

Abstract
Background
The role of endothelial nitric oxide synthase (eNOS) in isoflurane postconditioning (IsoPC)-elicited cardioprotection is poorly understood. We addressed this issue using eNOS-/- mice.
Methods
In vivo or Langendorff-perfused mouse hearts underwent 30 min of ischemia followed by 2 h of reperfusion in the presence and absence of postconditioning produced with isoflurane 5 min before ischemia and 3 min after reperfusion. Ca2+-induced mitochondrial permeability transition pore opening was assessed in isolated mitochondria. Echocardiography was used to evaluate ventricular function.
Results
Postconditioning with 0.5, 1.0, and 1.5 minimum alveolar concentrations of isoflurane decreased infarct size from 56 ± 10% (n = 10) in control to 48 ± 10%, 41 ± 8% (n = 8, P < 0.05), and 38 ± 10% (n = 8, P < 0.05), respectively and improved cardiac function in wild-type mice. Improvement in cardiac function by IsoPC was blocked by NG-nitro-L-arginine methyl ester (a nonselective NOS inhibitor) administered either prior to ischemia or at the onset of reperfusion. Mitochondria isolated from postconditioned hearts required significantly higher in vitro Ca2+ loading than control (78 ± 29 vs. 40 ± 25 μM CaCl2 mg protein-1, n = 10, P < 0.05) to open the mitochondrial permeability transition pore. Hearts from eNOS-/- mice displayed no marked differences in infarct size, cardiac function, and sensitivity of mitochondrial permeability transition pore to Ca2+, compared to the wild-type hearts. However, IsoPC failed to alter infarct size, cardiac function or the amount of Ca2+ necessary to open the mitochondrial permeability transition pore in mitochondria isolated from the eNOS-/- hearts compared to control hearts.
Conclusions
IsoPC protects mouse hearts from reperfusion injury by preventing MPT pore opening in an eNOS-dependent manner. Nitric oxide functions as both a trigger and a mediator of cardioprotection produced by IsoPC.
Introduction
Myocardial injury is attenuated in hearts subjected to brief episodes of ischemia/reperfusion (I/R) that interrupt the initial period of sustained reperfusion (ischemic postconditioning).1,2 This cardioprotective effect can also be achieved by pharmacological interventions instituted at the onset of reperfusion.3-5 We and others have shown that the volatile anesthetic, isoflurane, reduces myocardial damage when administered during the early phase of reperfusion (isoflurane postconditioning [IsoPC]).6-8 Whether cardioprotection produced by IsoPC is modified by genetic factors, such as deficiency of nitric oxide synthase (NOS) gene, remains unknown.
Pharmacological studies suggest that nitric oxide derived from L-arginine is involved in IsoPC-induced cardioprotection against I/R injury in rabbits and rats.9,10 Under physiological conditions, nitric oxide is predominantly produced by endothelial NOS (eNOS) in vascular endothelium.11 However, during myocardial I/R, nitric oxide may be produced from three distinct NOS isoforms expressed in cardiac myocytes: neuronal NOS, inducible NOS, and eNOS.11,12 Since pharmacological inhibitors usually non-selectively inhibit all isoforms of NOS, it is unknown which isoform is responsible for IsoPC-induced cardioprotection. Gene knockout mice for the eNOS isoform (eNOS-/- mice) have recently been used as a tool to examine the role of eNOS in physiological and pathological states such as ischemic preconditioning.13 Whether eNOS-/- mice are also useful for establishing the role of the eNOS isoform in IsoPC-induced cardioprotection remains unclear. Furthermore, the targets of nitric oxide in IsoPC are not yet identified. Growing evidence suggests that among potential candidates, the mitochondrial permeability transition (MPT) pore is a key target for nitric oxide-induced protection against I/R injury.14-17 Accordingly we investigated whether IsoPC protects myocardium against I/R injury by modulating MPT in wild-type (WT) and eNOS-/- mice.
Materials and Methods
Animals
Male C57BL/6J WT mice and eNOS-/- mice (weight: 25.3 ± 1.3 g; age: 9-12 weeks) were purchased from The Jackson Laboratory (Bar Harbor, ME). The animals were kept on a 12-h light-dark cycle in a temperature-controlled room. All experimental procedures used in this study were approved by the Animal Care and Use Committee of the Medical College of Wisconsin (Milwaukee, Wisconsin) and conformed to the Guide for the Care and Use of laboratory Animals (NIH Publication No. 85-23, revised 1996).
Acute Myocardial Ischemia/Reperfusion Injury in Vivo
Surgical Preparation
We have previously described the mouse in vivo model of myocardial I/R injury.18,19 Briefly, mice were anesthetized by intraperitoneal injection of sodium pentobarbital (80 mg/kg) and ventilated with room air supplemented with 100% oxygen at a rate of ∼104 breaths/min with a tidal volume of ∼0.5 ml using a rodent ventilator (Harvard Apparatus, South Natick, MA). Arterial blood gas tensions and acid-base status were maintained within a physiological range (pH between 7.35 and 7.45, PaCO2 between 35 and 45 mmHg, and PaO2 between 120 and 180 mmHg) by adjusting the respiratory rate or tidal volume. Myocardial ischemia was produced by occluding the left anterior descending coronary artery, and reperfusion was initiated by loosening the suture. Body temperature was maintained between 36.8°C and 37.5°C throughout the experiment by using a heating pad (Model TC-1000, CWE Inc.; Ardmore, PA). The infarct area was delineated by perfusing the coronary arteries with 2,3,5-triphenyltetrazolium chloride via the aortic root, and the area at risk was delineated by perfusing phthalo blue dye (Heucotech Ltd., Fairless Hill, PA) into the aortic root after tying the coronary artery at the site of previous occlusion. As a result of these procedures, the nonischemic portion of the left ventricle (LV) was stained dark blue. Viable myocardium within the area at risk was stained bright red, and infarcted tissue was light yellow.
Experimental Protocol
In vivo IsoPC experiments followed two different protocols (fig. 1). The concentration-dependent effects of IsoPC were determined in C57BL/6J mice randomly assigned to 4 experimental groups (8-10 mice/group): control, ISO0.5, ISO1.0, and ISO1.5 (fig. 1A). After instrumentation was completed, all mice were stabilized for 30 min and subjected to 30 min of coronary occlusion followed by 2 h of reperfusion. Isoflurane was administered via an isoflurane-specific vaporizer (Ohio Medical Instruments, Madison, WI). Postconditioning was produced with 0.5, 1.0, or 1.5 minimum alveolar concentrations (MAC) of isoflurane (1.0 MAC = 1.40% in the mouse) administered during the last 5 min of coronary occlusion and first 3 min of reperfusion. Control mice received no isoflurane. Arterial blood pressure was measured with a Millar pressure catheter (Model SPR-1000, Millar Instruments, Houston, TX) inserted into right carotid artery and connected to a pressure transducer (ADInstruments, Sydney, Australia), as we have previously described.20 Heart rate was monitored from the electrocardiogram. Area at risk and infarct size were determined following each experiment. The effects of disruption of the eNOS gene on infarct size were measured in C57BL/6J or eNOS-/- mice without and with IsoPC (fig. 1B).
Figure 1.

Schematic diagram depicting the experimental protocols. A: effects of different concentrations of isoflurane on myocardial infarct size in WT mice following I/R; B: effects of disruption of the eNOS gene on infarct size; C: effects of different concentrations of isoflurane on cardiac function in Langendorff-perfused WT hearts following I/R; D: role of nitric oxide in the cardioprotective effect produced by isoflurane; E: effects of isoflurane on MPT and cardiac function in WT and eNOS-/- mice. eNOS-/- = endothelial nitric oxide synthase gene knockout; I/R = ischemia/reperfusion; ISO0.5 = 0.5 MAC isoflurane; ISO1.0 = 1.0 MAC isoflurane; ISO1.5 = 1.5 MAC isoflurane; L-NAME = NG-nitro-L-arginine methyl ester; MAC = minimum alveolar concentration; MPT = mitochondrial permeability transition; WT = wild-type.
Transthoracic Echocardiography
C57BL/6J and eNOS-/- mice were sedated by the inhalation of isoflurane (1.50 %) and oxygen and anchored to a warming platform in the supine position. Transthoracic echocardiography was performed with a VisualSonics Vevo 770 High-resolution Imaging System (Toronto, Canada) equipped with a 30 MHz transducer (Scanhead RMV 707). As described previously,21 LV dimensions and fractional shortening were measured by two-dimension guided M-mode method. Pulsed Doppler waveforms recorded in the apical-4-chamber view were used for the measurements of the peak velocities of mitral E (early mitral inflow) and A (late mitral inflow) waves, E wave acceleration velocity, E wave acceleration time, E wave deceleration velocity, and E wave deceleration time, isovolumic contraction time, ejection time, and isovolumic relaxation time of LV. Myocardial performance index was calculated with the following formula: myocardial performance index = (isovolumic contraction time + isovolumic relaxation time)/ejection time.
Ischemia/Reperfusion Injury in Isolated Hearts
Langendorff-perfusion of the Heart
The hearts were perfused at a constant pressure of 55 mmHg with Krebs-Henseleit buffer containing (in mM) NaCl 118, NaHCO3 25, KCl 4.7, MgCl2 1.2, CaCl2 2.5, KH2PO4,1.2, EDTA 0.5, and glucose 11.19,22 The buffer was continuously bubbled with a mixture of 95% oxygen/5% carbon dioxide via in-line filter (5 μm pore size). A fluid-filled plastic balloon was inserted into the chamber of LV via the mitral valve, and connected to a pressure transducer for continuous measurement of LV pressure. The hearts were immersed in perfusate maintained at 37.2 ± 0.3°C, and the balloon was inflated to a diastolic pressure of ∼5 to 10 mmHg. Coronary flow was monitored by an in-line flow probe connected to a flow meter (Transonics Systems Inc., Ithaca, NY). The LV pressure signal was monitored to obtain heart rate and LV dP/dt. The LV developed pressure (LVDP) was calculated as the difference between the systolic and end-diastolic LV pressure. Global I/R was produced by cessation of perfusion followed by reperfusion at a designated time.
Experimental Protocol
Langendorff-perfused hearts were used in three different protocols (fig. 1). The effects of isoflurane on cardiac function following I/R were measured in C57BL/6J mice (fig. 1C). All hearts were perfused for 30 min for stabilization, and baseline LV contraction and coronary flow were recorded. Hearts were then randomly assigned to 4 groups: control, ISO0.5, ISO1.0, and ISO1.5 (7-8 hearts/group) and subjected to 30 min of no-flow global ischemia and 2 h of reperfusion. Isoflurane was bubbled into the Krebs-Henseleit solution using an agent-specific vaporizer placed in the 95% oxygen/5% carbon dioxide gas mixture line, and isoflurane concentration was determined by gas chromatography in the coronary effluent. In the control group, oxygen/carbon dioxide-bubbled buffer was administered 5 min before and 3 min after reperfusion, whereas in the other groups, the buffer containing 0.5, 1.0, or 1.5 MAC isoflurane was administered 5 min before and 3 min after reperfusion. During global ischemia, buffer with isoflurane was perfused into the aorta by an infusion pump at a rate of 0.4 ml/min. LV contraction and coronary flow were continuously recorded during I/R. The role of nitric oxide as a trigger or mediator of IsoPC was evaluated in C57BL/6J mice (n = 7 mice/group) (fig. 1D). The hearts treated with 1.0 MAC isoflurane to produce postconditioning were perfused with 30 μM NG-nitro-L-arginine methyl ester (L-NAME, a nonselective NOS inhibitor) for 20 min either prior to ischemia (trigger group) or at the onset of reperfusion (mediator group). LVDP, +dP/dt (maximum rate of increase of LVDP), and −dP/dt (maximum rate of decrease of LVDP) at baseline and 30 and 60 min after reperfusion were determined. To evaluate the effects of IsoPC on MPT and cardiac function (fig. 1E), C57BL/6J and eNOS-/- mice were randomly assigned to 1 of the following 6 groups: (1) WT sham; (2) WT control; (3) WT ISO1.0; (4) eNOS-/- sham; (5) eNOS-/- control; and (6) eNOS-/- ISO1.0. After the stabilization for 30 min, control and ISO1.0 hearts were subjected to 30 min global ischemia and 30 min reperfusion without or with 1.0 MAC isoflurane. Sham hearts were not subjected to ischemia. Mitochondria were isolated from LV for the determination of Ca2+-induced MPT pore opening, as described below.
Detection of the Permeability Transition Pore Opening in Isolated Mitochondria
Preparation of Isolated Mitochondria
LVs were minced in ice cold isolation buffer containing 50 mM sucrose, 200 mM mannitol, 5 mM KH2PO4, 1 mM EGTA {ethylene glycol bis ([beta]-aminoethyl ether)-N,N,N′,N′-tetraacetic acid, Sigma-Aldrich, St. Louis, MO}, 5 mM 3-(N-morpholino)propanesulfonic acid (Sigma-Aldrich), and 0.1% bovine serum albumin (pH 7.3) and gently homogenized in the same buffer with T 25 disperser (IKA-Werke, Staufen, Germany) in the presence of protease from Bacillus licheniformis (0.5 mg/ml). The homogenate was differentially centrifuged, and the final mitochondrial pellet was resuspended in isolation buffer without EGTA.
Ca2+-induced MPT Pore Opening
Opening of the MPT pore after in vitro Ca2+ overload was assessed by following changes in the membrane potential (ΔΨm) using the fluorescent dye rhodamine 123 (50 nM; Invitrogen, Carlsband, CA) in the presence of pyruvate and malate (5 mM).23,24 Fluorescence was monitored with a PTI spectrofluorometer (Photon Technology International Inc. Birmingham, NJ). Excitation and emission wavelengths were set to 503 and 527 nm, respectively. Isolated mitochondria (0.5 mg/ml) were suspended in 1.0 ml recording buffer containing (in mM): 220 sucrose, 10 HEPES (4-[2-Hydroxyethyl]piperazine-1-ethanesulfonic acid, Sigma-Aldrich), 10 KH2PO4, pH = 7.3. At the end of the preincubation period, pulses of 10 μM CaCl2 were administered in 60-second intervals. After sufficient Ca2+ loading, MPT pore opening results in a sudden collapse of ΔΨm. To achieve complete mitochondrial depolarization, 1 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (Sigma-Aldrich) was added into the buffer at the end of the experiment. The amount of Ca2+ necessary to trigger this sudden collapse of ΔΨm is used as an indicator of the susceptibility of the MPT pore to Ca2+ overload.
Statistical Analysis
All data are expressed as mean ± SD. Statistical analysis of echocardiographic data was performed with two-way repeated measures ANOVA. Repeated measures ANOVA followed by Bonferroni's multiple comparison test was used to evaluate differences in heart rate, mean arterial blood pressure, and coronary flow over time between groups. One-way ANOVA followed by Bonferroni post-hoc test was used to analyze area at risk, infarct size, LVDP, +dP/dt, -dP/dt, LV end-diastolic pressure, and the amount of Ca2+ loading necessary to open the MPT pore. A value of P < 0.05 was considered as statistically different.
Results
IsoPC Protected Against Myocardial I/R Injury in WT mice
Heart rate at baseline was not different among 4 experimental groups (table 1). Coronary artery occlusion significantly decreased mean arterial blood pressure in all groups in comparison with baseline blood pressure. No significant differences in arterial blood pressure and heart rate were observed between groups during coronary occlusion and reperfusion.
Table 1. Systemic Hemodynamics during in vivo Experiments.
| Group | Baseline | Coronary occlusion | Reperfusion | |||
|---|---|---|---|---|---|---|
|
| ||||||
| 2 min | 30 min | 60 min | 120 min | |||
| Effects of isoflurane on heart rate and blood pressure during ischemia/reperfusion in the wild-type mice | ||||||
| Heart rate (beats/min) | ||||||
| Control | 402 ± 69 | 374 ± 60 | 429 ± 63 | 409 ± 53 | 398 ± 41 | 395 ± 56 |
| ISO0·5 | 411 ± 56 | 390 ± 34 | 400 ± 36 | 396 ± 31 | 382 ± 28 | 388 ± 17 |
| ISO1·0 | 416 ± 61 | 389 ± 48 | 402 ± 23 | 393 ± 39 | 374 ± 28 | 395 ± 31 |
| ISO1·5 | 406 ± 76 | 369 ± 31 | 431 ± 62 | 420 ± 42 | 389 ± 31 | 398 ± 55 |
| Mean arterial blood pressure (mmHg) | ||||||
| Control | 74 ± 15 | 57 ± 15* | 58 ± 15* | 60 ± 9* | 57 ± 12* | 55 ± 12* |
| ISO0·5 | 78 ± 11 | 59 ± 14* | 58 ± 11* | 59 ± 14* | 60 ± 14* | 61 ± 14* |
| ISO1·0 | 76 ± 14 | 58 ± 11* | 59 ± 11* | 61 ± 11* | 61 ± 12* | 60 ± 11* |
| ISO1·5 | 73 ± 14 | 50 ± 17* | 49 ± 16* | 52 ± 16* | 53 ± 16* | 55 ± 14* |
| Effects of ISO1.0 on heart rate during ischemia/reperfusion in the wild-type and eNOS-/- mice | ||||||
| WT Control | 403 ± 42 | 384 ± 41 | 391 ± 40 | 397 ± 37 | 401 ± 29 | 402 ± 52 |
| WT ISO1.0 | 424 ± 61 | 392 ± 50 | 399 ± 22 | 393 ± 42 | 379 ± 27 | 399 ± 29 |
| eNOS-/- Control | 397 ± 50 | 384 ± 39 | 416 ± 54 | 434 ± 32 | 400 ± 25 | 424 ± 34 |
| eNOS-/- ISO1.0 | 391 ± 55 | 383 ± 45 | 428 ± 46 | 403 ± 34 | 397 ± 31 | 392 ± 30 |
eNOS-/- = endothelial nitric oxide synthase gene knockout mice; ISO0·5 = 0.5 minimum alveolar concentration of isoflurane; ISO1·0 = 1.0 minimum alveolar concentration of isoflurane; ISO1·5 = 1.5 minimum alveolar concentration of isoflurane.
P < 0.05 versus baseline (n = 7-10 mice/group).
Area at risk and myocardial infarct size are shown in figure 2. There were no significant differences in area at risk among the 4 experimental groups (fig. 2A). Coronary occlusion followed by reperfusion resulted in infarct size of 56 ± 10% of area at risk (n = 10) in WT mice, which was significantly decreased to 41 ± 8% (P < 0.05, n = 8) and 38 ± 10% (P < 0.05, n = 8) by 1.0 and 1.5 MAC isoflurane administered 5 min before and 3 min after reperfusion, respectively. Infarct size was not altered by 0.5 MAC isoflurane treatment (48 ± 10%, NS, n = 8, fig. 2B).
Figure 2.

Concentration-dependent decreases in myocardial infarct size by isoflurane postconditioning (IsoPC) in wild-type mice subjected to 30 min of coronary occlusion followed by 2 h of reperfusion. A: area at risk expressed as a percentage of left ventricle area; B: myocardial infarct size expressed as a percentage of area at risk. IsoPC was produced by 0.5, 1.0, or 1.5 minimum alveolar concentration of isoflurane (ISO0.5, ISO1.0, or ISO1.5) administered during the last 5 min of ischemia and first 3 min of reperfusion. *P < 0.05 versus control (n = 8-10 mice/group).
Disruption of eNOS Gene Abolished IsoPC-elicited Decrease in Infarct Size
The effects of disruption of eNOS gene on area at risk and myocardial infarct size are shown in figure 3. There were no significant differences in area at risk among groups. Myocardial infarct size was decreased in the WT ISO1.0 group compared to the WT control group (40 ± 8% vs. 55 ± 10% respectively, n = 7, P < 0.05), but not significantly decreased by IsoPC in the eNOS-/- group (53 ± 8%; n = 7), suggesting that eNOS is obligatory for the cardioprotective effect of IsoPC. Furthermore, to investigate whether loss of the cardioprotective effect of IsoPC in the eNOS-/- mice arose from the changes in cardiac structure or function, we assessed the hearts of the eNOS-/- mice and WT mice with echocardiography. There were no significant differences between the eNOS-/- mice and WT mice in multiple cardiac parameters, including heart rate, LV internal diameters in diastole and systole, fractional shortening, peak velocity of mitral E and A waves, mitral E/A ratio, isovolumic contraction time, ejection time, isovolumic relaxation time, myocardial performance index, mitral E acceleration, E wave acceleration time, mitral E deceleration, and E wave deceleration time. Only LV wall thickness in diastole was found to be greater in the eNOS-/- mice than in the WT mice (table 2).
Figure 3.

Myocardial infarct size was decreased by isoflurane postconditioning (IsoPC) in wild-type mice but not in endothelial nitric oxide synthase knockout (eNOS-/-) mice subjected to 30 min of coronary occlusion followed by 2 h of reperfusion. IsoPC was produced by 1.0 minimum alveolar concentration of isoflurane (ISO1.0) administered during the last 5 min of ischemia and first 3 min of reperfusion. A: area at risk; B: infarct size; C: representative heart slices from a wild-type control mouse; D: heart slices from a wild-type ISO1.0 mouse; E: heart slices from an eNOS-/- control mouse; F: heart slices from an eNOS-/- ISO1.0 mouse. The hearts were stained with 2,3,5-triphenyltetrazolium chloride and phthalol blue dye to delineate area at risk (red plus light yellow areas) and infarct size (light yellow areas). *P < 0.05 versus control (n = 7 mice/group).
Table 2. Echocardiographic Parameters in Wild-type and eNOS-/- Mice.
| Wild-type mice | eNOS-/- mice | |
|---|---|---|
| Heart rate (beats/min) | 473 ± 49 | 454 ± 45 |
| AWd (mm) | 0.81 ± 0.12 | 0.98 ± 0.14* |
| AWs (mm) | 1.28 ± 0.24 | 1.34 ± 0.15 |
| PWd (mm) | 0.88 ± 0.10 | 1.04 ± 0.16* |
| PWs (mm) | 1.34 ± 0.30 | 1.37 ± 0.18 |
| LVIDd (mm) | 3.57 ± 0.29 | 3.35 ± 0.23 |
| LVIDs (mm) | 2.16 ± 0.40 | 2.22 ± 0.27 |
| Fractional shortening (%) | 39 ± 8 | 34 ± 8 |
| Peak E (cm/s) | 79 ± 16 | 75 ± 12 |
| Peak A (cm/s) | 48 ± 13 | 55 ± 11 |
| Mitral E/A ratio | 1.72 ± 0.37 | 1.40 ± 0.29 |
| IVCT (ms) | 14.1 ± 4.4 | 15.4 ± 3.4 |
| Ejection time (ms) | 42.2 ± 4.2 | 46.4 ± 4.0 |
| IVRT (ms) | 16.4 ± 2.3 | 15.9 ± 1.3 |
| MPI | 0.73 ± 0.14 | 0.68 ± 0.11 |
| Mitral E acceleration (cm/ms) | 8,303 ± 2,145 | 7,763 ± 1,325 |
| Eat (ms) | 9.1 ± 1.8 | 9.9 ± 0.6 |
| Mitral E deceleration (cm/ms) | 4,382 ± 1,656 | 4,962 ± 1,782 |
| Edt (ms) | 16.2 ± 6.9 | 16.0 ± 4.2 |
| LV mass (g) | 91 ± 15 | 99 ± 18 |
AWd = anterior wall (interventricular septum) at end diastole; AWs = anterior wall at end systole; Eat = mitral E wave acceleration time; Edt = E wave deceleration time; eNOS-/- mice = endothelial nitric oxide synthase gene knockout mice; fractional shortening = 100 × (LVIDd-LVIDs)/LVIDd; IVCT = isovolumic contraction time of LV; IVRT = isovolumic relaxation time of LV; LV = left ventricle; LVIDd = LV internal diameter at end diastole; LVIDs = LV internal diameter at end systole; MPI (myocardial performance index) = the ratio of the sum of isovolumic contraction and relaxation times to ejection time; Peak A = peak velocity of mitra A wave; Peak E = peak velocity of mitral E valve; PWd = posterior wall at end diastole; PWs = posterior wall at end systole.
P < 0.05 versus wild-type (n = 8 mice/group).
IsoPC Improved Functional Recovery in Perfused WT Hearts
The effects of different concentrations of isoflurane on cardiac function in the Langendorff-perfused WT hearts are shown in figure 4. Baseline values of LVDP, +dP/dt, -dP/dt, LV end-diastolic pressure, coronary flow, and heart rate were comparable among groups. During reperfusion, LVDP, +dP/dt, -dP/dt, coronary flow, and heart rate in all groups gradually increased, and LV end-diastolic pressure decreased. At all time points, there were no differences between the ISO0.5 group and control group. However, LVDP, +dP/dt, and -dP/dt were significantly increased from 30 min to 2 h after reperfusion in the ISO1.0 and ISO1.5 groups in comparison with the control group. In addition, LV end-diastolic pressure was decreased in the ISO1.5 group.
Figure 4.

Isoflurane postconditioning produced concentration-dependent increases in cardiac function in Langendorff-perfused wild-type hearts subjected to 30 min of global ischemia followed by 2 h of reperfusion. A: LVDP (left ventricular developed pressure); B: +dP/dt (maximum rate of increase of LVDP); C: -dP/dt (maximum rate of decrease of LVDP); D: LVEDP (LV end-diastolic pressure); E: coronary flow; F: heart rate. ISO0.5 = 0.5 minimum alveolar concentration of isoflurane; ISO1.0 = 1.0 minimum alveolar concentration of isoflurane; ISO1.5 = 1.5 minimum alveolar concentration of isoflurane. *P < 0.05 versus control (n = 7-8 hearts/group).
Nitric Oxide Functioned as both a Trigger and a Mediator of IsoPC in WT Hearts
The importance of nitric oxide in cardioprotection by IsoPC is shown in figure 5. No differences in baseline values of LVDP, +dP/dt, and −dP/dt were observed between experimental groups. At 30 and 60 min after reperfusion, increases in LVDP, +dP/dt, and −dP/dt produced by IsoPC were significantly attenuated by L-NAME administered either prior to ischemia or at the onset of reperfusion, suggesting that nitric oxide functions as both a trigger and a mediator of IsoPC cardioprotection. The effects of isoflurane and L-NAME on heart rate and coronary flow are shown in table 3. There were no significant differences among the 4 groups.
Figure 5.
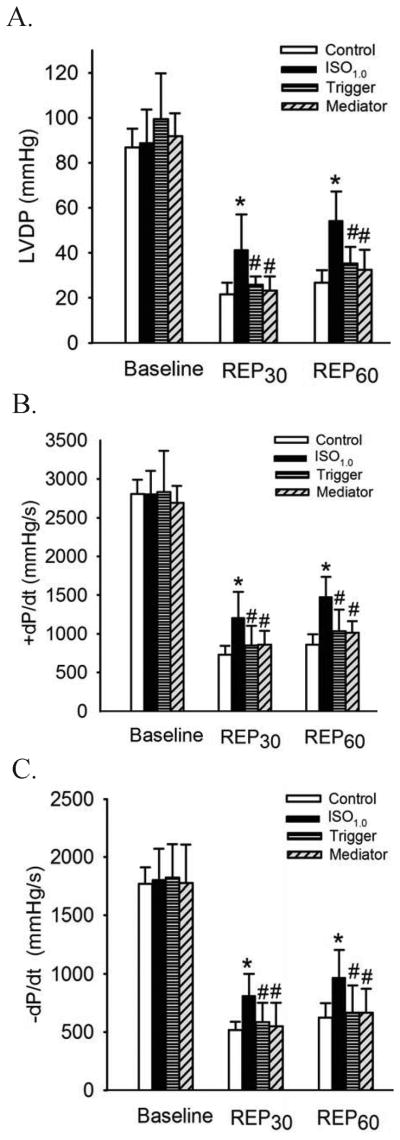
Nitric oxide is both a trigger and a mediator of isoflurane postconditioning-induced protection in wild-type hearts subjected to 30 min of ischemia followed by 60 min of reperfusion. A: LVDP (left ventricular developed pressure); B: +dP/dt (maximum rate of increase of LVDP); C: -dP/dt (maximum rate of decrease of LVDP). REP30 = 30 min after reperfusion; REP60 = 60 min after reperfusion. Buffer containing 1.0 minimum alveolar concentration of isoflurane (ISO1.0) was administered in the ISO1.0, trigger, and mediator groups. To inhibit nitric oxide synthesis before and after myocardial exposure to isoflurane, the hearts were perfused with 30 μM NG-nitro-L-arginine methyl ester (a non-selective endothelial nitric oxide synthase inhibitor) for 20 min either prior to ischemia (trigger group) or at the onset of reperfusion (mediator group). *P < 0.05 versus control; # P < 0.05 versus ISO1.0 (n = 7 hearts/group).
Table 3. Hemodynamics in the Langendorff-perfused Hearts.
| Baseline | Reperfusion | ||||
|---|---|---|---|---|---|
|
| |||||
| 2 min | 10 min | 30 min | 60 min | ||
| Effects of L-NAME on coronary flow and heart rate during ischemia/reperfusion in the WT hearts | |||||
| Heart rate (beats/min) | |||||
| Control | 294 ± 29 | 153 ± 32* | 240 ± 26 | 254 ± 37 | 233 ± 34* |
| ISO1.0 | 290 ± 26 | 167 ± 42* | 251 ± 45 | 264 ± 43 | 250 ± 56* |
| Trigger | 284 ± 30 | 187 ± 56* | 203 ± 34* | 222 ± 35* | 247 ± 19* |
| Mediator | 288 ± 34 | 155 ± 56* | 190 ± 43* | 220 ± 52* | 248 ± 55* |
| Coronary flow (ml/min) | |||||
| Control | 2.90 ± 0.63 | 1.79 ± 0.55* | 2.25 ± 0.61 | 2.34 ± 0.66 | 2.31 ± 0.63 |
| ISO1.0 | 2.92 ± 0.65 | 1.62 ± 0.42* | 2.37 ± 0.52 | 2.65 ± 0.77 | 2.48 ± 0.85 |
| Trigger | 2.78 ± 0.71 | 1.54 ± 0.35* | 1.83 ± 0.52* | 1.97 ± 0.51* | 1.81 ± 0.55* |
| Mediator | 3.11 ± 0.76 | 1.30 ± 0.63* | 1.94 ± 0.73* | 2.28 ± 0.44* | 2.34 ± 0.45* |
| Effects of ISO1.0 on heart rate and coronary flow during ischemia/reperfusion in the WT and eNOS-/- hearts | |||||
| Heart rate (beats/min) | |||||
| WT Sham | 295 ± 30 | 287 ± 29 | 284 ± 28 | 289 ± 21 | - |
| WT Control | 297 ± 32 | 158 ± 33*# | 220 ± 32*# | 241 ± 28*# | - |
| WT ISO1.0 | 290 ± 25 | 170 ± 40*# | 221 ± 53*# | 244 ± 35# | - |
| eNOS-/- Sham | 300 ± 29 | 292 ± 28 | 283 ± 27 | 278 ± 32 | - |
| eNOS-/- Control | 290 ± 22 | 168 ± 45*# | 243 ± 38* | 267 ± 33 | - |
| eNOS-/- ISO1.0 | 293 ± 25 | 160 ± 35*# | 233 ± 51* | 247 ± 38 | - |
| Coronary flow (ml/min) | |||||
| WT Sham | 2.93 ± 0.65 | 2.75 ± 0.53 | 2.77 ± 0.42 | 2.66 ± 0.46 | - |
| WT Control | 2.81 ± 0.67 | 1.60 ± 0.57*# | 2.08 ± 0.67 | 2.36 ± 0.64 | - |
| WT ISO1.0 | 3.06 ± 0.55 | 1.63 ± 0.58*# | 2.44 ± 0.37 | 2.60 ± 0.44 | - |
| eNOS-/- Sham | 2.85 ± 0.57 | 2.79 ± 0.53 | 2.75 ± 0.41 | 2.70 ± 0.43 | - |
| eNOS-/- Control | 2.87 ± 0.46 | 1.75 ± 0.58*# | 2.16 ± 0.69 | 2.37 ± 0.57 | - |
| eNOS-/- ISO1.0 | 2.72 ± 0.48 | 1.78 ± 0.47*# | 2.04 ± 0.45* | 2.26 ± 0.46 | - |
The hearts were treated with 1.0 minimum alveolar concentration of isoflurane (ISO1.0) without or with L-NAME (trigger and mediator groups) in wild-type (WT) or endothelial nitric oxide synthase knock-out (eNOS-/-) mice, as described in figures 5 and 7. L-NAME = NG-nitro-L-arginine methyl ester.
P < 0.05 versus baseline;
P < 0.05 versus WT sham (n = 7-10 hearts/group).
IsoPC Prevented MPT Pore Opening via an eNOS-dependent Mechanism
The impact of IsoPC on Ca2+-induced MPT pore opening of mitochondria is summarized in figure 6. In the WT sham group, the concentration of in vitro Ca2+ loading necessary to open the MPT pore was 148 ± 47 μM CaCl2 mg protein-1 (n = 10). This concentration was reduced to 40 ± 25 μM CaCl2 mg protein-1 (n = 10, P < 0.05) in mitochondria isolated from Langendorff-perfused WT hearts subjected to 30 min of global ischemia followed by 30 min of reperfusion (fig. 6). IsoPC significantly increased the amount of Ca2+ overload required for MPT pore opening to 78 ± 29 μM CaCl2 mg protein-1 (n = 10, P < 0.05 vs. I/R group), indicating that IsoPC delayed MPT pore opening. The concentration of Ca2+ required to trigger MPT pore opening was comparable between eNOS-/- (126 ± 38 μM CaCl2 mg protein-1, n = 10) and the WT sham groups (NS) and between eNOS-/- (33 ± 19 μM CaCl2 mg protein-1, NS, n = 10) and the WT control groups. IsoPC did not significantly alter the amount of Ca2+ overload required for MPT pore opening (41 ± 19 μM CaCl2 mg protein-1) compared to eNOS-/- control group, suggesting that isoflurane-induced inhibition of MPT pore opening is eNOS-dependent.
Figure 6.

Inhibition of the mitochondrial permeability transition pore opening by isoflurane postconditioning in wild-type (WT) hearts but not in endothelial nitric oxide synthase knockout (eNOS-/-) hearts subjected to 30 min of ischemia followed by 30 min of reperfusion. A: the amount of in vitro Ca2+ overload necessary to open the mitochondrial permeability transition pore in WT hearts and in eNOS-/- hearts; B: representative tracings showing the changes in membrane potential (ΔΨm) of mitochondria isolated from WT hearts after in vitro Ca2+ loading; C: tracings showing the changes in ΔΨm of mitochondria isolated from the eNOS-/- hearts after in vitro Ca2+ loading. Opening of the mitochondrial permeability transition pore was assessed after in vitro Ca2+ overload by following changes in ΔΨm using the fluorescent dye rhodamine 123. Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) was added at the arrows to depolarize mitochondria. *P < 0.05 versus sham; # P < 0.05 versus control (n = 10/group).
IsoPC Improved Cardiac Function in an eNOS-dependent Manner
The effects of IsoPC on cardiac function in WT and eNOS-/- hearts are shown in figure 7. There were no significant differences in baseline values of LVDP, +dP/dt, and −dP/dt among groups. Global ischemia for 30 min followed by reperfusion for 30 min caused significant decreases in LVDP, +dP/dt, and −dP/dt in WT and eNOS-/- hearts. IsoPC significantly increased LVDP, +dP/dt, and −dP/dt in WT hearts, but not in eNOS-/- hearts subjected to I/R insult, suggesting that eNOS plays an obligatory role in the cardioprotective effect produced by IsoPC. Heart rate and coronary flow during I/R are shown in table 3. There were no significant differences at all time points between WT sham and eNOS-/- sham, WT control and eNOS-/- control, and WT ISO1.0 and eNOS-/- ISO1.0 groups.
Figure 7.

Isoflurane postconditioning improved cardiac function in wild-type (WT) hearts but not in endothelial nitric oxide synthase knockout (eNOS-/-) hearts subjected to 30 min of global ischemia followed by 30 min of reperfusion. A: LVDP (left ventricular developed pressure) in WT hearts; B: LVDP in eNOS-/- hearts; C: +dP/dt (maximum rate of increase of LVDP) in WT hearts; D: +dP/dt in eNOS-/- hearts; E: -dP/dt (maximum rate of decrease of LVDP) in WT hearts; F: -dP/dt in eNOS-/- hearts. ISO1.0 = 1.0 minimum alveolar concentration of isoflurane; REP30 = 30 min after reperfusion. *P < 0.05 versus sham; # P < 0.05 versus control (n = 10/group).
Discussion
The present data demonstrate the dose-dependent effects of IsoPC on acute myocardial I/R injury in intact mice and in isolated hearts. Isoflurane administered during the last 5 min of ischemia and first 3 min of reperfusion reduced infarct size and was associated with improved recovery of LV systolic contractility and diastolic relaxation. These findings confirm other reports by Chiari and Feng et al.6,7, demonstrating that IsoPC decreases myocardial infarct size in intact rabbits and improves LV contractility in isolated rat hearts. Myocardial reperfusion after acute ischemia is a necessary process for salvage of viable myocardium and limitation of infarct size. However, restoration of coronary flow initiates a series of adverse events that also cause myocardial stunning, apoptosis, and necrosis,25 known as reperfusion injury.26-29 The present results indicate that isoflurane produces potent cardioprotective effects against acute reperfusion injury in mice.
IsoPC was produced in this investigation by administration of isoflurane 5 min before and 3 min after reperfusion. Our laboratory has previously shown that 1.0 MAC isoflurane administered upon reperfusion limited infarct size in rabbits.6 Using this same protocol in pilot experiments, we found that 1.0 MAC isoflurane did not significantly reduce myocardial infarct size in intact WT mice. Slighter longer durations of administration of isoflurane in mice versus rabbits were required to elicit a decrease in infarct size. This difference may be caused by smaller ventilatory volumes which delayed isoflurane to enter the myocardium in the mouse in vivo model.
Pharmacological studies suggest that nitric oxide generated by NOS may contribute to IsoPC-elicited cardioprotective effect in rabbits and rats.9,10 To examine whether eNOS is obligatory for IsoPC, we studied the effects of disruption of the eNOS gene on the cardioprotection produced by IsoPC. Myocardial infarct size was similar in eNOS-/- mice and WT mice subjected to a 30 min coronary occlusion followed by 2 h reperfusion. This result is in agreement with a recent study by Xuan et al.13, but inconsistent with another report by Jones et al.30 that myocardial infarct size was larger in eNOS-/- mice than in the WT mice. Nonetheless, myocardial infarct size was significantly decreased by 1.0 MAC IsoPC in WT mice, but not in the eNOS-/- mice. Furthermore, cardiac function was significantly improved by 1.0 MAC IsoPC in isolated WT hearts, but in the eNOS-/- hearts. Our data provide direct evidence that nitric oxide derived from eNOS (rather than other NOS isoforms) is critical for the cardioprotection by isoflurane against acute I/R injury. This is the first study to demonstrate the critical dependency of IsoPC on the eNOS isoform using eNOS-/- mice.
WT hearts treated with isoflurane to produce postconditioning were perfused with 30 μM L-NAME for 20 min either prior to ischemia or at the onset of reperfusion to inhibit nitric oxide synthesis before or after myocardial exposure to isoflurane, respectively. IsoPC-induced improvement in the recovery of cardiac function after I/R was blocked by L-NAME in both groups. This result suggests that nitric oxide functions as both a trigger and a mediator of IsoPC. Recently, we found that isoflurane could directly stimulate vascular endothelial cells to produce nitric oxide.31 In normal cardiac tissues, nitric oxide is predominantly derived from eNOS.11 Therefore, it is possible that nitric oxide derived from eNOS in coronary endothelial cells acts as a “trigger” to initiate intracellular events leading to IsoPC. Conversely, during I/R cardiac myocytes express eNOS and nitric oxide production is increased.11,32-35 We speculate that eNOS-dependent nitric oxide acting as a critical “mediator” of cardioprotection produced by IsoPC might be derived from both cardiac myocytes and coronary endothelial cells.
The MPT pore, a large and nonspecific pore spanning both inner and outer mitochondrial membranes, remains closed during myocardial ischemia but opens due to Ca2+ overload and excessive production of reactive oxygen species during reperfusion.36,37 Opening of the MPT pore results in collapse of the mitochondrial membrane potential, uncoupling of the respiratory chain, efflux of Ca2+ and cytochrome c, and matrix swelling. These events are thought to be pathognomonic for myocardial reperfusion injury.37-39 A previous study by Feng et al. reported that NAD+ content in WT rat myocardium was reduced by IsoPC,7 which suggests that IsoPC may prevent MPT pore opening since mitochondria possess 72-92% of the total cellular contents of NAD+.37 In the current study, we used a quantitative potentiometric approach to address the susceptibility of the MPT pore to open following Ca2+ loading in purified mitochondria that were directly isolated from ex vivo injured myocardium. Although the procedures for mitochondrial isolation might induce additional mitochondrial damage, isolated mitochondria are a unique tool to identify molecular entities that are modified by physiological, biochemical, or pharmacological processes.40-43 Mitochondria isolated from WT hearts after IsoPC required significantly higher in vitro Ca2+ loading than mitochondria without IsoPC to open the MPT pore. This suggests that IsoPC protects mitochondria by delaying MPT pore opening. It is very likely, therefore, that the beneficial actions of IsoPC on hearts subjected to I/R are related to mitochondrial protection. Interestingly, the protective effects of IsoPC on mitochondria were abolished in the eNOS-/- mice, suggesting that mitochondria are a target of eNOS-derived NO.14,16 Physiological levels of nitric oxide have been shown to be capable of inhibiting MPT pore opening.15 While the exact mechanisms by which nitric oxide inhibits MPT pore opening are unclear, eNOS-derived nitric oxide is involved in an indirect (through signaling pathways) and/or direct modulation of MPT pore opening in IsoPC.
The results of the present investigation should be interpreted within the constraints of several potential limitations. eNOS-/- mice are known to have a higher systemic blood pressure than WT mice.13,44 Because hypertension may initiate changes in cardiac structure and function, we used echocardiography to evaluate the hearts of the eNOS-/- and WT C57BL/6J mice at baseline conditions. Our results suggest that there is concentric hypertrophy of the LV in the eNOS-/- mice in comparison with the WT mice. This is consistent with a previous investigation by Yang et al.45, while another study by Ruetten et al. reported that LV morphology and function were similar between the eNOS-/- mice and WT mice.46 The increased LV wall thickness observed in the eNOS-/- mice in our study may be attributable to increased afterload. LV hypertrophy may alter cardiac function and therefore change myocardial I/R injury. However, no significant differences in multiple indices of LV function including systolic contractility (fractional shortening) and diastolic relaxation (ratio of mitral E/A and MVDT) were identified in eNOS-/- mice compared to WT mice (table 3). Therefore, it is unlikely that loss of the cardioprotective effect of isoflurane in the eNOS-/- mice can be attributed to changes in hemodynamics or cardiac function.
Five protocols of experiments were performed to examine cardioprotective effect produced by IsoPC and the role of eNOS in IsoPC in this study. A large number of comparisons were conducted and the error rate within each family of comparisons was meticulously controlled. One-way ANOVA followed by Bonferroni post-hoc test was used to analyze infarct size, LVDP, +dP/dt, and -dP/dt. A global adjustment of error rate was not performed, since these parameters were correlated to one another, rather than independent.
In summary, the present investigation demonstrates that brief administration of isoflurane during the late phase of ischemia and early phase of reperfusion reduces myocardial damage and improves cardiac function by preventing MPT pore opening. This cardioprotective effect is dependent on eNOS. Nitric oxide produced by eNOS serves not only as a trigger but also as a mediator of cardioprotection against I/R injury produced by IsoPC.
Final Boxed Summary Statement.
What we already know about this topic
-
*
Isoflurane protects the heart from ischemia reperfusion injury, when given upon reperfusion
-
*
Nitric oxide synthases are important to this effect, but its mechanisms are unclear
What this article tells us that is new
-
*
In mice, isoflurane protection depended on activation of the endothelial nitric oxide synthase subtype, and inhibited the mitochondrial permeability transition pore opening
-
*
Nitric oxide served as both a trigger and a mediator of myocardial protection in this setting
Acknowledgments
We thank John G. Krolikowski, B.A. and Amie R. Billstrom, B.A. (Research Technologists), David Schwabe, B.S. (Engineer), John Tessmer, B.A. (Manager) all from the Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, Wisconsin; and Chiaki Kwok, B.A. (Research Associate, Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, Wisconsin) for excellent technical assistance.
Sources of Funding: This work was supported in part by National Institutes of Health research grants HL 063705 (to Dr. Kersten), HL 054820 (to Dr. Warltier), and GM 066730 (to Drs. Kersten and Warltier) from the United States Public Health Services, Bethesda, Maryland.
References
- 1.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion. Comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–88. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- 2.Nishino Y, Webb IG, Davidson SM, Ahmed AI, Clark JE, Jacquet S, Shah AM, Miura T, Yellon DM, Avkiran M, Marber MS. Glycogen synthase kinase-3 inactivation is not required for ischemic preconditioning or postconditioning in the mouse. Circ Res. 2008;103:307–14. doi: 10.1161/CIRCRESAHA.107.169953. [DOI] [PubMed] [Google Scholar]
- 3.Tissier R, Waintraub X, Couvreur N, Gervais M, Bruneval P, Mandet C, Zini R, Enriquez B, Berdeaux A, Ghaleh B. Pharmacological postconditioning with the phytoestrogen genistein. J Mol Cell Cardiol. 2007;42:79–87. doi: 10.1016/j.yjmcc.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Penna C, Mancardi D, Raimondo S, Geuna S, Pagliaro P. The paradigm of postconditioning to protect the heart. J Cell Mol Med. 2008;12:435–58. doi: 10.1111/j.1582-4934.2007.00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sicard P, Jacquet S, Kobayashi KS, Flavell RA, Marber MS. Pharmacological postconditioning effect of muramyl dipeptide is mediated through RIP2 and TAK1. Cardiovasc Res. 2009;83:277–84. doi: 10.1093/cvr/cvp055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiari PC, Bienengraeber MW, Pagel PS, Krolikowski JG, Kersten JR, Warltier DC. Isoflurane protects against myocardial infarction during early reperfusion by activation of phosphatidylinositol-3-kinase signal transduction. Evidence for anesthetic-induced postconditioning in rabbits. Anesthesiology. 2005;102:102–9. doi: 10.1097/00000542-200501000-00018. [DOI] [PubMed] [Google Scholar]
- 7.Feng J, Lucchinetti E, Ahuja P, Pasch T, Perriard JC, Zaugg M. Isoflurane postconditioning prevents opening of the mitochondrial permeability transition pore through inhibition of glycogen synthase kinase 3β. Anesthesiology. 2005;103:987–95. doi: 10.1097/00000542-200511000-00013. [DOI] [PubMed] [Google Scholar]
- 8.Tsutsumi YM, Yokoyama T, Horikawa Y, Roth DM, Patel HH. Reactive oxygen species trigger ischemic and pharmacological postconditioning. In vivo and in vitro characterization. Life Sci. 2007;81:1223–7. doi: 10.1016/j.lfs.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krolikowski JG, Weihrauch D, Bienengraeber M, Kersten JR, Warltier DC, Pagel PS. Role of Erk1/2, p70s6K, and eNOS in isoflurane-induced cardioprotection during early reperfusion in vivo. Can J Anaesth. 2006;53:174–82. doi: 10.1007/BF03021824. [DOI] [PubMed] [Google Scholar]
- 10.Feng J, Fischer G, Lucchinetti E, Zhu M, Bestmann L, Jegger D, Arras M, Pasch T, Perriard JC, Schaub MC, Zaugg M. Infarct-remodeled myocardium is receptive to protection by isoflurane postconditioning. Role of protein kinase B/Akt signaling. Anesthesiology. 2006;104:1004–14. doi: 10.1097/00000542-200605000-00017. [DOI] [PubMed] [Google Scholar]
- 11.Schulz R, Kelm M, Heusch G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:402–13. doi: 10.1016/j.cardiores.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 12.Heinzel FR, Gres P, Boengler K, Duschin A, Konietzka I, Rassaf T, Snedovskaya J, Meyer S, Skyschally A, Kelm M, Heusch G, Schulz R. Inducible nitric oxide synthase expression and cardiomyocyte dysfunction during sustained moderate ischemia in pigs. Circ Res. 2008;103:1120–7. doi: 10.1161/CIRCRESAHA.108.186015. [DOI] [PubMed] [Google Scholar]
- 13.Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Bolli R. Endothelial nitric oxide synthase plays an obligatory role in the late phase of ischemic preconditioning by activating the protein kinase Cε-p44/42 mitogen-activated protein kinase-pSer-signal transducers and activators of transcription1/3 pathway. Circulation. 2007;116:535–44. doi: 10.1161/CIRCULATIONAHA.107.689471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotta Y, Otsuka-Murakami H, Fujita M, Nakagawa J, Yajima M, Liu W, Ishikawa N, Kawai N, Masumizu T, Kohno M. Protective role of nitric oxide synthase against ischemia-reperfusion injury in guinea pig myocardial mitochondria. Eur J Pharmacol. 1999;380:37–48. doi: 10.1016/s0014-2999(99)00531-2. [DOI] [PubMed] [Google Scholar]
- 15.Brookes PS, Salinas EP, Darley-Usmar K, Eiserich JP, Freeman BA, Darley-Usmar VM, Anderson PG. Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J Biol Chem. 2000;275:20474–9. doi: 10.1074/jbc.M001077200. [DOI] [PubMed] [Google Scholar]
- 16.Rakhit RD, Mojet MH, Marber MS, Duchen MR. Mitochondria as targets for nitric oxide-induced protection during simulated ischemia and reoxygenation in isolated neonatal cardiomyocytes. Circulation. 2001;103:2617–23. doi: 10.1161/01.cir.103.21.2617. [DOI] [PubMed] [Google Scholar]
- 17.West MB, Rokosh G, Obal D, Velayutham M, Xuan YT, Hill BG, Keith RJ, Schrader J, Guo Y, Conklin DJ, Prabhu SD, Zweier JL, Bolli R, Bhatnagar A. Cardiac myocyte-specific expression of inducible nitric oxide synthase protects against ischemia/reperfusion injury by preventing mitochondrial permeability transition. Circulation. 2008;118:1970–8. doi: 10.1161/CIRCULATIONAHA.108.791533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Black RG, Jr, Guo Y, Ge ZD, Murphree SS, Prabhu SD, Jones WK, Bolli R, Auchampach JA. Gene dosage-dependent effects of cardiac-specific overexpression of the A3 adenosine receptor. Circ Res. 2002;91:165–72. doi: 10.1161/01.res.0000028007.91385.ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, Auchampach JA. Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther. 2006;319:1200–10. doi: 10.1124/jpet.106.111351. [DOI] [PubMed] [Google Scholar]
- 20.Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M, Gutterman DD. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension. 2009;53:532–8. doi: 10.1161/HYPERTENSIONAHA.108.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson TJ, Ge ZD, Van Orman J, Barron M, Rudy-Reil D, Hacker TA, Misra R, Duncan SA, Auchampach JA, Lough JW. Improved cardiac function in infarcted mice after treatment with pluripotent embryonic stem cells. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:1216–24. doi: 10.1002/ar.a.20388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wan TC, Ge ZD, Tampo A, Mio Y, Bienengraeber MW, Tracey WR, Gross GJ, Kwok WM, Auchampach JA. The A3 adenosine receptor agonist CP-532,903 [N6-(2,5-dichlorobenzyl)-3′-aminoadenosine-5′-N-methylcarboxamide] protects against myocardial ischemia/reperfusion injury via the sarcolemmal ATP-sensitive potassium channel. J Pharmacol Exp Ther. 2008;324:234–43. doi: 10.1124/jpet.107.127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cesura AM, Pinard E, Schubenel R, Goetschy V, Friedlein A, Langen H, Polcic P, Forte MA, Bernardi P, Kemp JA. The voltage-dependent anion channel is the target for a new class of inhibitors of the mitochondrial permeability transition pore. J Biol Chem. 2003;278:49812–8. doi: 10.1074/jbc.M304748200. [DOI] [PubMed] [Google Scholar]
- 24.Mio Y, Shim YH, Richards E, Bosnjak ZJ, Pagel PS, Bienengraeber M. Xenon preconditioning. The role of prosurvival signaling, mitochondrial permeability transition and bioenergetics in rats. Anesth Analg. 2009;108:858–66. doi: 10.1213/ane.0b013e318192a520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79:609–34. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- 26.Braunwald E, Kloner RA. Myocardial reperfusion. A double-edged sword? J Clin Invest. 1985;76:1713–9. doi: 10.1172/JCI112160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumura K, Jeremy RW, Schaper J, Becker LC. Progression of myocardial necrosis during reperfusion of ischemic myocardium. Circulation. 1998;97:795–804. doi: 10.1161/01.cir.97.8.795. [DOI] [PubMed] [Google Scholar]
- 28.Gross GJ, Auchampach JA. Reperfusion injury. Does it exist? J Mol Cell Cardiol. 2007;42:12–8. doi: 10.1016/j.yjmcc.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 30.Jones SP, Girod WG, Palazzo AJ, Granger DN, Grisham MB, Jourd'Heuil D, Huang PL, Lefer DJ. Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am J Physiol. 1999;276:H1567–73. doi: 10.1152/ajpheart.1999.276.5.H1567. [DOI] [PubMed] [Google Scholar]
- 31.Amour J, Brzezinska AK, Weihrauch D, Billstrom AR, Zielonka J, Krolikowski JG, Bienengraeber MW, Warltier DC, Pratt PF, Jr, Kersten JR. Role of heat shock protein 90 and endothelial nitric oxide synthase during early anesthetic and ischemic preconditioning. Anesthesiology. 2009;110:317–25. doi: 10.1097/ALN.0b013e3181942cb4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michel T, Feron O. Nitric oxide synthases. Which, where, how, and why? J Clin Invest. 1997;100:2146–52. doi: 10.1172/JCI119750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Depre C, Fierain L, Hue L. Activation of nitric oxide synthase by ischaemia in the perfused heart. Cardiovasc Res. 1997;33:82–7. doi: 10.1016/s0008-6363(96)00176-9. [DOI] [PubMed] [Google Scholar]
- 34.Brahmajothi MV, Campbell DL. Heterogeneous basal expression of nitric oxide synthase and superoxide dismutase isoforms in mammalian heart. Implications for mechanisms governing indirect and direct nitric oxide-related effects. Circ Res. 1999;85:575–87. doi: 10.1161/01.res.85.7.575. [DOI] [PubMed] [Google Scholar]
- 35.Tsang A, Hausenloy DJ, Yellon DM. Myocardial postconditioning. Reperfusion injury revisited. Am J Physiol Heart Circ Physiol. 2005;289:H2–7. doi: 10.1152/ajpheart.00091.2005. [DOI] [PubMed] [Google Scholar]
- 36.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307:93–8. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–5. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 38.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–9. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 39.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 40.Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277:34793–9. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 41.Javadov SA, Clarke S, Das M, Griffiths EJ, Lim KH, Halestrap AP. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J Physiol. 2003;549:513–24. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M. Postconditioning inhibits mitochondrial permeability transition. Circulation. 2005;111:194–7. doi: 10.1161/01.CIR.0000151290.04952.3B. [DOI] [PubMed] [Google Scholar]
- 43.Clarke SJ, Khaliulin I, Das M, Parker JE, Heesom KJ, Halestrap AP. Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circ Res. 2008;102:1082–90. doi: 10.1161/CIRCRESAHA.107.167072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–42. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 45.Yang XP, Liu YH, Shesely EG, Bulagannawar M, Liu F, Carretero OA. Endothelial nitric oxide gene knockout mice. Cardiac phenotypes and the effect of angiotensin-converting enzyme inhibitor on myocardial ischemia/reperfusion injury. Hypertension. 1999;34:24–30. doi: 10.1161/01.hyp.34.1.24. [DOI] [PubMed] [Google Scholar]
- 46.Ruetten H, Dimmeler S, Gehring D, Ihling C, Zeiher AM. Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovasc Res. 2005;66:444–53. doi: 10.1016/j.cardiores.2005.01.021. [DOI] [PubMed] [Google Scholar]