

Abstract
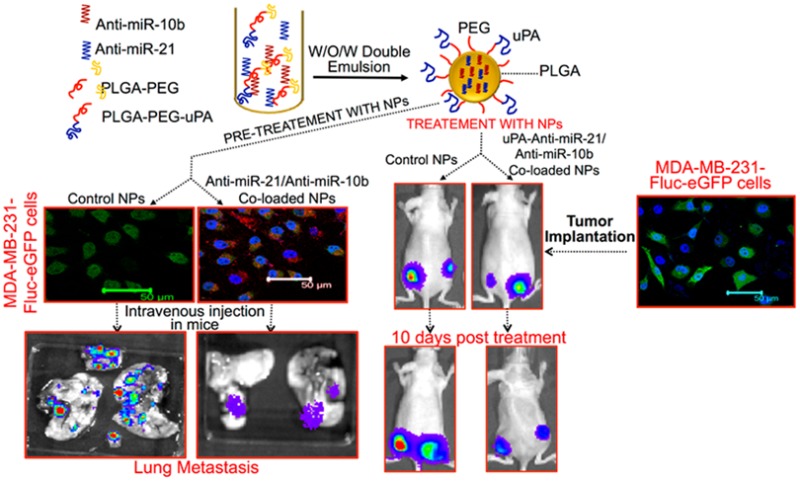
The current study shows the therapeutic outcome achieved in triple negative breast cancer (TNBC) by simultaneously antagonizing miR-21-induced antiapoptosis and miR-10b-induced metastasis, using antisense-miR-21-PS and antisense-miR-10b-PS delivered by polymer nanoparticles (NPs). We synthesized the antisense-miR-21 and antisense-miR-10b loaded PLGA-b-PEG polymer NPs and evaluated their cellular uptake, serum stability, release profile, and the subsequent synchronous blocking of endogenous miR-21 and miR-10b function in TNBC cells in culture, and tumor xenografts in living animals using molecular imaging. Results show that multitarget antagonization of endogenous miRNAs could be an efficient strategy for targeting metastasis and antiapoptosis in the treatment of metastatic cancer. Targeted delivery of antisense-miR-21 and antisense-miR-10b coloaded urokinase plasminogen activator receptor (uPAR) targeted polymer NPs treated mice showed substantial reduction in tumor growth at very low dose of 0.15 mg/kg, compared to the control NPs treated mice and 40% reduction in tumor growth compared to scramble peptide conjugated NPs treated mice, thus demonstrating a potential new therapeutic option for TNBC.
Keywords: microRNA, antisense-miRNAs, anti-miRs, cancer therapy, PLGA, nanoparticles, targeted delivery, in vivo molecular imaging, bioluminescence
Oncogenesis, in part, has been associated with dysregulated expression of various microRNAs (miRNAs or miRs).1−5 Inhibition or restoration of endogenous miRs is a unique and effective strategy to restore cellular homeostasis, and is considered to be a promising new generation of molecularly targeted anticancer therapeutics.6,7 Recent findings support targeting miRNAs as a potentially useful antimetastatic therapeutics strategy. The antisense-miRNAs or anti-miRNAs are a novel class of chemically modified oligonucleotides of 18–22 bases used to block the functions of endogenous miRNAs that regulate various genes involved in major cellular processes, such as cell proliferation, differentiation, and apoptosis.8,9 Antagonizing miR-10b by antisense-miR-10b has been reported to be effectively inhibited metastatic spread to the lungs, without affecting the growth of previously metastasized tumors in a mouse model of human breast cancer.10,11 Antisense-RNA mediated knockdown of endogenous miR-21 has been reported to impair tumor cell growth, induce apoptosis, and reduce the migration and invasion of cancer cells expressing miR-21 at high levels.12−18 We hypothesize that antagonizing multiple endogenous miRNAs could simultaneously affect target genes of different miRNAs and result in additive or enhanced therapeutic effect.
Despite improvement in the stability of sense- and antisense-miRNAs by modification of the nucleic acid structure, therapeutic sense- and antisense-miRNAs require effective delivery systems in order to become useful agents. Several delivery systems have been designed and developed to protect nucleic acids from degradation.19−27 Cell-penetrating peptide conjugated, anti-miR-155-loaded PLGA NPs effectively inhibited endogenous miR-155 function and significantly reduced the growth of leukemia and lymphoma (pre-B-cell) tumors in vivo in mice models at the dose of 1.5 mg/kg body weight.25 However, these NPs showed significantly lower antitumor efficacy when the dose was reduced down to 0.5 mg/kg body weight.25 Antisense-miR-10b-loaded polylysine NPs inhibited the invasive property of human breast cancer cells in vitro in cell culture for up to 4 weeks, as measured by the wound-healing assay.28 MiR-29b loaded cationic lipoplexes successfully delivered miR-29b to A549 nonsmall-cell lung carcinoma (NSCLC) cells and significantly inhibited tumor growth in animals.27 Even though most of these above-mentioned NPs were able to deliver sense- and antisense-miRNAs and achieve different levels of functional effect, specific delivery of miRNA-based therapeutics to tumor cells in vivo remains a challenge due to degradation of miRNAs by serum nucleases, off-target effects, poor cellular uptake, and rapid renal clearance after administration.29 In addition, poor miRNA loading efficiency is another major limiting factor, which necessitates the injection of higher percentage of carrier for miRNAs delivery that in turn causes carrier associated toxicity. Moreover, the miRNAs release profile from NPs and the stability of NP loaded miRNAs in serum at physiological conditions are other important phenomena lacking experimental support in the literature, but are essential to address for optimal in vivo delivery. To advance beyond current strategies, serum stable, tumor targeted, improved delivery systems with higher loading efficiency and sustained-release properties for miRNAs are much needed.
NPs synthesized from a synthetic polymer, PLGA, have been extensively studied for anticancer drugs delivery.22,30 Compared to most other types of delivery systems, PLGA has many advantages, such as (1) FDA approved for drug delivery in humans,31,32 (2) biodegradability, (3) sustained-release efficacy ranging from days to weeks in physiological conditions, (4) long-term stability of loaded bioactive molecules, (5) the ability to entrap hydrophobic and/or hydrophilic drugs, and (6) extensive functionalization options.22 PEG is another well-known water-soluble, biocompatible polymer extensively used for coating a wide variety of drugs to improve encapsulation efficiency, and circulation time and bioavailability in animals, that also protects NPs from immune surveillance.22,33 Here, we report an efficient delivery and antimetastatic and antitumor potentials of a specific combination of antisense-miRNAs or anti-miRNAs (antisense-miR-21 or anti-miR-21 and antisense-miR-10b or anti-miR-10b) utilizing NPs prepared from biodegradable PLGA-b-PEG copolymers and uPA peptide conjugated NPs targeting breast cancer specific uPA-receptor (uPAR) in triple negative breast cancer (TNBC) tumors.
Results and Discussion
Nanoparticle Preparation and Characterization
PLGA-b-PEG and uPA-peptide conjugated PLGA-b-PEG-NPs loaded with antisense-miRNAs were formulated using water-in-oil-in-water (w/o/w) double emulsion method (Supporting Information (SI) Scheme S1, Figures S1–S2). Two different emulsifiers are crucial for w/o/w multiple emulsion stabilization, one with a low hydrophile–lipophile balance (HLB) for the w/o interface, and the second one with a high HLB for the o/w interface. Tween 80 (HLB = 15) is often used in combination with Span 80 (HLB = 4.3) in multiple w/o/w emulsions because of similarity in their chemical structure.34 We used spermidine as a counterion35,36 for encapsulating various sense- and antisense-miRNAs (SI Table S1), lipophilic surfactant Span 80 for stabilizing the first emulsion (w/o), and hydrophilic surfactant Tween 80 for stabilizing the second emulsion (Figure 1A). Dynamic light scattering (DLS) of prepared NPs showed a size range of 100 to 200 nm (Figure 1B) with a polydispersity index (PDI) of 0.09–0.264. The zeta potential of NPs was in the range of −22 to −46 mV in ultrapure water (SI Tables S2–S3). The highly anionic nature of antisense-miRNAs should increase the negative zeta potential of NPs. As anticipated, the antisense-miRNAs loaded NPs showed much higher negative zeta potential compare to control NPs (SI Tables S2–S3). Morphology and size of NPs were further confirmed by transmission electron microscopy after staining with 1% phosphotungstic acid (Figure 1C). The entrapment efficiency of various antisense-miRNAs loaded in NPs was calculated using Quant-iT RNA Assay kit, as well as by optical CCD camera imaging based quantification for the coloaded Cy5-antisense-miR-21 after resolving the NPs by agarose gel electrophoresis (Figure 1D). The average number of antisense-miRNAs encapsulated in various NP formulations was estimated to be in the range of 400 to 1000 molecules/NP (SI Tables S2–S3). Moreover, we also evaluated the distribution of antisense-miR-21 and antisense-miR-10b in the coloaded nanoparticles by qRT-PCR analysis. The results indicate that coloading of antisense-miR-21 and antisense-miR-10b in PLGA-b-PEG-NPs is found almost at equimolar concentration in NPs prepared in different batches (SI Figure S3). The antisense-miRNAs extracted from the equimolar mixture of NPs formulated with each antisense-miRNAs (antisense-miR-21 and antisense-miR-10b) separately was used as control.
Figure 1.

Nanoparticle preparation and characterization. (A) Schematic illustration of nanoparticle formulation. (B) Hydrodynamic size of antisense-miRNA encapsulated PLGA-b-PEG NPs measured by dynamic light scattering (DLS). (C) TEM image of antisense-miRNA encapsulated PLGA-b-PEG NPs taken after staining with 1% phosphotungstic acid (scale bar, 100 nm). (D) Evaluation of coloaded Cy5-labeled antisense-miR-21 (10%) from the encapsulated PLGA-b-PEG NPs after being resolved in 3% agarose gel electrophoresis by optical CCD-camera imaging with the excitation of 570 nm and emission filter at 660 nm. (E,F) PLGA-b-PEG NPs loaded miRNA-21 release profile evaluated after seeding the coloaded NPs in PBS at physiological pH for 8 days of incubation at 37 °C by qRT-PCR analysis: (E) miR-21 fraction present in NPs different time points after incubation. (F) miR-21 fraction released from NPs different time points after incubation.
MicroRNA Stability and Release in Cell Culture Medium
Evaluation of in vitro stability and the release of loaded antisense-miRNAs in cell culture medium and in serum are important for predicting the potential of developed NPs for therapeutic applications. There is not many reports available in the literature except a single report where it describes the in vitro release profile and serum stability of miRNAs loaded in protamine sulfate-nanodiamond by a semiquantitative gel electrophoresis method.37 Since, no data available on the complete release profile and serum stability of miRNAs loaded in NPs using an absolute quantitation method, in this study, we evaluated the release profile of miR-21 loaded in PLGA-b-PEG-NPs (SI Tables S1–S2) using TaqMan-qRT-PCR. The results indicate that ∼4% of loaded miR-21 was released into the medium in the first 3 days, and the release was down to ∼1–2% at the later time points (Figure 1E,F). The reduction in the amount of miR-21 observed in the released fraction at later time points may be partially associated with the stability of released miR-21 in the medium. While the released fraction significantly dropped over time, the miR-21 fractions extracted from the NPs were significantly higher even 8 days after incubation (∼75% of initial spiked miR-21 was found in the NP-fraction) (Figure 1E,F). These results clearly indicate that miRNAs loaded in PLGA-b-PEG-NPs are stable in a normal growth medium for extended period of times.
MicroRNA Cellular Uptake and Serum Stability
Mitchell et al. reported that synthetic naked miRNAs are rapidly degraded in plasma compared to various endogenous miRNAs isolated from human samples.38 It could be possibly due to protein complexes associated with the endogenous microRNAs, which are in general lacking for the delivered synthetic therapeutic miRNAs. We tested the stability of PLGA-b-PEG-NPs encapsulated miR-21 delivered in MDA-MB-231 cells and spiked in mouse serum by TaqMan-qRT-PCR analysis (Figure 2). MDA-MB-231 cells were treated with miR-21 loaded PLGA-b-PEG-NPs for 24 h, and the total microRNAs extracted from the cells after a thorough wash to remove the extracellular NPs were quantitatively measured for the presence of intact miR-21 by qRT-PCR. The cells without any NP treatment, and treated with control NPs were used as controls. The results show that in cells treated with 10 and 50 pmols of miR-21-NPs, the intracellular miR-21 level was ∼836 ± 188 and ∼11322 ± 1486 fold higher, respectively, compared to controls (Figure 2A). The cells treated with control NPs showed a minor but not significant drop in the endogenous miR-21 level compared to untreated control.
Figure 2.

(A–G) Sense- and antisense-miRNA loaded PLGA-b-PEG NPs delivery in MDA-MB-231 cells, and the PLGA-b-PEG NPs loaded miRNAs stability in serum studied after incubation for various time points (0–48 h) at 37 °C by Taqman-qRT-PCR. (A) Evaluation of miR-21 levels in MDA-MB-231 cells delivered by control NP and PLGA-b-PEG NPs loaded with miR-21 in different concentrations (10 and 50 pmols) by qRT-PCR analysis. (B) Endogenous expression level of miR-21, miR-10b, and RNU66 in MDA-MB-231 cells (relative expression fold of various miRNAs compared to miR-10b). (C) Ct values measured for miRNAs expressions in (B) (Relative fluorescent intensity by Taqman probe). (D,E) Confocal fluorescent microscope images of MDA-MB-231-Fluc-eGFP cells treated with control-PLGA-b-PEG NPs and PLGA-b-PEG NPs coloaded with Cy5-antisense-miR-21 (0.5 nmols), antisense-miR-21 (9.5 nmols) and antisense-miR-10b (10 nmols), for 24 h at 37 °C. (F–H) Serum stability of naked miR-21 and miR-21 loaded in PLGA-b-PEG NPs evaluated at different time points after initial spiking (0, 12, 24, and 48 h). (F) Fluorescence intensity graph used for measuring Ct-values for serum spiked with naked miR-21. (G) Fluorescence intensity graph used for measuring Ct-values for serum spiked with miR-21 loaded PLGA-b-PEG NPs. (H) Relative miR-21 levels measured from serum spiked with naked and PLGA-b-PEG NPs loaded miR-21 over time.
To further evaluate the endogenous basal level expression of miR-10b and miR-21, the two therapeutic microRNA targets selected for the study, in MDA-MB-231 cells, the total microRNAs extracted from the cells were measured by qRT-PCR. The results demonstrate ∼943 ± 76 fold greater endogenous miR-21 expression in MDA-MB-231 cells, when compared to miR-10b. We used RNU66 as control miRNA, and the level of RNU66 was estimated to be ∼26.9 ± 2 fold higher compared to miR-10b level (Figure 2B,C). To track the cellular uptake and to monitor the clearance of the antisense-miRNAs in MDA-MB-231 cells, PLGA-b-PEG-NPs coloaded with Cy5-labeled-antisense-miR-21 (0.5 nmols), antisense-miR-21 (9.5 nmols) and antisense-miR-10b (10 nmols) combination was used. Confocal fluorescence microscope images of MDA-MB-231 cells treated with the coloaded NPs show a significant accumulation of NPs inside the cells (Figure 2D,E). The results found, the NPs were localized inside the cells in the periplasmic regions at the early time points (4 h), and, later, they accumulated in the cytoplasm (20 h) (SI Figures S5–S6). There was not much accumulation observed in the nucleus (SI Figures S4–S6). To test whether there is any endosomal involvement in the transport and accumulation of PLGA-b-PEG-NPs in cells, MDA-MB-231 cells were immunostained for EEA1, an early endosomal marker antibody, at different time points after exposure to antisense-miRNA coloaded NPs. The results show no colocalization of EEA1 staining with the Cy5-antisense-miR-21-NPs (SI Figure S7). Another batch of cells exposed to similar conditions was monitored at different time points for its cellular uptake, distribution and clearance for up to 120 h by fluorescent microscopy. The results show a constant increase in the NP accumulation over time up to 72 h, and the Cy5 signal started dropping slowly after 72 h (SI Figure S4). Dynamic uptake of Cy5-anti-miR-21 loaded PLGA-b-PEG-NP was studied in MDA-MB-231 cells stably expressing Fluc-eGFP reporter fusion protein using a time-lapse microscopic imaging system under a controlled (5% CO2 and 37 °C) incubation condition for 16 h, with snapshot images taken once every 10 min. The results show that the NP-uptake started as early as 30 min from the onset, and saturation was reached at around 6 h (SI Movie S1). However, we observed cell death after 16 h of incubation, which was mainly due to the instrument setup that was not optimal for prolonged incubation of cells. In another experiment, the MDA-MB-231 cells were incubated with various concentrations of PLGA-b-PEG-NPs loaded with Cy5-antisense-miR-21 for 24 h, and assessed the cellular uptake of NPs by FACS analysis (SI Figure S8A). The results show a concentration-dependent increase in the intracellular accumulation of NPs in the cells, with maximum fluorescence intensity at 25 nM. In addition, cells treated with NPs coencapsulated with Cy5-antisense-miR-21-antisense-miR10b showed similar fluorescence intensity, further confirming that the coencapsulation is not affecting the intracellular entry of Cy5-antisense-miR-21 (SI Figure S8B).
To further test the stability of miR-21 in serum, miR-21-PS loaded in PLGA-b-PEG-NPs or equal amount of free miR-21-PS was spiked into 100 μL of mouse serum and measured for intact miR-21 at 0, 12, 24, and 48 h after incubation at 37 °C. After specified incubation times, miRNA was extracted from the serum sample and analyzed for the presence of intact miR-21 by TaqMan-qRT-PCR assay. The results indicate that more than 95% of free miR-21 degraded within the first 24 h of incubation. Interestingly, more than 50% of miR-21 loaded in NPs was detected even 48 h after incubation (Figure 2F–H). These results indicate that the miR-21 loaded in PLGA-b-PEG NPs was well protected from the action of serum nucleases.
Cell Viability Evaluation in TNBC Cells Treated with Antisense-miRNAs Loaded PLGA-b-PEG-NPs
PLGA-b-PEG NPs loaded with and without antisense-miRNAs were evaluated for induced cytotoxicity in MDA-MB-231 TNBC cells by MTT assay. The concentration dependent toxicity analysis in MDA-MB-231 cells shows that polymer equivalent of up to 50 μg/mL of PLGA-b-PEG NPs in media could be used without significant toxic effect. Hence, in all other antisense-miRNA therapeutic studies, NPs at PLGA-b-PEG polymer concentrations below 50 μg/mL was used. The MDA-MB-231 cells treated with 0–25 pmols of antisense-miRNA loaded NPs were evaluated for cell viability 24 h after treatment. The results show no toxicity in cells treated with control NPs. In contrast, the cells treated with antisense-miRNA loaded NPs show dose dependent reduction in cell viability, with the highest one resulting from coloaded-NPs with antisense-miRNAs concentrations (antisense-miR-21 and antisense-miR-10b) of 25 pmols each (17 ± 2% less viability compared to control). The cells treated with 25 pmols of antisense-miR-21 and antisense-miR-10b loaded NPs show 15 ± 2% and 13 ± 1% reduction in cell viability, respectively (Figure 3A–D). Time dependent cell viability analysis indicate that there was significant growth reduction without much cell death (assessed by PI staining-FACS) observed in cells exposed to antisense-miR-21 loaded NPs, and antisense-miR-21 and antisense-miR-10b coloaded NPs, as compared to cells treated with control NPs or antisense-miR-10b loaded NPs (Figure 3E–H). We also compared the effect of antisense-miRNAs delivered by PLGA-b-PEG-NPs with liposome-mediated transfection in cells. The results indicate significant cytotoxicity (P < 0.05) in MDA-MB-231 and MCF7 cells when cells were transfected with a combination of antisense-miRNA-21 and antisense-miRNA-10b by liposome (SI Figure S9) compared to cells treated with PLGA-b-PEG-NPs coloaded with a combination of antisense-miR-21 and antisense-miR-10b (Figure 3A–H). Moreover, a significant reduction in cell viability was observed in cells cotransfected with antisense-miRNA-21 and antisense-miRNA-10b combination by liposome compared to control cells (P = 0.0158 [antisense-miR-21:28 ± 3% reduction in cell viability] vs 0.0108 [antisense-miR-21 + antisense-miR-10b: 32 ± 2% reduction in cell viability]).
Figure 3.

Cytotoxicity evaluation of various antisense-miRNAs loaded PLGA-b-PEG-NPs in MDA-MB-231-Fluc-eGFP cells by MTT assay. (A–D) Cells treated with 0 to 25 pmol/mL miRNA equivalent of control NPs, antisense-miR-21 and antisense-miR-10b individually-and coloaded NPs for 24 h and assessed for cytotoxicity by MTT assay. (E–H) Cells treated with 12.5 and 25 pmol/mL miRNA equivalent of control NPs, antisense-miR-21 and antisense-miR-10b individually-and coloaded NPs for various time points (24–72 h) and assessed for cytotoxicity by MTT assay. Error bars are SEM of three determinants (*p < 0.05).
In Vitro Cell Growth and Invasion Studies
Since miR-10b has been mainly associated with anti-invasive and antimetastatic properties, and miR-21 has been associated with antiapoptotic property in breast cancer cells,39,40 we evaluated the change in cells migration and invasive property after simultaneously antagonizing miR-21 and miR-10b in MDA-MB-231 cells by using the transwell-Matrigel cell migration and invasion assay. MDA-MB-231 cells stably expressing Fluc-eGFP reporter gene was used for this assay for easy visualization and quantitation of GFP positive cells in Matrigel. The cells were treated for 48 h with the respective NPs and assayed for cell migration in Matrigel transwell chamber. We found ∼23 ± 5% reduction in cell migration in cells treated with NPs loaded with antisense-miR-21, and antisense-miR-10b, and coloaded with antisense-miR-21 and antisense-miR-10b as compared to control NP treated cells (SI Figure S10A). We also found similar results when measured by wound healing assay (SI Figure S10B).
MicroRNA Functional Studies
To test bioactivity of antisense-miRNAs delivered by PLGA-b-PEG-NPs, we treated TNBC cells with 25 pmols of antisense-miR-21 and antisense-miR-10b either individually or coloaded NPs for 48 h and measured mRNA (mRNA) levels of target genes PTEN, PDCD4, and HoxD10 by RT-PCR analysis. The expressions were normalized to the house keeping control gene β-Actin. The results show upregulation of PDCD4 expression in cells treated with NPs loaded with antisense-miR-21 and coloaded with antisense-miR-21 and antisense-miR-10b combination. Similarly, upregulation of HoxD10 was observed in cells treated with NPs loaded with antisense-miR-10b and coloaded with antisense-miR-21 and antisense-miR-10b combination (Figure 4A; SI Table S4). The respective protein levels were measured by immunoblot analysis. An increase in the target protein expression was also observed in cells treated with respective antisense-miRNAs loaded NPs (Figure 4B). Interestingly, we observed that PTEN mRNA and protein levels increase when cells were treated with antisense-miR-10b. Similarly, confocal microscopic imaging of cells immunostained for miR-21 target PDCD4 showed higher level of PDCD4, with disintegrated nuclear structure in cells treated with NPs loaded with antisense-miR-21 and coloaded with antisense-miR-10b- and antisense-miR-21 combination, and not with control NPs or NPs loaded with antisense-miR-10b (Figure 4C). To further test the functional role of antisense-miRNAs in blocking the endogenous miRNAs, downregulation of miR-21-target genes maspin and PDCD4 were indirectly monitored in cells by expressing firefly luciferase reporter gene containing the 3′-UTR sequences derived from maspin or PDCD4 gene.41 We found a significant upregulation of luciferase signal in cells treated with NPs loaded with antisense-miR-21 and coloaded with antisense-miR-21 and antisense-miR-10b combination, as compared to cells treated with NPs loaded with the scrambled antisense-miRNA or antisense-miR-10b (SI Figure S11A–D).
Figure 4.

(A–C) Effect of antisense-miRNAs delivered by PLGA-b-PEG-NPs in blocking the function of endogenous miR-21 and miR-10b, and subsequent downstream regulation of target gene (miR-21: PTEN and PDCD4; miR-10b: HoxD10) expression in MDA-MB-231-Fluc-EGFP cells. (A) RT-PCR analysis for the expression of target genes of miR-21 and miR-10b (PTEN, PDCD4 and HoxD10) in MDA-MB 231 cells after delivering antisense-miRNAs by PLGA-b-PEG-NPs. (B) Immunoblot analysis for the expression of miR-21 and miR-10b (PTEN, PDCD4 and HoxD10) target proteins in MDA-MB-231 cells after treating with antisense-miRNAs delivered by PLGA-b-PEG-NPs (GAPDH as internal control). (C) Immunofluorescent staining for PDCD4 expression in MDA-MB 231 cells after treating with antisense-miRNAs delivered by PLGA-b-PEG-NPs. (D–G) Evaluation of metastatic properties of MDA-MB-231-Fluc-eGFP cells after treatment with control-NPs, and antisense-miR-21 and antisense-miR-10b coloaded NPs by bioluminescence imaging in mice. (D) Bioluminescence imaging of animals tail vein injected with MDA-MB-231-Fluc-eGFP cells after pretreatment by control-NPs, and NPs coloaded with antisense-miR-21 and antisense-miR-10b combination, for the identification of metastatic tumor growth. (E) Quantitation of bioluminescence signal in animals tail-vein injected with MDA-MB-231-Fluc-eGFP cells pretreated by control-NPs (red), and NPs coloaded with antisense-miR-21 and antisense-miR-10b combination (blue). Error bars are SEM of three determinants (* p < 0.05). (F) Ex-vivo bioluminescence imaging of lung tissues excised from the animals 6 weeks after the initial injection of MDA-MB-231-Fluc-eGFP cells pretreated by control-NPs (A1 and A2) and NPs coloaded with antisense-miR-21 and antisense-10b combination (A3 and A4). Each bioluminescent spot represents one metastatic tumor nodule. (G) H&E staining analysis of lung tissues (A2: section of lung tissue excised from animal injected with MDA-MB-231-Fluc-eGFP cells pretreated by control-NPs; A3: section of lung tissue excised from animal injected with MDA-MB-231-Fluc-eGFP cells pretreated by NPs coloaded with antisense-miR-21 and antisense-miR-10b combination).
Antitumor and Antimetastatic Properties of Antisense-miRNAs Delivered by PLGA-b-PEG-NPs in TNBC Tumor Xenograft in Living Animals
The growth, metabolic rates, and the metastatic properties of tumor cells, in response to the treatment of PLGA-b-PEG NPs loaded with and without antisense-miRNAs were measured in living mice by optical bioluminescence imaging. Subcutaneous tumor xenografts of MDA-MB-231-Fluc-eGFP cells were established on either side of the lower flanks of nude mice. One group of animals (n = 5) was implanted with cells pretreated for 48 h with control PLGA-b-PEG-NPs, and the second group (n = 5) was implanted with cells pretreated for 48 h with PLGA-b-PEG NPs coloaded with antisense-miR-21, Cy5-antisense-miR-21 and antisense-miR-10b combination. Animals were imaged every other day by optical CCD camera for bioluminescence signal and tumor volume measure using digital Vernier caliper. The results demonstrate that growth of tumors established by cells pretreated with NPs coloaded with antisense-miR-21 and antisense-miR-10b combination was significantly slower (∼35 ± 10%; p < 0.05) when compared to tumors implanted from cells pretreated with control NPs (SI Figure S12A,B). The aim of this part of the study was to test the sustained release of functional antisense-miRNAs by the delivered NPs. To confirm the sustained release and the presence of delivered antisense-miRNAs in cells, the animals were imaged by fluorescence imaging for the presence of the coloaded Cy5-antisense-miR-21. A significant amount of Cy5-signal was visualized in the tumors even 8 days after the implantation of tumor (SI Figure S13). Three weeks after repetitive imaging, the tumors were collected and analyzed ex vivo for tumor tissue architecture by H&E staining, and for tumor cell apoptosis by TUNEL assay. The results show a significant amount of apoptotic cells in the tumors of animals implanted with cells pretreated with NPs coloaded with the antisense-miR-21 and antisense-miR-10b combination, as compared to control NPs treated cells (SI Figure S14).
To confirm the downregulation of endogenous miR-21 and miR-10b function in tumor cells owing to the delivery of antisense-miR-21 and antisense-miR-10b coloaded NPs compared to control NPs, and to study their effect on regulation of tumor cell metastasis, 1 × 106 MDA-MB-231-Fluc-eGFP cells pretreated for 48 h either with control-NP or NPs coloaded with antisense-miR-21 and antisense-miR-10b combination were injected by tail-vein into 4 weeks old female nude mice. The animals were optically imaged for the expression of firefly luciferase for 6 weeks. The results indicate that the cells pretreated with NPs coloaded with antisense-miR-21 and antisense-miR-10b combination were less effective in triggering the development of metastatic tumors in the lungs and other organs, when compared to that of cells pretreated with control NPs (Figure 4D–F). After 6 weeks, lung tissues from the animals were collected and imaged ex vivo by optical bioluminescence imaging for metastatic tumor nodules. The results demonstrate ∼10-fold decrease in metastatic tumor nodules in animals injected with cells pretreated with NPs coloaded with antisense-miR-21 and antisense-miR-10b combination, compared to animals injected with cells pretreated with control NPs (Figure 4E). The lung tissues were histologically analyzed to identify tumor nodules by H&E staining. The results indicate that tumor nodules colocalized with regions emitting bioluminescence signals (Figure 4G).
Antitumor Effect of Systemically Injected PLGA-b-PEG NPs Coloaded with Antisense-miR-21 and Antisense-miR-10b Combination in Living Mice
After confirming the antitumor effect of antisense-miR-21 and antisense-miR-10b combinations in tumor xenografts of cells pretreated with PLGA-b-PEG coloaded NPs in living animals, we further tested the effect of the same NPs when systemically injected in animals bearing MDA-MB-231-Fluc-eGFP tumor xenografts. Subcutaneous tumor xenografts of MDA-MB-231-Fluc-eGFP cells were established on either side of the lower flanks of nude mice (Nu/Nu, n = 10). After 15 days, when the tumors size reached 100–300 cubic mm, the first group of mice (n =10, 5 animals bearing two tumors each) were intravenously injected with saline (200 μL) and the second set of mice (n = 10, 5 animals bearing two tumors each) were injected with PLGA-b-PEG-NPs coloaded with antisense-miR-21 and antisense-miR-10b combination on Day 0, Day 3, and Day 6. The tumors growth was monitored by measuring the tumor volume and by bioluminescence imaging over time (SI Figure S15). The results indicate that in both groups the tumor growth showed no significant difference from Day 0 through Day 4. However, starting from Day 6 onward, in the saline treated group the tumor growth rapidly increased compared to animals treated with antisense-miRNAs coloaded NPs. The bioluminescence imaging signals measured from these animals correlated with the tumor volumes (SI Figure S15A–C).
Antitumor Effect of Systemically Delivered Urokinase Plasminogen Activator Receptor (uPAR) Targeted PLGA-b-PEG-NPs Coloaded with Antisense-miR-21 and Antisense-miR-10b Combination in Living Animals
In this section we further evaluated the therapeutic effect of systemically injected PLGA-b-PEG-NPs coloaded with antisense-miRNAs in TNBC tumor xenografts in nude mice. We used uPA peptide conjugated PLGA-b-PEG-NPs targeting tumor specific uPAR receptor to efficiently deliver antisense-miRNAs at increased amounts, in addition to NPs delivered to tumors through inherent enhanced permeabilization and retention (EPR) effect. We used PLGA-b-PEG-NPs conjugated with scrambled (sc or scrb)-uPA peptide and coloaded with antisense-miR-21 and antisense-miR-10b combination as matched control. The PLGA-b-PEG copolymers were conjugated with uPA and scrambled-uPA peptides using standard chemistry (SI Scheme S1), and the conjugation was confirmed by 1H NMR and MALDI-TOF mass spectrometry (SI Figures S1–S2). The NPs prepared using PLGA-b-PEG copolymers with uPA and Scrambled-uPA were loaded with antisense-miR-21 and antisense-miR-10b individually and in combinations (SI Table S3). The NPs were first tested for the loaded antisense-miRNAs delivery and functional effect in MDA-MB-231 cells in culture by treating with 25 pmols of each antisense-miRNAs. The results show significant antiproliferative effect in cells treated with uPA and Scrb-uPA conjugated PLGA-b-PEG-NPs coloaded with antisense-miR-21 and antisense-miR-10b combinations compared to cells treated with other NPs (SI Figure S16). The cells treated at similar conditions for 36 h were used for evaluating the functional effect of delivered antisense-miRNAs on target genes expression (PDCD4 and HoxD10) by TaqMan qRT-PCR. The results indicate that the mRNA level of miR-21 target PDCD4 increased when cells were treated by NPs loaded with antisense-miR-21 (uPA-antisense-miR-21, uPA-antisense-miR-21-antisense-miR-10b and Sc-uPA-antisense-miR-21-antisense-miR-10b), compared to control and NPs loaded with antisense-miR-10b (SI Figure S17A). In contrast, HoxD10, the target of miR-10b, expression increased in cells treated by NPs loaded with antisense-miR-10b (uPA-antisense-miR-10b, uPA-antisense-miR-21-antisense-miR-10b and Sc-uPA-antisense-miR-21-antisense-miR-10b) (SI Figure S17B). Since these in vitro results confirmed the functional efficiency of the synthesized targeted NPs, we used them for antitumor studies in animals for systemic delivery experiments.
The antitumor effect of systemically injected uPAR targeted NPs loaded with different combination of antisense-miRNAs was studied in tumor xenografts of MDA-MB-231 cells stably expressing Fluc-eGFP reporter fusion gene. The targeted delivery of NPs loaded with antisense-miR-21 and antisense-miR-10b, and coloaded with antisense-miR-21 and antisense-miR-10b combination, injected at 0.15 mg/kg, was measured for tumor size and bioluminescence imaging signal over time. The scrambled-uPA-peptide conjugated PLGA-b-PEG-NPs coloaded with antisense-miR-21 and antisense-miR-10b, and control PLGA-b-PEG-NPs injected animals served as controls. The mouse TNBC tumor xenograft model was developed by injecting 0.1 mL of MDA-MB-231-Fluc-eGFP cell suspension (10 × 106) into the left and right flanks of female nude mice (Nu/Nu) using 50% (v/v) Matrigel. After 15 days, when the tumors size reached 100–300 cubic mm, the animals [n = 25, 5 animals with two tumors each (n = 10 for each treatment group) for each treatment group] were intravenously injected with the NPs on Day 0, Day 3, and Day 6 (Figure 5A). The tumor development and growth was monitored over time (up to Day 9) by measuring tumor volume and bioluminescence imaging. After the completion of imaging on Day 9, the tumors were excised for ex vivo histological analysis. The results indicate that the tumor growth of animals treated by NPs loaded with both antisense-miR-10b and antisense-miR-21 individually and in combinations showed significant reduction in tumor growth, compared to control-NPs treated animals, with highest effect being detected in animals treated by uPA–PLGA-b-PEG-NPs coloaded with antisense-miR-10b and antisense-miR-21 combinations. The antisense-miR-21-10b coloaded-uPA NPs treated mice shows 40% more tumor reduction compared to antisense-miR-21-10b coloaded NPs with scrambled-uPA, signifying the uPA targeting effects on MDA-MB-231 tumors (Figure 5A). The bioluminescence imaging signal also showed similar level of signal drop in animals treated with antisense-miRNAs loaded NPs compared to control-NPs treated group (Figure 5A-C). The ex vivo analysis of kidney and liver tissues showed no sign of toxicity in animals treated by NPs with or without antisense-miRNAs (SI Figure S18A–C). The ex vivo TUNEL staining of tumor tissues showed significant amount of apoptotic cells in animals treated with antisense-miRNAs loaded NPs, with the highest level being detected in animals treated with uPA-targeted NPs coloaded with both antisense-miR-10b and antisense-miR-21 combination (Figure 5D, SI Figure S18D).
Figure 5.

In vivo tumor growth analysis and bioluminescence imaging of mice (n = 25) bearing MDA-MB-231 tumors stably expressing Fluc-eGFP that are treated with antisense-miR-21 and antisense-miR-10b loaded or coloaded with uPA–PLGA-b-PEG and Sc-uPA–PLGA-b-PEG NPs. (A) Tumors growth volume (mm3) measured in different treatment groups over time. (B) Optical bioluminescence images of animals (n = 10, 5 animals bearing two tumors each for each treatment group) treated with different NPs over time. (C) Quantitative graph showing the bioluminescence signals quantitated from animals shown in (B). (D) TUNEL staining of tumor tissues of animals treated with different NPs.
In this study we successfully developed and optimized protocols for encapsulating antisense-miRNAs in PLGA-b-PEG-NPs with higher loading efficiency compared to previously reported methods.22,35,42 The resulted NPs can be potentially used for achieving prolonged therapeutic efficiency with lower dose with minimum off target toxicity. The antisense-miRNA targets selected in this study for inhibitions are the widely studied miR-21 and miR-10b. The role of miR-2112−18,43 and miR-10b10,11,39,43 in tumor growth, invasion, metastasis, and drug resistance in breast cancer is well-documented.11,44 However, the utility of antisense-miRNAs in therapeutic intervention is largely dependent on effective delivery and sustained efficacy of the internalized antisense-miRNAs following treatment. Systemically delivered miRNAs require an efficient system of ensuring intracellular transport, specificity, and metabolic stability to succeed as therapeutic options.
The synthesized PLGA-b-PEG-NPs in cell viability studies indicate an absence of significant toxic effects at relatively high concentrations of NPs. Simultaneous codelivery of antisense-miR-21 and antisense-miR-10b combinations in cell migration, metastasis, and invasion assays show a significant and incremental decrease in motility and invasiveness in vitro, and also indicates a cumulative effect in reducing TNBC xenograft tumor growth in animals. The sustained release of antisense-miR-21 and antisense-miR-10b from delivered NPs over 15 days achieved an approximately 40% reduction in tumor growth in animal model, and a mouse metastasis model indicated a significant decrease in lung and organ metastasis following treatment.
Targeted delivery of drugs and small RNAs loaded in NPs can overcome problems displayed by naked small RNAs and free drugs, comprising poor serum stability, lack of selectivity, and rapid renal clearance after administration, which leads to nonspecific toxicity to normal cells and constrains adequate dose administration to eliminate the cancer cells.21 The uPAR has been recognized to be involved in multiple steps in cancer progression.45 We synthesized the uPAR targeting peptide conjugated, antisense-miR-21 and antisense-miR-10b coloaded, PLGA-b-PEG-NPs. We envisioned that PEGylation of PLGA would increase the circulation time of NPs, owing to EPR effect in mice and uPA peptide targeting the tumor cells. Our results indicated that the uPA peptide conjugated NPs efficiently delivered the antisense-miRNAs and achieved highest tumor reduction in animals where NPs codelivered antisense-miR-21 and antisense-miR-10b combination, whereas scrambled NPs treated mice showed 40% less tumor reduction compared to uPAR targeting NPs. We consider that significant tumor reduction in animals treated with scrb-NPs is because of the EPR effect of PEG group in the NPs.
Conclusion
In summary, we have successfully developed an efficient protocol for coloading antisense-miRNAs in PLGA-b-PEG-NPs. The simultaneous delivery of antisense-miR-21 and antisense-miR-10b has a cumulative effect in reducing breast cancer cell proliferation in vitro in culture and in vivo tumor xenografts. The sustained release of antisense-miR-21 and antisense-miR-10b from NPs over 15 days achieved a 40% reduction in tumor growth in animal model. These results suggest that antagonizing multiple miRNA activities could be an efficient and enhanced therapeutic strategy to block metastatic properties of tumor cells, in addition to inducing apoptosis, in cancer therapy. Targeted delivery of NPs showed a substantial reduction in tumor growth at very low dose of antisense-miRNAs (0.15 mg/kg), compared to the control NPs, and 40% reduction in tumor growth compared to scrambled peptide conjugated NPs treated mice, suggesting a potential therapeutic strategy for TNBC tumors. This strategy can be combined with other therapeutic drugs to achieve improvement in chemotherapy at lower doses of drugs. Further studies are in progress to study the effects of therapeutic drugs and miRNAs coloaded NPs in breast tumors.
Materials and Methods
Materials
All chemical reagents used for the study were of analytical grade or above. Poly(d,l-lactide-co-glycolide) (50/50) with acid terminated (PLGA, inherent viscosity 0.16–0.24 dL/g, MW 7000–17 000), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), 4-dimethylaminopyridine (DMAP) and diisopropylethylamine (DIPEA) were obtained from Sigma-Aldrich (St. Louis, MO, USA). NH2-PEG-COOH (MW 3400) was purchased from JenKem Technology, USA (Allen, TX) and NOF Corporation, USA (Irvine, CA). Antisense-miRNAs were custom synthesized by PAN Facility at Stanford, at purity above 90% (SI Table S1 for sequences of miRs and antisense-miRNAs with various modifications). Urokinase plasminogen activator receptor (uPAR) targeted uPA-peptide (VSNKYFSNIHWGC) and scrambled (sc or scrb) peptide (SGLITPGILPIHGCSF) were synthesized using solid phase peptide synthesizer (CS Bio Company Inc., Menlo Park, California).
Synthesis of PLGA-b-PEG-COOH Polymer
To activate acid terminated PLGA, a solution of PLGA (500 mg) in dry dichloromethane (DCM) (5 mL), N-hydroxysuccinimide (NHS) (80 mg) and 1-ethyl-3-[3-(dimethylamino)propyl] carbodiimide (EDC) (48 mg) were combined at room temperature and stirred for 4 h. The reaction mixture was then added dropwise into cold diethyl ether/MeOH (1:1) (40 mL), and the resultant precipitate was centrifuged (5000 rpm for 5 min), the supernatant was decanted, and the resulted pellet was further washed (three times). The precipitate of PLGA-NHS was dried at high vacuum overnight. The PLGA-NHS (360 mg) and HCl-NH2-PEG-COOH (102 mg) were dissolved in chloroform (5 mL), and diisopropylethylamine (DIPEA) (37 μL) was added at room temperature and stirred for 20 h. The reaction mixture was then added dropwise into cold diethyl ether/MeOH (1:1) (40 mL), and the resultant precipitate was centrifuged (5000 rpm for 5 min), the supernatant was decanted, and the pellet was further washed (three times) and dried under high vacuum afforded the PLGA-b-PEG-COOH (342 mg). The efficiency of the coupling reaction was confirmed by 1H NMR (Agilent 400 MHz NMR, CDCl3) (SI Figure S1).46
Synthesis of uPAR-Targeted Peptide (uPA) and Scrambled Peptide Conjugated PLGA-b-PEG-COOH Polymers
Peptide conjugation was achieved using Steglich esterification reaction conditions with slight modifications. A solution of synthesized PLGA-b-PEG-COOH copolymer (1 equiv) in dry DMF and targeted peptide sequence (VSNKYFSNIHWGC) (1.5 equiv) in dry DMF (1.5 mL), 4-dimethylaminopyridine (2 equiv) were combined at RT. The reaction mixture was cooled to 0 °C using ice base, and 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide (EDC) (10 equiv) in dry DMF was added to it dropwise and stirred for 10 min. The ice base was removed, and the reaction mixture was stirred at RT for 6 h. The reaction mixture was then added dropwise into cold diethyl ether/MeOH (1:1) (10 mL), and the resultant precipitate was centrifuged (5000 rpm for 5 min), the supernatant was decanted, and the pellet was further washed (three times) to remove the excess free peptide, EDC, dried under a high vacuum, and afforded the PLGA-b-PEG-uPA peptide (88% yield). The efficiency of the peptide conjugation was confirmed by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF-MS) mass spectrometry using AB Sciex 5800 TOF/TOF System (SI Figure S2). The scramble peptide sequence (SGLITPGILPIHGCSF) conjugation to PLGA-b-PEG-COOH was achieved using similar procedure.
Formulation of Antisense-miRNAs Loaded PLGA-b-PEG and Peptide Conjugated PLGA-b-PEG NPs
PLGA-b-PEG nanoparticles loaded with antisense-miRNAs/spermidine were formulated by double emulsion solvent evaporation technique with minor modifications.35,36 In all coencapsulations 95% antisense-miR-21 with 5% Cy5-antisense-miR-21 was used to maintain the consistency in the formulation across different experiments, and 20% peptide conjugated PLGA-b-PEG and 80% PLGA-b-PEG polymers were used for targeted peptide conjugated nanoparticle synthesis. Antisense-miRNAs (10 nmol) were complexed with spermidine in an N/P ratio of 15:1 at room temperature for 15 min in DNase/RNAase free water.35,36 The antisense-miRNA-spermidine complex was added dropwise to the stirred solution of PLGA-b-PEG polymer (10 mg) in dichloromethane (1 mL) containing 3% Span-80, followed by sonication for 60 s at 40% amplitude in ice bath to form the first emulsion. To this primary emulsion, 5 mL of emulsifying water (Tween-80, 1% w/v) was added and sonicated at 40% amplitude in ice bath for 60 s to form second emulsion, followed by stirring for 3 h to allow the dichloromethane to evaporate. The NPs were sterile filtered using 0.45 μm syringe filter (Whatman PURADISC 25 AS, PES, GE Healthcare), and excess surfactants and free antisense-miRNAs were removed by an ultracentrifuge filter device with 100 kDa MWCO Membrane (Millipore, USA) by centrifuging at 3000 rpm. The concentrated nanoparticles were washed and centrifuged several times with DNase/RNAase free water (Invitrogen, USA). The concentrated nanoparticles were diluted to a known volume, estimated for their particle size, Zeta potential and the loaded antisense-miRNAs level, prior to use in different experiments.
In Vivo Tumor Growth Studies with PLGA-b-PEG-uPA and Scrambled-uPA NPs
The mouse TNBC xenograft tumor model was developed by injecting 0.1 mL of MDA-MB-231-Fluc-eGFP cell suspension (10 × 106) into the left and right flank of a female nude mice (nu/nu) using 50% (v/v) Matrigel. After 15 days, when the tumors grew to 100–300 cubic mm, the animals were randomized and selected for various treatment groups (1: Control-NP, 2: uPA-antisense-miR-21-NP, 3: uPA-antisense-miR-10b-NP, 4: uPA-antisense-miR-21 and antisense-mioR-10b-NP and 5: Scrb-uPA-antisense-miR-21 and antisense-miR-10b-NP). In each group, 5-animals (total 25 animals) were chosen for treatment. Prior to NPs administration, animals were anesthetized and injected with 100 μL of d-luciferin (30 mg/mL in PBS) via I.P., and assessed by IVIS-Spectrum Imaging system for bioluminescence from tumors. At every time point, tumor volumes were measured and imaged using IVIS Spectrum; after imaging, animals were intravenously administered with control NPs, different antisense-miRNAs loaded NPs on Day 0, Day 3, and Day 6. All animals were also imaged on Day 1, 4, 7, and 9. On Day 9, all animals were sacrificed and tumors were collected for ex vivo histological analysis.
Acknowledgments
This work was partially supported by the National Institutes of Health (NIH Grant R01 CA161091) to R. Paulmurugan. The authors acknowledge the use of the SCi3 Core Facility. The authors thank Y. Mo, South Illinois University School of Medicine, Springfield, Illinois, for Luciferase constructs with miR-21 targets from Maspin and PDCD4, S. Ramakrishnan for technical assistance, and S.S. Gambhir, Chairman, Department of Radiology, Stanford University, for his constant support. We also thank Canary center at Stanford University for facilities and support. R. Devulapally synthesized and characterized uPAR targeted peptide, PLGA-b-PEG-copolymer and uPAR targeted peptide conjugated polymer NPs. R. Devulapally, N. M. Sekar synthesized the nontargeted NPs. T. V. Sekar performed Western blot analysis. K. Foygel performed histology studies. R. Devulapally performed miRNAs release, dynamic uptake and serum stability experiments. R. Devulapally, R. Paulmurugan performed qRT-PCR studies. R. Devulapally, T. V. Sekar performed in vivo nontargeted delivery and uPAR-targeted delivery studies in mice. N. M. Sekar., T. V. Sekar performed in vivo studies in mice with pretreated cells. R. Devulapally, R. Paulmurugan designed the experiments, analyzed the data and wrote the manuscript, which was reviewed by all authors. T. F. Massoud, J. K. Willmann helped with experimental design and manuscript writing. R. Paulmurugan directed the study.
Supporting Information Available
Supplementary Scheme S1, Tables S1–S4, Figures S1–S18, Movie S1 and Methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Contributions
† R.D., N.M.S., and T.V.S. contributed equally to this work.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Akao Y.; Nakagawa Y.; Hirata I.; Iio A.; Itoh T.; Kojima K.; Nakashima R.; Kitade Y.; Naoe T. Role of Anti-Oncomirs MiR-143 and -145 in Human Colorectal Tumors. Cancer Gene Ther. 2010, 17, 398–408. [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A.; Slack F. J. Oncomirs—MicroRNAs with a Role in Cancer. Nat. Rev. Cancer 2006, 6, 259–269. [DOI] [PubMed] [Google Scholar]
- Kitade Y.; Akao Y. MicroRNAs and Their Therapeutic Potential for Human Diseases: MicroRNAs, MiR-143 and -145, Function as Anti-Oncomirs and the Application of Chemically Modified MiR-143 as an Anti-Cancer Drug. J. Pharmacol Sci. 2010, 114, 276–280. [DOI] [PubMed] [Google Scholar]
- Krutovskikh V. A.; Herceg Z. Oncogenic MicroRNAs (Oncomirs) as a New Class of Cancer Biomarkers. Bioessays 2010, 32, 894–904. [DOI] [PubMed] [Google Scholar]
- Reshmi G.; Pillai M. R. Beyond HPV: Oncomirs as New Players in Cervical Cancer. FEBS Lett. 2008, 582, 4113–4116. [DOI] [PubMed] [Google Scholar]
- Kota J.; Chivukula R. R.; O’Donnell K. A.; Wentzel E. A.; Montgomery C. L.; Hwang H. W.; Chang T. C.; Vivekanandan P.; Torbenson M.; Clark K. R.; et al. Therapeutic MicroRNA Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model. Cell 2009, 137, 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Kelnar K.; Liu B.; Chen X.; Calhoun-Davis T.; Li H.; Patrawala L.; Yan H.; Jeter C.; Honorio S.; et al. The MicroRNA MiR-34a Inhibits Prostate Cancer Stem Cells and Metastasis by Directly Repressing CD44. Nat. Med. 2011, 17, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutzfeldt J.; Rajewsky N.; Braich R.; Rajeev K. G.; Tuschl T.; Manoharan M.; Stoffel M. Silencing of MicroRNAs In Vivo with ‘Antagomirs’. Nature 2005, 438, 685–689. [DOI] [PubMed] [Google Scholar]
- Ross J. S.; Carlson J. A.; Brock G. Mirna: The New Gene Silencer. Am. J. Clin Pathol 2007, 128, 830–836. [DOI] [PubMed] [Google Scholar]
- De Palma M.; Naldini L. Antagonizing Metastasis. Nat. Biotechnol. 2010, 28, 331–332. [DOI] [PubMed] [Google Scholar]
- Ma L.; Reinhardt F.; Pan E.; Soutschek J.; Bhat B.; Marcusson E. G.; Teruya-Feldstein J.; Bell G. W.; Weinberg R. A. Therapeutic Silencing of MiR-10b Inhibits Metastasis in a Mouse Mammary Tumor Model. Nat. Biotechnol. 2010, 28, 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani I. A.; Rasheed S. A.; Nikolova D. A.; Leupold J. H.; Colburn N. H.; Post S.; Allgayer H. MicroRNA-21 (MiR-21) Post-Transcriptionally Downregulates Tumor Suppressor Pdcd4 and Stimulates Invasion, Intravasation and Metastasis in Colorectal Cancer. Oncogene 2008, 27, 2128–2136. [DOI] [PubMed] [Google Scholar]
- Chan J. A.; Krichevsky A. M.; Kosik K. S. MicroRNA-21 is an Antiapoptotic Factor in Human Glioblastoma Cells. Cancer Res. 2005, 65, 6029–6033. [DOI] [PubMed] [Google Scholar]
- Cheng A. M.; Byrom M. W.; Shelton J.; Ford L. P. Antisense Inhibition of Human MiRNAs and Indications for an Involvement of MiRNA in Cell Growth and Apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffler D.; Brocke-Heidrich K.; Pfeifer G.; Stocsits C.; Hackermuller J.; Kretzschmar A. K.; Burger R.; Gramatzki M.; Blumert C.; Bauer K.; et al. Interleukin-6 Dependent Survival of Multiple Myeloma Cells Involves the Stat3-Mediated Induction of MicroRNA-21 through a Highly Conserved Enhancer. Blood 2007, 110, 1330–1333. [DOI] [PubMed] [Google Scholar]
- Papagiannakopoulos T.; Shapiro A.; Kosik K. S. MicroRNA-21 Targets a Network of Key Tumor-Suppressive Pathways in Glioblastoma Cells. Cancer Res. 2008, 68, 8164–8172. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Li Z.; Gao C.; Chen P.; Chen J.; Liu W.; Xiao S.; Lu H. MiR-21 Plays a Pivotal Role in Gastric Cancer Pathogenesis and Progression. Lab. Invest. 2008, 88, 1358–1366. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Wu H.; Wu F.; Nie D.; Sheng S.; Mo Y. Y. MicroRNA-21 Targets Tumor Suppressor Genes in Invasion and Metastasis. Cell Res. 2008, 18, 350–359. [DOI] [PubMed] [Google Scholar]
- Su J.; Baigude H.; McCarroll J.; Rana T. M. Silencing MicroRNA by Interfering Nanoparticles in Mice. Nucleic Acids Res. 2011, 39, e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Schwind S.; Yu B.; Santhanam R.; Wang H.; Hoellerbauer P.; Mims A.; Klisovic R.; Walker A. R.; Chan K. K.; et al. Targeted Delivery of MicroRNA-29b by Transferrin-Conjugated Anionic Lipopolyplex Nanoparticles: A Novel Therapeutic Strategy in Acute Myeloid Leukemia. Clin. Cancer Res. 2013, 19, 2355–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley C. E.; Carnes E. C.; Phillips G. K.; Padilla D.; Durfee P. N.; Brown P. A.; Hanna T. N.; Liu J. W.; Phillips B.; Carter M. B.; et al. The Targeted Delivery of Multicomponent Cargos to Cancer Cells by Nanoporous Particle-Supported Lipid Bilayers. Nat. Mater. 2011, 10, 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devulapally R.; Paulmurugan R. Polymer Nanoparticles for Drug and Small Silencing RNA Delivery to Treat Cancers of Different Phenotypes. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2014, 6, 40–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeylath S. C.; Ganta S.; Iyer A. K.; Amiji M. Combinatorial-Designed Multifunctional Polymeric Nanosystems for Tumor-Targeted Therapeutic Delivery. Acc. Chem. Res. 2011, 44, 1009–1017. [DOI] [PubMed] [Google Scholar]
- Fattal E.; Barratt G. Nanotechnologies and Controlled Release Systems for the Delivery of Antisense Oligonucleotides and Small Interfering RNA. Br. J. Pharmacol. 2009, 157, 179–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babar I. A.; Cheng C. J.; Booth C. J.; Liang X.; Weidhaas J. B.; Saltzman W. M.; Slack F. J. Nanoparticle-Based Therapy in an In Vivo MicroRNA-155 (miR-155)-Dependent Mouse Model of Lymphoma. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E1695–E1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y.; Kang C. S.; Yuan X. B.; Zhou X.; Xu P.; Han L.; Wang G. X.; Jia Z.; Zhong Y.; Yu S.; et al. Co-Delivery of As-miR-21 and 5-FU by Poly(amidoamine) Dendrimer Attenuates Human Glioma Cell Growth In Vitro. J. Biomater. Sci., Polym. Ed. 2010, 21, 303–314. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Crawford M.; Mao Y.; Lee R. J.; Davis I. C.; Elton T. S.; Lee L. J.; Nana-Sinkam S. P. Therapeutic Delivery of MicroRNA-29b by Cationic Lipoplexes for Lung Cancer. Mol. Ther.—Nucleic Acids 2013, 2, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H.; Yu Y.; Chrisler W. B.; Xiong Y.; Hu D.; Lei C. Delivery of MicroRNA-10b with Polylysine Nanoparticles for Inhibition of Breast Cancer Cell Wound Healing. Breast Cancer 2012, 6, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot C. V.; Calin G. A.; Coleman R. L.; Lopez-Berestein G.; Sood A. K. RNA Interference in the Clinic: Challenges and Future Directions. Nat. Rev. Cancer 2011, 11, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Fernandez A.; Manchanda R.; McGoron A. J. Theranostic Applications of Nanomaterials in Cancer: Drug Delivery, Image-Guided Therapy, and Multifunctional Platforms. Appl. Biochem. Biotechnol. 2011, 165, 1628–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J. M.; Wang X.; Marin-Muller C.; Wang H.; Lin P. H.; Yao Q.; Chen C. Current Advances in Research and Clinical Applications of PLGA-Based Nanotechnology. Expert Rev. Mol. Diagn. 2009, 9, 325–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundargi R. C.; Babu V. R.; Rangaswamy V.; Patel P.; Aminabhavi T. M. Nano/Micro Technologies for Delivering Macromolecular Therapeutics Using Poly(d,l-Lactide-Co-Glycolide) and Its Derivatives. J. Controlled Release 2008, 125, 193–209. [DOI] [PubMed] [Google Scholar]
- Harris J. M.; Chess R. B. Effect of Pegylation on Pharmaceuticals. Nat. Rev. Drug Discovery 2003, 2, 214–221. [DOI] [PubMed] [Google Scholar]
- Schmidts T.; Dobler D.; Nissing C.; Runkel F. Influence of Hydrophilic Surfactants on the Properties of Multiple W/O/W Emulsions. J. Colloid Interface Sci. 2009, 338, 184–192. [DOI] [PubMed] [Google Scholar]
- Woodrow K. A.; Cu Y.; Booth C. J.; Saucier-Sawyer J. K.; Wood M. J.; Saltzman W. M. Intravaginal Gene Silencing Using Biodegradable Polymer Nanoparticles Densely Loaded with Small-Interfering RNA. Nat. Mater. 2009, 8, 526–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulmurugan R.; Sekar N. M.; Sekar T. V. Biodegradable Polymer Nanocarriers for Therapeutic Antisense MicroRNA Delivery in Living Animals. Proc. SPIE 2012, 8232, 823208. [Google Scholar]; Colloidal Nanocrystals for Biomedical Applications VII.
- Cao M.; Deng X.; Su S.; Zhang F.; Xiao X.; Hu Q.; Fu Y.; Yang B. B.; Wu Y.; Sheng W.; et al. Protamine Sulfate-Nanodiamond Hybrid Nanoparticles as a Vector for MiR-203 Restoration in Esophageal Carcinoma Cells. Nanoscale 2013, 5, 12120–12125. [DOI] [PubMed] [Google Scholar]
- Mitchell P. S.; Parkin R. K.; Kroh E. M.; Fritz B. R.; Wyman S. K.; Pogosova-Agadjanyan E. L.; Peterson A.; Noteboom J.; O’Briant K. C.; Allen A.; et al. Circulating MicroRNAs as Stable Blood-Based Markers for Cancer Detection. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L. Role of MiR-10b in Breast Cancer Metastasis. Breast Cancer Res. 2010, 12, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.; Teruya-Feldstein J.; Weinberg R. A. Tumour Invasion and Metastasis Initiated by MicroRNA-10b in Breast Cancer. Nature 2007, 449, 682–688. [DOI] [PubMed] [Google Scholar]
- Sekar T. V.; Mohanram R. K.; Foygel K.; Paulmurugan R. Therapeutic Evaluation of MicroRNAs by Molecular Imaging. Theranostics 2013, 3, 964–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C. J.; Saltzman W. M. Polymer Nanoparticle-Mediated Delivery of MicroRNA Inhibition and Alternative Splicing. Mol. Pharmaceutics 2012, 9, 1481–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyevleva A. G.; Kuligina E.; Mitiushkina N. V.; Togo A. V.; Miki Y.; Imyanitov E. N. High Level of MiR-21, MiR-10b, and MiR-31 Expression in Bilateral vs. Unilateral Breast Carcinomas. Breast Cancer Res. Treat. 2012, 131, 1049–59. [DOI] [PubMed] [Google Scholar]
- Yan L. X.; Wu Q. N.; Zhang Y.; Li Y. Y.; Liao D. Z.; Hou J. H.; Fu J.; Zeng M. S.; Yun J. P.; Wu Q. L.; et al. Knockdown of MiR-21 in Human Breast Cancer Cell Lines Inhibits Proliferation, In Vitro Migration and In Vivo Tumor Growth. Breast Cancer Res. 2011, 13, R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulisse S.; Baldini E.; Sorrenti S.; D’Armiento M. The Urokinase Plasminogen Activator System: A Target for Anti-Aancer Therapy. Curr. Cancer Drug Targets 2009, 9, 32–71. [DOI] [PubMed] [Google Scholar]
- Gu F.; Langer R.; Farokhzad O.. Formulation/Preparation of Functionalized Nanoparticles for In Vivo Targeted Drug Delivery. In Micro and Nano Technologies in Bioanalysis; Foote R. S., Lee J. W., Eds.; Humana Press: New York, 2009; Vol. 544, pp 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.