with panitumumab [17]. Therefore, these EGFR mutations need to be better understood in the context of anti-EGFR treatment re-challenge.
It is notable that newly detected KRAS and EGFR mutations were present at a substantially lower level in ctDNA than pre-existing KRAS mutations, which suggests that the acquired KRAS mutations may not fully or permanently repopulate the entirety of the tumor, although direct quantification of the percent of the tumor repopulated with the mutant clone is not possible with the current assay. Indeed, given the appearance of multiple mechanisms of resistance in individual patients, it is possible that these mechanisms may co-exist with other previously reported mechanisms [11, 18, 19]. Additionally, recent reports have noted that paracrine mechanisms arising from resistant clones may propagate resistance through several proposed mechanisms. Hober et al. demonstrated that TGF-α and amphiregulin secreted from resistant cells induced resistance to EGFR inhibition while Troiani et al. identified TGF-α as an activator of the MET and EGFR axis, resulting in cetuximab resistance [20, 21]. In the absence of continued selective pressure from EGFR inhibition, the prevalence of these KRAS and EGFR mutant clones may decline, as suggested by the trend in mutant read frequency over time that was seen in our population. These data appear to be consistent with a recent report of clinical benefit seen with cetuximab retreatment in previously sensitive patients who were off of EGFR mAb for an extended interval, and suggest future opportunities for ctDNA-guided EGFR mAb rechallenge studies [22].
In conclusion, we confirm that low-frequency KRAS mutations detected by high-sensitivity assays are associated with reduced benefit from anti-EGFR mAb therapy and that these rare clones are the dominant clones identified in ctDNA at the time of progression. Analysis of ctDNA in the plasma provides opportunities to interrogate mechanisms of resistance and should be integrated into future prospective studies as a validation measure. Our study shows that, in contrast to the model of a single dominant clone, there are multiple dynamic genetic changes that may develop and lead to resistance in a given patient under treatment pressure [23]. This insight may lead to future efforts to better tailor anti-EGFR re-challenge clinical trials and therapeutic efforts to reverse resistance to EGFR inhibitors [24].
funding
This work was supported in part by an MD Anderson Cancer Center Institutional Research Grant; by the National Institutes of Health grants CA136980, Cancer Center Support Grant CA16670, and CA172670; by CPRIT RP110584 and 1R01CA172670.
disclosure
FD and PA have leading position at Sysmex Inostics. No conflict of interest for the other authors.
Supplementary Material
references
- 1.Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009; 360: 1408–1417. [DOI] [PubMed] [Google Scholar]
- 2.Montagut C, Dalmases A, Bellosillo B, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med 2012; 18: 221–223. [DOI] [PubMed] [Google Scholar]
- 3.Diaz LA, Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012; 486: 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012; 486: 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014; 6: 224ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tougeron D, Lecomte T, Pages JC, et al. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann Oncol 2013; 24: 1267–1273. [DOI] [PubMed] [Google Scholar]
- 7.Molinari F, Felicioni L, Buscarino M, et al. Increased detection sensitivity for KRAS mutations enhances the prediction of anti-EGFR monoclonal antibody resistance in metastatic colorectal cancer. Clin Cancer Res 2011; 17: 4901–4914. [DOI] [PubMed] [Google Scholar]
- 8.Nougaret S, Rouanet P, Molinari N, et al. MR volumetric measurement of low rectal cancer helps predict tumor response and outcome after combined chemotherapy and radiation therapy. Radiology 2012; 263: 409–418. [DOI] [PubMed] [Google Scholar]
- 9.Diehl F, Li M, He Y, et al. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods 2006; 3: 551–559. [DOI] [PubMed] [Google Scholar]
- 10.Esposito C, Rachiglio AM, La Porta ML, et al. The S492R EGFR ectodomain mutation is never detected in KRAS wild-type colorectal carcinoma before exposure to EGFR monoclonal antibodies. Cancer Biol Ther 2013; 14: 1143–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorich MJ, Wiese MD, Rowland A, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015; 26: 13–21. [DOI] [PubMed] [Google Scholar]
- 12.Santini D, Loupakis F, Vincenzi B, et al. High concordance of KRAS status between primary colorectal tumors and related metastatic sites: implications for clinical practice. Oncologist 2008; 13: 1270–1275. [DOI] [PubMed] [Google Scholar]
- 13.Tong JH, Lung RW, Sin FM, et al. Characterization of rare transforming KRAS mutations in sporadic colorectal cancer. Cancer Biol Ther 2014; 15: 768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Roock W, Jonker DJ, Di Nicolantonio F, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010; 304: 1812–1820. [DOI] [PubMed] [Google Scholar]
- 15.Imamura Y, Lochhead P, Yamauchi M, et al. Analyses of clinicopathological, molecular, and prognostic associations of KRAS codon 61 and codon 146 mutations in colorectal cancer: cohort study and literature review. Mol Cancer 2014; 13: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loupakis F, Ruzzo A, Cremolini C, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer 2009; 101: 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newhall K, Price T, Peeters M, et al. O-0011Frequency of S492R mutations in the epidermal growth factor receptor: analysis of plasma DNA from metastatic colorectal cancer patients treated with panitumumab or cetuximab monotherapy. Ann Oncol 2014; 25: ii109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 2013; 3: 658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobs B, De Roock W, Piessevaux H, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol 2009; 27: 5068–5074. [DOI] [PubMed] [Google Scholar]
- 20.Hobor S, Van Emburgh BO, Crowley E, et al. TGF-alpha and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clin Cancer Res 2014; 20: 6429–6438. [DOI] [PubMed] [Google Scholar]
- 21.Troiani T, Martinelli E, Napolitano S, et al. Increased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res 2013; 19: 6751–6765. [DOI] [PubMed] [Google Scholar]
- 22.Santini D, Vincenzi B, Addeo R, et al. Cetuximab rechallenge in metastatic colorectal cancer patients: how to come away from acquired resistance? Ann Oncol 2012; 23: 2313–2318. [DOI] [PubMed] [Google Scholar]
- 23.Ciardiello F, Normanno N, Maiello E, et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: findings from the CAPRI-GOIM trial. Ann Oncol 2014; 25: 1756–1761. [DOI] [PubMed] [Google Scholar]
- 24.De Mattos-Arruda L, Weigelt B, Cortes J, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol 2014; 25: 1729–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.