

Abstract
Disease processes provoke a balancing act between tissue damage and repair. In the 1980s, the discovery that tumor necrosis factor (TNF)-α is a general mediator of disease-related injury led to the development of novel therapeutics to neutralize its activity. In contrast, identification of potential mediator(s) of tissue repair remained elusive. Studies performed over the last 15 years have documented that the type 1 cytokine erythropoietin (EPO), produced by cells within surrounding regions subjected to injury, acts as a master regulator, controlling both damage and repair. The transducer of these activities is the previously unrecognized innate repair receptor (IRR), which is comprised of the EPO receptor and β common receptor subunits. Notably, although proinflammatory cytokines upregulate the IRR, EPO and proinflammatory cytokines inhibit each other’s production, resulting in a relative underproduction of EPO. Although exogenous EPO attenuates disease activity in many preclinical models, its clinical utility is limited by serious hematopoietic and thrombotic adverse effects. To circumvent this problem, novel compounds engineered from the structure of EPO have been developed as selective ligands of the IRR. These compounds possess no hematopoietic activity, yet are fully tissue-protective and reparative. The lead molecule of this development effort (the 11–amino acid peptide ARA290) tips the balance toward healing in diverse preclinical models of disease and is currently under evaluation in advanced clinical trials as a disease-modifying agent in painful neuropathy and diabetes.

By definition, disease processes injure tissues and organs. Although attention generally has focused on the damage done, injury has another critical consequence in the subacute timeframe: the activation of healing and repair. The outcome for an individual afflicted by disease depends on the balance of these two processes. Tony Cerami’s investigations carried out over the last four decades have been instrumental in understanding the mediators and mechanics of this biological balancing act. On the damage side of the process, identification of tumor necrosis factor (TNF)-α and understanding its role in cachexia started a flurry of productive work in the field of the proinflammatory cytokines. These advances led directly to the development of novel therapeutics for a number of serious chronic diseases (for example, rheumatoid arthritis) for which there had previously been no truly effective treatment. Tony’s contribution to understanding the repair side of the injury process only came much later, but interestingly cycled back to his earlier work to complete the picture. In the end, these processes appear to be two sides of the same coin.
The discovery of proinflammatory cytokines generated a conundrum for Tony. Specifically, proinflammatory processes are self-amplifying. The dramatic pathological state of sepsis leading to cardiovascular collapse and death is one clear example. How is it possible that mild to moderate injuries do not usually spin out of control? Obviously, an endogenous mechanism must exist to reign in inflammation. Although Tony and his team searched diligently for the mediator of this control, it eluded identification for decades.
In 1998, Tony came to Yale, where I was an endocrinologist, and delivered an inspiring lecture on the discovery of TNF-α. Our common roots of the graduate program at the Rockefeller University had led to our paths crossing a number of times since the mid-1970s. In catching up after the lecture, Tony invited me to come visit the Kenneth S. Warren Laboratory, a new nonprofit endeavor co-founded by Tony and his daughter Carla. After discussion, he invited me to join him in the pursuit of “interesting scientific questions.” At that time, erythropoietin (EPO), the hormone that mediates red cell production, was being developed as a treatment for anemia in patients with cancer. During the course of clinical studies, clinicians had noticed that patients frequently “perked up” cognitively, in advance of any increase in red cell number. The company marketing EPO to this group of patients (Orthbiotech) gave Tony, Carla and me a challenge (and, importantly, a grant) to determine whether EPO directly affects cognitive function and well-being. At the time, this concept was not consistent with the accepted mechanism of action of EPO, which was viewed as being restricted to the bone marrow and the production of red cells. It was known, however, that cancer patients often exhibited elevated circulating levels of proinflammatory cytokines (for example, TNF-α), which inhibited EPO production and caused anemia. Additionally, proinflammatory cytokines were known to degrade cognitive function and general well-being. We speculated that EPO administered to these patients might reduce TNF-α production and hence improve cognition. Alternatively, EPO could directly affect cognition.
My doctoral work at the Rockefeller University was in the area of neurobiology and behavior. During this work, I spent two instructive years with Neal Miller and Barry Dworkin in the area of behavioral neuroscience (that is, how central nervous system activity relates to behavior, and vice versa). The challenge was to determine whether EPO (a large protein that, according to the scientific paradigm at that time, could not possibly cross the blood–brain barrier) had direct effects on the central nervous system. In discussion, we decided that the phenomenon of conditioned taste aversion would be an ideal methodology to evaluate whether EPO possessed any cognitive activity.
Conditioned taste aversion is a robust form of learning in which a gustatory stimulus paired with illness is subsequently vigorously avoided. This learning is hypothesized to have evolved to avoid the effects of repeated food poisoning. In an experimental paradigm, illness is typically produced by the administration of lithium chloride, which, in rodents, causes illness and retching behavior. When lithium is then paired with a distinctive taste (for example, vanilla), the test subjects assiduously avoid any further ingestion of vanilla. It was also known that, similar to other toxins, lithium chloride administered systemically stimulates the wide-spread production of TNF-α.
Our hypothesis was that the EPO would inhibit lithium-induced TNF-α production and, by this mechanism, would attenuate the establishment of conditioned taste aversion. To test this, mice were trained to obtain their full daily water requirements over a single 10-min period. The volume of water ingested each day was recorded. After the animals became trained to the protocol, we injected all animals with lithium via the intraperitoneal route and also administered EPO to half the group. At the same time, we added vanilla extract to the water for the first time. Of course, lithium made the animals sick, and none would drink vanilla water for a day or two. But in contrast to the animals that had received saline, the EPO animals avoided vanilla water for additional days, becoming very dehydrated before finally drinking. EPO, contrary to our expectations, had amplified the learning experience (1). This outcome was reminiscent of the results observed after injection of nerve growth factor directly into the brain, since nerve growth factor is not active when given systemically. Clearly, the EPO we administered must have passed the blood–brain barrier to affect cognition. Direct measurements of time-dependent appearance of EPO into the cerebral spinal fluid after intraperitoneal injection confirmed this concept (1).
This observation stimulated a paradigm shift for us. EPO, a highly glycosylated protein of molecular weight ~30 kDa, penetrated the blood–brain barrier. To follow up on this concept, the direct effect of EPO on cognitive function was evaluated by using a Morris water maze, a common methodology to evaluate learning. In these experiments, mice were placed in a large water bath with a platform on which to stand, hidden below the surface of the water. The water was made opaque by adding a dye. If the animals could identify the location of the platform, they could rest from swimming. To help identify where the platform was located, distinctive visual cues were placed around the edges of the water tank, so that the animals could learn by trial and error. Remarkably, animals administered EPO learned the location of the platform more rapidly than those receiving saline. A direct effect of peripherally administered EPO on cognitive function was confirmed.
At this time, we were also aware of reports from Professor Sasaki’s laboratory in Japan that direct injection of EPO into rodent brain was protective of hypoxia (2). Furthermore, injection of a soluble hematopoietic EPO receptor (that complexed any endogenous EPO present) greatly amplified injury. On the basis of our results from the conditioned taste aversion experiments, we now predicted that peripherally administered EPO would be protective of this type of brain injury. Our experiments using an ischemic stroke model confirmed this hypothesis (3). Remarkably, EPO administered even 6 h after a stroke was effective in significantly reducing the lesion size. Follow-up studies have shown that EPO administered as late as several days after experimental stroke is beneficial.
Additionally, in this work, there was a hint of the mechanism of action of the neuroprotective activity of EPO. By using an in vitro system of neuronal injury, the Sasaki group also showed that EPO added to the culture medium could directly antagonize the toxic effects of an excitatory amino acid neuro-transmitter (4). The particulars of the experiment suggested that the programmed cell death that excitotoxins activated was antagonized by EPO. Tony and I considered the stroke model and realized that the so-called penumbra surrounding the central core of the lesion was composed of cells that were not irrevocably injured, but rather were programmed to undergo apoptosis. Similar to the function of EPO in the bone marrow—the rescue of red cell progenitors that otherwise undergo apoptosis—EPO was turning off programmed cell death in otherwise functional cells. Tony’s unique perspective on the biology of proinflammatory cytokines equipped him to verbalize a biological pathway to make sense of this observation. In his mind, a primary evolutionary role of proinflammatory cytokines, for example, TNF-α, was to control the invasion of parasites. In this setting, TNF-α generation by tissue surrounding the pathogen acts to sterilize the surrounding environment by killing normal tissue by activating in part programmed cell death. In this manner, the invader was localized and destroyed.
Clearly, the temporal response to injury at the tissue level was quite stereotyped. First, injury elicited the local generation of proinflammatory cytokines. Then these cytokines diffused away from the initiating focus, programming nearby cells for death. In the experimental stroke model, cells within the penumbra could be rescued by EPO. Although exogenous EPO was effective, what was its source in vivo? The kidney was not a reasonable hypothesis. The answer, again, came from Sasaki’s group, which reported that glial cells derived from the nervous system produced EPO in response to hypoxia (5).
The paradigm at that point became obvious: EPO was made in the tissue surrounding the injury site. It was the concentration profiles of EPO into the region and TNF-α out of the region that defined the lesion size. Further, a receptor for EPO was specifically expressed only by the cells within the penumbra. Therefore, unstressed tissues that did not express this receptor were unresponsive to the EPO produced. Implicit in this hypothesis was the concept that this molecular yin-yang occurred for any type of injury, not just ischemic. Subsequent work has confirmed and extended this idea. It became clear, for example, that similar to the well-known suppressive effects of inflammation on renal EPO production, and therefore on red cell production, TNF-α also inhibited local production of EPO. This result provided the rationale for administration of exogenous EPO as a substitute for depressed local synthesis. We also suspected that the reverse was true: EPO could inhibit proinflammatory cytokine production.
One of our experiments to test this hypothesis was a traumatic brain injury model performed in the mouse. In this protocol, a hydraulic piston delivered a calibrated impact to an anesthetized mouse, causing a well-defined brain injury. Untreated, this resulted in a large cavitary lesion that developed within 10 d after trauma. Treatment with EPO before or up to 6 h after the injury resulted in a dramatic reduction of injury (Figure 1). Examining these tissues histologically revealed a crucial finding: brains from the EPO-treated animals possessed few inflammatory cells in the vicinity of the lesion, compared with the damaged brains of saline-treated animals, which exhibited abundant leukocyte infiltration. Clearly, EPO also had a marked effect on the inflammatory response.
Figure 1.

EPO protects the brain from traumatic injury. Blunt trauma delivered to the left side of the cerebral cortex of a mouse produces a large cavitary lesion densely populated by inflammatory cells by 7–10 d after injury (top). Administration of EPO after injury (bottom) markedly reduces the size of the lesion. Few inflammatory cells can be found within and surrounding the area of damage. Image reproduced from (3) (copyright © 2000, The National Academy of Sciences).
The results of further studies confirmed that EPO could markedly reduce injury-related proinflammatory cytokine production in a variety of models (6). Much later, abundant work has shown that immune-competent cells—for example, macrophages, microglia (the macrophage equivalent of the central nervous system), lymphocytes, as well as others—expressed a receptor for EPO and therefore could be modulated by EPO.
As the experimental work progressed, we made a serendipitous, crucial observation: surgical wounds in the experimental animals receiving EPO appeared to heal much faster. Even the fur shaved to perform surgery grew back more quickly. We quantified this and confirmed that EPO accelerated the healing process (7). There were also observations by others that in vitro EPO acted as a trophic molecule supporting neurite growth. The “aha” moment occurred when we realized that the function of locally produced EPO was not only to turn off excessive inflammation, but also to activate healing, restorative processes. In this setting, EPO exhibited direct regenerative effects, for example, stimulating new blood vessel growth or recruitment of stem cells into the damaged region (8), which then differentiate into adult phenotypes. Additionally, EPO also stimulated the production of other trophic molecules, for example, brain-derived neurotrophic factor (9). In this view, EPO functioned as a “master regulator,” controlling both arms of the injury-repair response (Figure 2).
Figure 2.

EPO is a master regulator of injury and repair. EPO produced locally in response to injury or metabolic stress prevents injury and attenuates inflammation, reducing the cascade of proinflammatory cytokines. Simultaneously, EPO activates tissue repair. This process includes the recruitment of stem cells into the region of injury, for example, progenitor vascular endothelial cells that participate in angiogenesis, leading to tissue regeneration.
However, one conceptual issue with this idea was the potential cross-talk between circulating, hormonal EPO of renal origin and locally produced paracrine/autocrine EPO. This dilemma was resolved by the identification of two characteristics by which these systems differed radically. One involved the EPO molecule itself; the other involved the receptor mediating the biological effects. Together, these factors assured that, under normal circumstances, there could be no cross-talk between these two systems.
First, to function in its hematopoietic role, EPO contains sugars terminated by sialic acids. This feature provides for a relatively long half-life in the circulation (~4–5 h). In contrast, the EPO made in vitro by glial cells is poorly sialated (5). This form of EPO can be produced by enzymatically desialating recombinant human EPO. As previously known, desialated EPO (asialo-EPO) has a short circulating half-life (~2 min) and does not activate erythropoiesis in vivo. In spite of this inactivity in the bone marrow, we showed that asialo-EPO was fully effective in a tissue-protective mode (10). For example, in a spinal cord injury model, asialo-EPO performed as well as recombinant human EPO (rhEPO) in protecting tissue from injury and restoring function.
A second protection from cross-talk occurred at the level of the receptor. Additional work in Sasaki’s lab had shown that although an EPO receptor (EPOR) was expressed by neural cells, it exhibited a lower affinity and differed biochemically from the erythroid EPOR (11). To us, this obviously suggested the presence of a receptor isoform. EPO is a member of the type 1 cytokine family. Receptors for the family members are typically multimeric and constructed from distinct subunits. Review of the literature of erythropoiesis revealed that the β common receptor (also known as CD131, a component of the receptors for granulocyte-macrophage colony-stimulating factor, interleukin [IL]-3 and IL-5), appeared to be physically associate with EPOR in some experimental systems. However, CD131 knockout mice were subsequently developed and showed unequivocally that the β common receptor was not involved in erythropoiesis.
We were intrigued by the possibility that an EPOR-CD131 receptor underpinned the tissue-protective/restorative activities of EPO. Examination of tissues and cells using molecular methods confirmed that EPOR and CD131 were physically linked via cysteine residues (12). But to confirm this hypothesis, we needed to neutralize the biological activity of the β common receptor. The CD131 knockout mouse was an obvious choice. Tony, in his typical direct manner, quickly contacted the originators of this mouse and obtained some breeding pairs for us to study. As predicted, tissue injury models performed in these mice were not sensitive to the administration of EPO (12). To distinguish this receptor from the hematopoietic EPOR, we have designated it as the innate repair receptor (IRR), since its activation attenuates injury while initiating healing.
Although rhEPO proved to be markedly active in injury models and in healing, including a proof of concept clinical trial in ischemic stroke (13), the required dosages are higher than those used to treat anemia. Unfortunately, large doses of rhEPO are associated with obvious adverse effects. For example, in addition to mediating erythropoiesis, rhEPO induces vascular endothelial cells to express selectins and promotes platelet production (both of which are markedly prothrombotic actions). Indeed, results of large clinical trials performed using rhEPO showed a beneficial effect of EPO on survival, but with the side effect of clinically significant thrombosis (14).
Although the beneficial effects of rhEPO are impressive in a wide range of preclinical models, is there another way forward? Asialo-EPO was not a true solution, since that molecule interacted with both receptor types, not completely eliminating potential adverse effects when used for chronic dosing. To us, the alternate receptor suggested that molecules could be designed to preferentially interact with only the IRR.
We began this process by examining the structure of the hematopoietic receptor. Fortunately, the precise recognition sites of EPO and EPOR for each other were understood to the level of the interacting amino acids. In considering the amino acids responsible for binding to the receptor, Tony reached back to his earlier work in sickle cell disease and suggested that we could carbamylate the EPO molecule. This chemical modification would change the positively charged lysines involved in the binding site into the neutral amino acid homocitrulline. Carbamylated EPO did not bind to the hematopoietic receptor. Would this modified EPO indeed retain tissue-protective/restorative properties?
Evaluation of a wide range of models showed that carbamylated EPO fully retained its beneficial effects in injury but exhibited no hematopoietic potency in vivo or in vitro (15). At that time, we worked with H. Lunbeck A/S (Denmark) to translate this technology into the clinic. Marcel Leist, Thomas Sager, Lars Torup and a host of other colleagues (especially Carla Cerami, Qiaowen Xie and Thomas Coleman from our own group) worked productively together to explore the biology and practical issues of development of this compound. There were a number of other ways to chemically modify EPO, and some of these methods also proved to be effective innate repair activators. Similarly, genetic manipulation of the hematopoietic receptor recognition sites within the EPO molecule also proved to be a successful approach to produce specific ligands for the IRR.
The signaling pathways involved in the IRR activation (Figure 3) are multiple and overlap in part with those used in hematopoiesis (16). Similar to other type 1 cytokines signaling, the initial step is phosphorylation of Janus kinase 2. Thereafter, three principal pathways have been identified, with some variation between different tissues. Like erythropoiesis, the signal transducer and activator of transcription (STAT) pathway is involved in antiapoptosis. The phosphatidylinositol 3-kinase pathway modulates apoptosis as well as stem cell migration and regional blood flow that underpins healing and tissue repair. Finally, the mitogen-activated protein (MAP) kinase pathway reduces inflammation and edema as well as mediates antiapoptosis.
Figure 3.

The IRR mediates tissue-protective and -reparative activities via multiple intracellular pathways. Occupancy of the EPOR-β common receptor heteromer results in Janus kinase-2 phosphorylation that subsequently leads, via diverse pathways, to a decrease in injury (red outlined boxes) and to regeneration (blue outlined boxes). Image reproduced from (16).
Unfortunately, in terms of pharmaceutical manufacturing as a protein, EPO is a biological compound. This designation means that drug development is exceeding costly. Additionally, the EPO variants that we produced had some other less-than-optimum characteristics, including long-term stability issues requiring a cold storage chain and the potential for antibody formation. Therefore, in spite of our successes, we continued to think about the structure of EPO with the goal to derive additional IRR ligands. In this work, it was clear from the three-dimensional structure of EPO—a four-helix globular protein—that the helices provided a relatively rigid structure to the molecule. Therefore, the exterior portions of these helices were always oriented toward the aqueous medium. Three of these helices interacted with the two binding sites of the hematopoietic EPOR homodimer (dashed boxes 1 and 2 in Figure 4). The only one that did not was helix B. Interestingly, this region of EPO is relatively highly evolutionarily conserved (17) (as are the regions that recognize the binding sites of the hematopoietic receptor), suggesting that it plays a biologically important function. Was this the portion of the EPO molecule recognized by the IRR?
Figure 4.
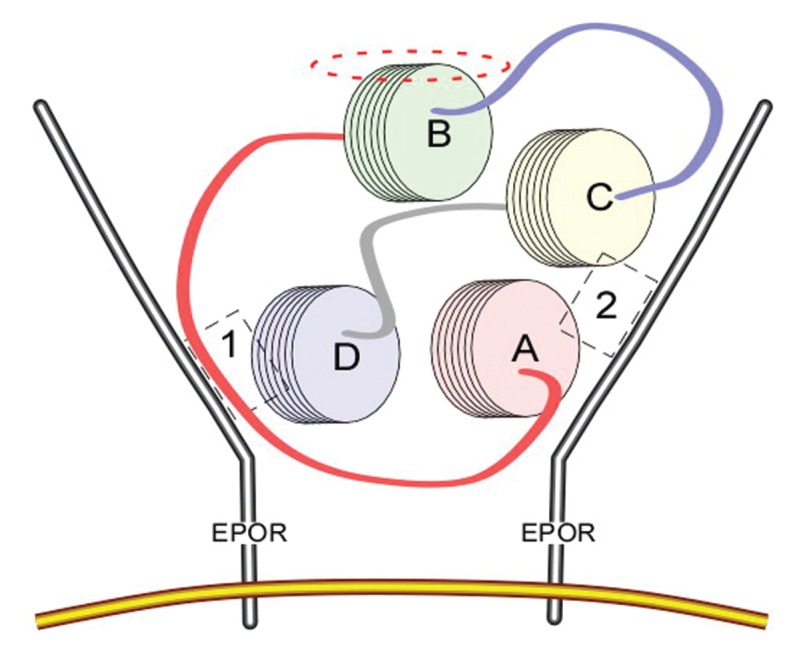
EPO binding the homodimeric receptor mediating red cell production via binding sites located on each receptor monomer (sites 1 and 2, as indicated in the dashed boxes). The portions of the EPO molecule interacting with the receptor consist of portions of helices A, C and D. In contrast, helix B faces away from the receptor. The exterior portion of helix B (dashed ellipse) is highly conserved from an evolutionary perspective and binds to the IRR, a heteromeric receptor consisting of EPOR and β common receptor subunits. Image reproduced from (18) (copyright © 2008 by The National Academy of Sciences of the USA).
Following this line of thought, we speculated that if helix B interacted with the receptor, the responsible amino acids would be those constituting the external face. Using the three-dimensional structural data available in the protein data bank, we synthesized an 11–amino acid peptide consisting of these amino acids in a linearized form. This peptide, with no primary sequence homology with EPO but recapitulating its three-dimensional exterior structure, proved to be as active as EPO in a wide variety of model systems (18). As predicted for a small peptide (1,257 daltons), the plasma half-life was short, similar to asialo-EPO (~2 min). Despite this short half-life, but similar to signal transduction by other cytokines, triggering of the IRR activated a self-sustained biological cascade that did not require continued receptor occupancy.
Preclinical toxicology showed that this compound (helix B surface peptide; ARA290) raised no safety issues. Favorable results of phase I studies performed in normal volunteers also encouraged further development. On the basis of his past experience in selection of clinical studies to support clinical development, Tony understood that the particular clinical endpoint chosen for evaluation was critical for success in translating a new molecule such as ARA290 from preclinical promise to clinical utility. With the almost endless possibilities a master regulator of tissue injury and repair offers, selection of an appropriate endpoint proved especially important as well as challenging.
Several years ago, Albert Dahan at Leiden University Medical Center brought to our attention the unmet medical need of the condition of painful neuropathy. This is a fascinating syndrome where injury to a nerve, often minor, produces dramatic, incapacitating pain that is resistant to treatment. In subsequent preclinical studies, ARA290 proved remarkably effective in preventing development of pain (19) and additionally showed efficacy in accelerating nerve regeneration. ARA290 had no effect when the same experiment was performed using β common knockout mice that lack a functional IRR. Additionally, the model used is characterized by intense inflammation within the spinal cord at level of entry of the injured peripheral nerve. ARA290 administration dramatically reduced this inflammatory process as well (20).
One type of neuropathic pain arises from damage and loss of small nerve fibers of the sensory and autonomic nervous systems, termed “small fiber neuropathy” (SFN). This condition is a relatively newly recognized clinical entity that is difficult to diagnose because of its protean symptoms. Yet SFN commonly occurs in a wide spectrum of diseases. One condition that has a high prevalence of SFN with currently no treatment options is the orphan disease sarcoidosis. Studies carried out in this patient population have provided promising results in terms of alleviating pain and other symptoms due to small nerve fiber damage and loss, as well as evidence supportive of nerve repair and re-growth (21,22). Studies are ongoing in this group of patients.
Diabetes is a disease that has a major worldwide public health impact and has been one focus of Tony’s work. The underlying pathophysiology of the disease involves, in part, progressive damage to the pancreatic islets that secrete insulin. With our colleague Claus-Goran Ostenson in Stockholm, we have shown in an animal model that ARA290 is protective of islet injury and, further, can improve islet function. Additionally, hyperglycemia causes damage to multiple tissues, including the nervous system. Another collaborator, Robert Schmidt, showed that in a murine model of diabetic autonomic neuropathy (a form of SFN) that ARA290 reverses neuronal injury (23). Because SFN is one prominent problematic issue with patients that have had longstanding glucose intolerance, we are currently studying patients with painful neuropathy and type 2 diabetes to determine the effects of ARA290 on metabolic control and neuropathic symptoms. In these studies, a primary endpoint is hemoglobin A1c. It is a remarkable fact that one of Tony’s earlier clinical triumphs presently constitutes an essential endpoint of this latest clinical development program.
From an intellectual perspective, the voyage we have undertaken into this area of investigation has been fascinating. The identification of a master regulator of injury and repair and the development of specific analogs offers the opportunity to positively affect treatment of many different diseases currently untreated or undertreated. It has been instructive and inspiring to observe how Tony has always jumped forward by being able to go back and retrieve information from his previous work (and critical contacts) that proved to be instrumental for success. As in most successful collaborations, our individual abilities have proved to be synergistic of each other. In a parallel situation to the physical and mathematical sciences, what initially seemed to be completely separate fields—inflammation and healing—has actually proven to be integrally related. Finally, in addition to being rewarding, the road taken has proven to be an enjoyable adventure.
ACKNOWLEDGMENTS
As can easily be observed by looking at the publications arising from this area of research over the years, I have been especially blessed by an abundance of collaborators of the highest caliber from around the world. Unfortunately, there are too many to mention here in the concern that I will inadvertently fail to acknowledge someone. It has always amazed me how many individuals take considerable time to meet and discuss topics that are often somewhat outside of their area of core expertise. Without this collegiate collaboration, this work could not have progressed as rapidly or as far as it has. I also sincerely thank the many volunteers and patients, as well as their families, for agreeing to participate in our clinical research programs.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
M Brines is an officer of Araim Pharmaceuticals Inc., which is developing ARA290 for clinical uses.
REFERENCES
- 1.Ehrenreich H, et al. Erythropoietin: a candidate compound for neuroprotection in schizophrenia. Mol. Psychiatry. 2004;9:42–54. doi: 10.1038/sj.mp.4001442. [DOI] [PubMed] [Google Scholar]
- 2.Sakanaka M, et al. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci U S A. 1998;95:4635–4640. doi: 10.1073/pnas.95.8.4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brines ML, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci U S A. 2000;97:10526–31. doi: 10.1073/pnas.97.19.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience. 1997;76:105–16. doi: 10.1016/s0306-4522(96)00306-5. [DOI] [PubMed] [Google Scholar]
- 5.Masuda S, et al. A novel site of erythropoietin production: oxygen-dependent production in cultured rat astrocytes. J Biol Chem. 1994;269:19488–93. [PubMed] [Google Scholar]
- 6.Brines M, Cerami A. Erythropoietin-mediated tissue protection: reducing collateral damage from the primary injury response. J Intern Med. 2008;264:405–32. doi: 10.1111/j.1365-2796.2008.02024.x. [DOI] [PubMed] [Google Scholar]
- 7.Erbayraktar Z, et al. Nonerythropoietic tissue protective compounds are highly effective facilitators of wound healing. Mol Med. 2009;15:235–41. doi: 10.2119/molmed.2009.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaneko N, Kako E, Sawamoto K. Enhancement of ventricular-subventricular zone-derived neurogenesis and oligodendrogenesis by erythropoietin and its derivatives. Front Cell Neurosci. 2013;7:235. doi: 10.3389/fncel.2013.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viviani B, et al. Erythropoietin protects primary hippocampal neurons increasing the expression of brain-derived neurotrophic factor. J Neurochem. 2005;93:412–21. doi: 10.1111/j.1471-4159.2005.03033.x. [DOI] [PubMed] [Google Scholar]
- 10.Erbayraktar S, et al. Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc Natl Acad Sci U S A. 2003;100:6741–6. doi: 10.1073/pnas.1031753100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masuda S, et al. Functional erythropoietin receptor of the cells with neural characteristics: comparison with receptor properties of erythroid cells. J Biol Chem. 1993;268:11208–16. [PubMed] [Google Scholar]
- 12.Brines M, et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci U S A. 2004;101:14907–12. doi: 10.1073/pnas.0406491101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehrenreich H, et al. Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med. 2002;8:495–505. [PMC free article] [PubMed] [Google Scholar]
- 14.Corwin HL, et al. Efficacy and safety of epoetin alfa in critically ill patients. N Engl J Med. 2007;357:965–76. doi: 10.1056/NEJMoa071533. [DOI] [PubMed] [Google Scholar]
- 15.Leist M, et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004;305:239–42. doi: 10.1126/science.1098313. [DOI] [PubMed] [Google Scholar]
- 16.Brines M, Cerami A. The receptor that tames the innate immune response. Mol Med. 2012;18:486–96. doi: 10.2119/molmed.2011.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brines M, Cerami A. Erythropoietin and engineered innate repair activators. Methods Mol Biol. 2013;982:1–11. doi: 10.1007/978-1-62703-308-4_1. [DOI] [PubMed] [Google Scholar]
- 18.Brines M, et al. Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc Natl Acad Sci U S A. 2008;105:10925–30. doi: 10.1073/pnas.0805594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swartjes M, et al. ARA290, a peptide derived from the tertiary structure of erythropoietin, produces long-term relief of neuropathic pain: an experimental study in rats and beta-common receptor knockout mice. Anesthesiology. 2011;115:1084–92. doi: 10.1097/ALN.0b013e31822fcefd. [DOI] [PubMed] [Google Scholar]
- 20.Swartjes M, et al. ARA 290, a peptide derived from the tertiary structure of erythropoietin, produces long-term relief of neuropathic pain coupled with suppression of the spinal microglia response. Mol Pain. 2014;10:13. doi: 10.1186/1744-8069-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dahan A, et al. ARA 290 improves symptoms in patients with sarcoidosis-associated small nerve fiber loss and increases corneal nerve fiber density. Mol Med. 2013;19:334–45. doi: 10.2119/molmed.2013.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heij L, et al. Safety and efficacy of ARA 290 in sarcoidosis patients with symptoms of small fiber neuropathy: a randomized, double-blind pilot study. Mol Med. 2012;18:1430–6. doi: 10.2119/molmed.2012.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt RE, et al. Effect of insulin and an erythropoietin-derived peptide (ARA290) on established neuritic dystrophy and neuronopathy in Akita (Ins2 Akita) diabetic mouse sympathetic ganglia. Exp Neurol. 2011;232:126–35. doi: 10.1016/j.expneurol.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]