

Abstract
Despite the tremendous hurdles presented by the complexity of the liver’s structure and function, advances in liver physiology, stem cell biology and reprogramming, and the engineering of tissues and devices are accelerating the development of cell-based therapies for treating liver disease and liver failure. This State of the Art Review discusses both the near and long-term prospects for such cell-based therapies and the unique challenges for clinical translation.
Introduction
Liver disease and the subsequent loss of liver function is an enormous clinical challenge, and is currently the twelfth most frequent cause of death in the United States and the fourth most frequent for middle-aged adults (1). The situation is progressively worsening, prompted by several factors including the emergence of new liver diseases such as non-alcoholic fatty liver disease (NAFLD) and steatohepatitis, the lack of a hepatitis C vaccine, and an aging population of hepatitis patients at risk for progression to hepatocellular carcinoma (2, 3). Liver transplantation is the primary treatment for liver failure and is the only therapy shown to directly alter mortality. In order to expand the supply of available livers for transplant, numerous surgical options have been pursued, including split liver transplants and living-related partial donor procedures (4). In spite of these surgical advances and improvements in organ allocation, organ shortages remain acute, suggesting that it is unlikely that liver transplantation procedures alone will ever meet the increasing demand. Cell-based therapies have long held promise as an alternative to organ transplantation. In this State of the Art Review, we will describe both near and long-term prospects for cell-based treatments, including the use of stem cells and other non-hepatocyte sources and tissue engineering, within the context of clinical manifestations of liver disease. We will discuss the unique potential and big challenges that exist for cell-based approaches and will provide an overview of fundamental biological questions, technological tools, and future directions for the field.
The Liver in Health and Disease
The liver is the largest internal organ in the body, accounting for 2–5% of body weight, and performs a complex array of over 500 functions including metabolic, synthetic, immunologic, and detoxification processes. The liver also exhibits a unique capacity for regeneration, with the potential for full restoration of liver mass and function even after massive damage in which less than one-third of the cells remain uninjured (5, 6). In fact, procedures such as partial liver transplants take advantage of this significant regenerative potential combined with the body’s finely tuned homeostatic regulation of liver mass. However, the potential for liver regeneration is often difficult to predict clinically and criteria for identifying patients that may resolve liver failure complications due to regenerative responses remain poorly defined. As a result, efforts have been made towards the development of liver support technologies that could provide temporary function for patients with liver failure, thereby enabling sufficient time for regeneration of the native liver tissue or serving as a bridge to transplantation. These measures include extracorporeal support devices that act in a manner analogous to kidney dialysis systems, processing the blood or plasma of liver failure patients (7, 8). Initial designs based on non-biological exchange/filtering systems have showed limited clinical success, likely due to the insufficient level of hepatocellular functions exhibited by these devices. In order to provide a larger complement of important liver functions, including synthetic and regulatory processes, support devices incorporating living hepatic cells have been developed, although these systems remain primarily experimental to date (9). In addition to temporary extracorporeal platforms, the development of cell-based therapies aimed at the replacement of damaged or diseased liver tissue is an active area of research. For instance, the transplantation of isolated liver cell types, such as mature hepatocytes, has been extensively explored (10) and has potential as an attractive therapeutic option particularly for inherited single gene metabolic deficiencies. Moreover, liver tissue engineering approaches, wherein preformed cellular constructs are implanted as therapeutics, are under development. Finally, these engineered tissues are also being explored as in vitro model systems for fundamental and applied studies of liver function in healthy and diseased states.
The development of liver cell-based therapies poses unique challenges, largely stemming from the scale and complexity of liver structure and function. The organ displays a repeated, multicellular architecture, in which hepatocytes, the main parenchymal cell of the liver, are arranged in cords that are sandwiched by extracellular matrix in the space of Disse (Figure 1). The space between cords is also home to a multitude of supporting cell types such as sinusoidal endothelial cells, Kupffer cells, biliary ductal cells, and stellate cells. Due to this architectural arrangement and cellular heterogeneity, the hepatocytes are exposed to gradients of nutrients, hormones, and growth factors delivered via the combined blood supply of the portal vein and hepatic artery. In particular, a major challenge that has hindered the advancement of cell-based therapeutic strategies is the propensity of hepatocytes to lose liver-specific functions and the ability to replicate when isolated from the normal in vivo microenvironment. Microenvironmental signals including soluble factors, extracellular matrix components, and heterotypic cell-cell interactions have all been implicated in the regulation of hepatocyte survival and phenotypic stability. The success of cell-based therapies hinges on the capacity to replace or overcome such necessary signals in order to promote the survival and function of hepatocytes transferred to patients in a clinical setting. Research efforts to achieve this goal have included studies at the interface of microtechnology and cell biology, and have led to the ability to manipulate the hepatocyte phenotype by the controlled presentation of environmental cues. Such systems exhibit long-term stabilized hepatocyte function and also enable fundamental studies investigating drug-induced liver injury and hepatotrophic infections, in addition to providing insight into how clinically transplanted hepatocytes might be supported in an in vivo setting. The continued evolution of in vitro liver platforms through the incorporation of additional liver cell types and capabilities, such as perfusion and control over tissue architecture in three dimensions, will be critical for the improved understanding of mechanisms underlying hepatocyte processes and the enhanced functionality of cell-based therapeutics.
Figure 1. Structure of the Liver.

The liver is the largest internal organ in the body and performs over 500 functions, including numerous metabolic, synthetic, immunologic, and detoxification processes. (A) The liver exhibits a hierarchical structure consisting of repeated functional tissue units (liver lobules). Within a lobule, oxygenated blood enters through branches of the hepatic artery and portal vein, and flows in specialized sinusoidal vessels towards the central vein. Bile, that is produced and excreted by hepatocytes, flows in the counter direction towards the intrahepatic bile duct. (B) Hepatocytes are polarized epithelial cells that interact closely with a number of non-parenchymal cells types along the sinusoidal tracts of the liver lobule. Collectively, these cellular components and multiscale tissue structures contribute to the diverse functional roles of the liver.
Another obstacle in the progress of cell-based approaches is the limited availability of human hepatocytes. Only a small supply of human hepatocytes is currently available from organs determined to be inappropriate for transplantation. Despite the significant proliferative capacity exhibited during regeneration in vivo, as noted above, mature human hepatocyte proliferation in culture is limited. Hence, the elucidation of molecular mediators that regulate hepatocyte proliferation and that could potentially promote expansion in vitro is an active area of investigation. In addition, significant research efforts are focused on the potential of alternative cell sources, most notably leveraging stem cell differentiation and reprogramming. Based on knowledge of developmental mechanisms, recent advances in pluripotent stem cell differentiation protocols have illustrated that highly proliferative pluripotent stem cells can give rise to hepatocyte-like cells derived from a single donor (i.e. normal/disease genotype) (11–14). Yet it remains to be seen whether it will be feasible to produce these cells on a clinically-relevant scale and whether lingering safety concerns will be overcome for transplantation purposes.
In the case of hepatic failure, in particular hepatic encephalopathy, which is classified by graded alterations in mental status, the liver community faces a greater challenge – at least in some ways – than in other cases of organ failure, in that the clinical priorities for achieving functional improvement (ie. cardiac output for heart, filtration rate for kidney), are not available due to our lack of fundamental understanding of the pre-existing disease state. That is, we have limited ‘biomarkers’ that are predictive of clinical response in a liver failure patient. For example, despite the use of metabolic readouts and parameters, such as timing of jaundice, bilirubin levels, prothrombin time and age, the causes of hepatic encephalopathy are not completely understood, with distinct clinical indicators in acute versus chronic liver disease and marked patient-to-patient variability. Thus, any animal models used to assess candidate therapeutics must be very carefully chosen given that etiologies of liver failure from trauma, metabolic liver disease, and cirrhotic disease are each very distinct, and any clinical trial design must be tailored, based on the particular clinical setting in question. Despite the many challenges that remain, substantial progress has been made in the understanding of liver development, regeneration, and the development of instructive animal models. These insights, together with recent innovations in engineered in vitro culture platforms and in vivo transplantation approaches, form a strong foundation for future advancements and the ultimate implementation of cell-based therapeutics for liver disease.
Clinical manifestations of liver disease
The vast majority of liver functions are mediated by the hepatocyte, the functional metabolic unit of the liver that constitutes about two-thirds of its cell mass (15). Though liver disease does not routinely result in abdominal pain, it does lead to a variety of life-threatening metabolic and physiologic abnormalities. For example, the absence of these functions leads to bleeding abnormalities, accumulation of neurotoxins causing altered consciousness, low blood sugar and accumulation of serum ammonia, and jaundice from elevation of serum bilirubin. Unfortunately, although patients with liver disease can be medically supported through therapies targeted at features such as portal hypertension and coagulopathy, there are no therapeutic strategies that collectively augment the range of impacted functions, and thus an organ transplant has been the only permanently successful therapy to date. This approach contrasts with other organ systems, such as the heart and the kidney, in which patients with failing tissues can be given ionotropes to improve contractility, or diuretics to improve fluid balance, respectively, without the need for immediate transplantation.
The most appropriate approaches to future treatment design for patients with liver damage depend largely on the particular etiology of the organ damage in an individual case. Types of liver disease can be broadly grouped into three categories: chronic liver disease due to metabolic dysfunction, i.e., in the absence of trauma or tissue scarring; acute liver failure that does not damage normal tissue architecture but is associated with direct injury and loss of hepatocytes; and chronic liver failure that is accompanied by widespread tissue damage and scar-based remodeling, or cirrhosis (Figure 2). Whereas there will always be circumstances that call for an alternate approach to clinical intervention, these three categories are typically best targeted by distinct therapeutic strategies, as outlined below.
Figure 2. The Liver in Health and Disease.

Mechanisms that lead to hepatocyte damage and reduce liver function include drug-mediated toxicity, alcohol-induced and nonalcoholic fatty liver disease, hepatotrophic infections, and hereditary disorders. Fatty liver disease, resulting from both chronic alcohol exposure as well as nonalcoholic mechanisms, is increasingly common and leads to the chronic accumulation of fat droplets within the liver. Liver cirrhosis can be caused by hepatitis virus infection, autoimmune processes, chronic alcohol abuse, as well as chronic inflammation and fat accumulation. Cirrhosis is characterized by alterations in the sinusoidal structure and function of the liver and the accumulation of extracellular matrix, which is commonly referred to as scarring. These alterations lead to a reduction in hepatic function that can progress to hepatic failure and increased risk of hepatocellular carcinoma.
Metabolic-based chronic liver disease
Patients with life-threatening, inborn liver-based metabolic disorders, often caused by defects in single enzymes or transport proteins, are not typically accompanied by changes in liver architecture. However, even in the absence of structural change to the liver, these inborn deficits often cause injury to other organ systems, such as the brain. This is the case with urea cycle disorders, Crigler-Najjar syndrome,, and phenylketonuria. In oxalosis, the liver-based genetic abnormality leads to accumulation of oxalate crystals in the kidney and renal failure. To date, although patients suffering from this category of liver diseases are treated with organ transplantation, their clinical manifestations offer an ideal scenario for cell transplantation therapy. Given that the tissue architecture is unscarred and patients can be identified early and treated over time – prior to an acute episode - transferred hepatocytes may have both time and opportunity to engraft and replace endogenous, functionally inadequate cells.
Acute liver injury
Acute liver failure is defined as a rapid deterioration of liver function over a period of less than 26 weeks in patients who have no preexisting liver disease (16). Acute injury initially leads to necrosis of hepatocytes, which causes release of liver-specific enzymes, such as alanine transaminase (ALT) and aspartate transaminase (AST) into the blood. An increase in ALT and AST indicate hepatocyte injury but are not indicators of hepatic failure in and of themselves. Acute liver failure carries a very high mortality in many, but not all, cases, thus it is important to identify its cause as some forms can be treated or will resolve spontaneously. Clinically, the dominant findings are onset of fatigue and jaundice, a transient increase in serum concentrations of ALT and AST, followed by progressive increases in serum bilirubin, worsening coagulation, and development of altered consciousness (hepatic encephalopathy), leading to coma and brain swelling. Histologically, the liver architecture is intact except for loss or necrosis of the hepatocytes. As it is often difficult to know which patients will recover spontaneously, liver transplantation is currently the only available treatment option for severe, even if transient, acute hepatic failure. It remains unclear if hepatocyte transplantation procedures could be sufficiently optimized such that cell engraftment would occur on a clinically relevant time scale due to the urgent, acute status. The advent of extracorporeal artificial liver devices may eventually address this need (see next section) in that they could offer a clinical ‘bridge’ to address acute symptoms during a period in which the endogenous liver could be given an opportunity to recover spontaneously.
Chronic liver failure with accompanying cirrhosis
Liver failure more commonly occurs in the setting of cirrhosis, as an acute decompensation or abrupt loss of liver function, in patients with chronic liver disease. Cirrhosis of the liver caused approximately 1 million deaths worldwide in 2010 (17). Histologically, cirrhosis is characterized by expansion of the extracellular matrix with capillarization of the sinusoidal endothelium and loss of fenestrae with production of regenerative hepatic nodules (18) (Figure 2). In addition to producing symptoms of hepatic failure, the scarring from cirrhosis results in resistance to flow in the portal circulation. The increased pressure in what is normally a low pressure conduit leads to gastrointestinal bleeding, severe accumulation of abdominal ascites, and can lead to secondary dysfunction of the kidneys (hepatorenal syndrome) and lungs (hepatopulmonary syndrome). Some patients with cirrhosis are completely asymptomatic, whereas others with a similar histological picture exhibit severe symptoms of hepatic failure. Cirrhosis may be caused by hepatitis B or C infection, autoimmune processes, chronic alcohol abuse, or inflammation and fat accumulation from chronic metabolic syndromes. Additionally, in contrast to examples discussed above, some inborn errors of metabolism can lead to structural liver damage with cirrhosis and liver failure. Examples include alpha-1-antitrypsin deficiency, hemochromatosis, Wilson’s disease, hereditary tyrosinemia, and cystic fibrosis. As is currently the case for acute liver failure, the only definitive therapy for end-stage cirrhosis and liver failure is orthotopic (i.e. in the natural position) liver transplantation. While it may one day be possible for these non-acute patients to benefit from cell transplantation or implantable tissue engineered grafts, given that their symptoms should provide sufficient time for both engraftment and vascularization, there are significant barriers to overcome that may preclude these therapeutic advances from being applied. Specifically, neither the site of implantation – in this case the high-pressure portal system or the scarred microenvironment – may be amenable to accepting infiltrating cells or engineered grafts. Thus, ectopic sites of transplantation are being considered for these sorts of interventions.
Design and development of existing cell-based therapeutic interventions
Clinical effectiveness of hepatocyte transplantation
In the clinical scenarios of liver damage presented above, there are a variety of cases in which hepatocyte transplantation – either in the context of an extracorporeal device, a tissue engineered graft, or as individually engrafted cells – may offer a clinical alternative to orthotopic organ transplantation (Figure 3). Since the development of techniques for isolation of individual hepatocytes by collagenase digestion (19), investigators have studied whether hepatocyte transplantation could be used to treat liver diseases, first in the laboratory and then in patients. As hepatocyte transplant would classically involve simple infusion of isolated cells into the liver through the portal vein, this form of therapy is far less invasive than orthotopic liver transplantation and could be performed safely in severely ill patients. In the presence of normal host liver architecture, some fraction of the transplanted cells should cross the endothelium to integrate into the host liver (20–22). Because the native liver is not removed, the transplanted hepatocytes only need to provide replacement of defective enzyme activity or enough hepatic function to overcome the liver dysfunction. Other sites of transplantation have been explored in animal models including the mesentery, spleen and the renal capsule (23–25). In these settings, the survival and persistence of cells at ectopic sites seems to depend minimally upon nutrient availability through local vascular beds; however, cells may not require perfusion specifically with portal blood containing ‘hepatotrophic’ factors, as was once believed. Unexpectedly, the lymph node, a highly vascularized site has been a useful location for promoting remarkable growth and function of transplanted hepatocytes in animal models (26), adding further support for the potential clinical utility of adult hepatocyte transplantation. Regardless, in acute liver failure approximately 40% of patients with advanced symptoms recover spontaneously with only medical management (27). As there is no effective means to distinguish patients who will survive without transplantation from those who will not (16), treatment with transplanted hepatocytes could potentially obviate the need to perform an irreversible organ transplant on a patient who could recover spontaneously. However, in an acute setting, it remains to be established whether functional engraftment could be accomplished on a clinically relevant time scale.
Figure 3. Organ transplantation and cell-based therapies.

Currently, liver transplantation is the primary treatment for liver failure and the only therapy shown to improve survival in patients with liver failure. Due to the limited number of livers suitable for transplantation, advanced surgical procedures including split liver and partial donor transplants have been pursued clinically. Additionally, a diverse range of cell-based therapies are currently being explored to treat liver disease and liver failure. These include the transplantation of various adult and stem cell-derived cell populations, the development of extracorporeal bioartificial liver devices, and the implantation of engineered tissues.
Numerous studies in rodents done over the last 30 years indicate that adult hepatocyte transplantation can reverse hepatic failure and can correct various metabolic deficiencies of the liver (28). Although clinical trials of hepatocyte transplantation have demonstrated the long-term safety of the procedure, only partial correction of metabolic disorders has been achieved, and the degree to which donor hepatocytes have restored failing livers has not yet been adequate to circumvent the need for organ replacement (10, 29, 30). Because of the limited availability of fresh donor hepatocytes, or effective alternatives, such as cryopreserved hepatocytes or in vitro expanded cells, trials have been limited to case reports or anecdotes involving few patients and no untreated control patients (23, 24, 31–36). However, this limited clinical experience with hepatocyte transplantation has helped to identify barriers to its successful application and potential pathways for overcoming these challenges. One explanation for the failure to translate laboratory studies to the clinic is the limited number of animal models that fully recapitulate human disease and the challenges of mapping animal model data to clinical outcomes and action plans. A re-evaluation of the results of cell transplantation in animal studies has shown that attaining adequate levels of engraftment and technical issues such as cell availability will need further attention. In addition, immunological graft rejection is more difficult to manage in patients than expected because it is not possible to diagnose rejection of donor hepatocytes by conventional biopsy techniques. This gap most likely explains why successful correction of metabolic diseases by transplantation has been transient, rarely lasting more than a year or two. Through the development of improved animal models, pretreatment strategies, and engineered delivery platforms, ongoing efforts in the field aim to build on these preliminary findings in patients towards an improvement in transplantation efficiency and clinical efficacy.
Mature hepatocytes as sources for cellular therapies
Despite the lack of correlation between some animal model experiments and the minimal success of hepatocyte transfer to patients, there is sufficient support for their clinical potential to prompt the field to discover strategies to obtain, maintain and expand human hepatocytes. Access to sufficient functional cell numbers is a major hurdle despite evidence from serial transplantation using genetically marked, mature donor hepatocytes showing that such cells have a remarkable ability to replicate in vivo, many times more than that seen following partial hepatectomy (37). Thus, unlike many other fully differentiated cell types, hepatocytes have the intrinsic capacity for robust replication, at least under a set of poorly defined circumstances. Still, despite decades of research and partial successes with culture conditions, hepatocytes in vitro rapidly lose their differentiated characteristics and proliferate poorly (38, 39). It is currently not feasible to routinely obtain hepatocytes from healthy human donor livers, amplify the cell populations in vitro while maintaining their functional capacities, and obtain sufficient cells for transplantation or use in extracorporeal devices. This situation will be helped by further research on the hepatocyte microenvironment including the roles of extracellular signaling, matrix, cell-cell interactions, physical forces and soluble factors (40, 41), in order to explore diverse and potentially unexpected ways to generate a replicative environment for the hepatocyte.
A different approach to controlling hepatocyte replication is to manipulate the intrinsic cellular mechanisms that either keep hepatocytes quiescent in the liver or allow the cells to enter the cell cycle after loss of tissue mass (42). In the past decade, substantial inroads have been made in identifying how different cellular networks converge on the proteins and complexes that govern the hepatocyte cell cycle during hepatic regeneration (43). The FoxM1b transcription factor (44) activates Cdc25B, which in turn is needed for Cdk1 activation and entry into mitosis after partial hepatectomy. The anaphase promoting complex restrains hepatocytes in vivo from entering the cell cycle (45) and the Hippo/YAP signaling pathway promotes cell cycle re-entry (46). Recently, the kinase MKK4 was found to restrain hepatocyte proliferation, such that experimental inactivation of MKK4 stimulated hepatocyte regeneration in mouse models of acute and chronic liver failure (47). These disparate findings and others could be brought to bear in a united fashion to better understand how the hepatocyte cell cycle can be controlled.
Extracorporeal bioartificial liver devices
With improved understanding of the signals needed to expand and/or maintain long term function of isolated hepatocytes, it may be possible to provide appropriate architecture-based and soluble-based signals to populate extracorporeal liver devices. Extracorporeal liver devices are principally aimed at providing temporary support to patients with liver failure. Early attempts at developing extracorporeal support technologies were based on non-biological mechanisms such as hemodialysis, hemoperfusion, hemodiabsorption, plasmapheresis, and plasma exchange (48), in part due to the lack of available hepatocytes needed to populate such devices. More recent configurations of artificial, non-biological support systems have focused on the elimination of albumin-bound toxins utilizing a method termed albumin dialysis. These devices, such as the Molecular Adsorbent Recirculating System (MARS; Gambro, Sweden) and the Prometheus (Fresenius Medical Care, Germany) platform have been shown to be effective for the reduction of plasma bilirubin, bile acids, and other albumin-bound molecules (49, 50). Although some reports point to the potential utility of these approaches as bridge treatments prior to organ transplantation (51–53), improvements in patient outcome have not been fully demonstrated (54, 55), suggesting that additional trials are required to fully evaluate their effectiveness. In particular, initial clinical studies have illustrated beneficial effects on organ function without an associated improvement in transplant-free survival (55, 56), which further underscores the complexity of liver failure events and the need for controlled clinical trials. In light of the broad range of vital synthetic functions carried out by the liver and the lack of a liver ‘biomarker’ around which to design these devices, much of the field has gravitated towards cellular rather than purely device-based solutions.
Accordingly, to provide a more complete array of synthetic and biochemical functions, which is lacking in strictly artificial support systems, considerable efforts have been placed in the development of bioartificial liver (BAL) devices containing hepatic cells. Following the initial studies, such as the study by Matsumura et al. in 1987 (57), a broad range of BAL device designs have been reported, and have highlighted the importance of several criteria in the development of an effective device (Figure 4). These include issues of cell sourcing, maintenance of cell viability and hepatic functions, sufficient bidirectional mass transport, and scalability to therapeutic levels. As discussed earlier, the sourcing of hepatic cells is a fundamental challenge for all liver cell-based therapies. As a result, xenogeneic sources (primarily porcine) or transformed/immortalized human hepatocyte cell lines have formed the basis for the majority of BAL devices (which typically include approximately 1 × 1010 cells) tested in the clinic to date (58–61). However, lack of hepatocyte function in transformed cell lines (62) and cryopreserved cells, and the potential risk for porcine endogenous retroviruses have hampered attempts to demonstrate efficacy. More recently, studies have begun to incorporate primary liver cells by exploiting advances in cryopreservation, fetal cell procurement, and stem cell differentiation (60, 63–66). Magnifying the cell sourcing challenge are the scalability requirements for translation.
Figure 4. Extracorporeal bioartificial liver devices.

Extracorporeal bioartificial liver devices incorporate functional liver cells and aim to provide an array of important liver functions (detoxification, metabolic, synthetic) for a patient by processing the patient’s blood/plasma outside of the body. This approach could serve as a temporary support and bridge until a liver becomes available for transplantation. Currently, such devices are either in the clinical trial or exploratory research stage. Liver cell-based bioreactor designs primarily fall into four general categories based on device configuration. These include hollow fiber devices, packed beds, flat plate systems, and encapsulation-based reactors. The majority of current clinical trials utilize a hollow fiber design in which cells are positioned outside the fibers and the patient’s blood/plasma is perfused through the fiber lumen. Cell sources include three categories; currently existing tumor cell lines, porcine and human primary hepatocytes, and hepatic cells derived from embryonic stem cells (ESC), induced pluripotent stem cells (iPSC), or reprogrammed from other cell types. Due to their increased availability compared to primary human hepatocytes, primary porcine hepatocytes are the most common cellular component of current bioartificial liver devices. Device design characteristics have been shown to affect the functional stability of the cellular components. Many design challenges exist including the balanced delivery of oxygen and nutrients to the cells, preventing mechanical shear forces from damaging the cells, and clinically relevant scale-up.
Furthermore, the design of an effective BAL device is dependent on the incorporation of the appropriate environmental and organizational cues that enable maximal survival and function of the hepatocellular component. Hollow fiber devices are the most common BAL design and contain hepatic cells within cartridge units (67), with the hollow fiber membranes serving as a scaffold for cell attachment and compartmentalization (Figure 4). A range of modifications aimed at optimizing cellular performance have been explored. In particular, due to the enhanced function of hepatocyte aggregates relative to single cell suspensions, many device configurations contain either attached or encapsulated hepatocellular spheroids (67–71). In the modular extracorporeal liver support (MELS) system (Charite, Germany), hepatocytes are aggregated in co-culture with liver non-parenchymal cells resulting in the formation of tissue-like organoid structures (72). Furthermore, exposure of hepatocytes to plasma of a sick patient may necessitate specific alterations in hepatocyte culture conditions. For example, the supplementation of plasma with amino acids has been shown to increase albumin and urea synthesis (73), and preconditioning with physiological levels of insulin (lower than standard culture medium), has been demonstrated to prevent abnormal lipid accumulation in hepatocytes (74). Overall, environmental conditions within a BAL device, such as oxygen tension and fluid shear forces can significantly affect hepatocyte functions (75). In addition, both the convective and diffusive properties of the systems must be optimized to provide vital nutrients to the cells while simultaneously allowing export of therapeutic cellular products. Currently, although clinical efficacy of BAL devices remains limited, improvements in device and trial design continue to be implemented. It is anticipated that parallel progress towards the development of highly functional in vitro platforms will provide a reciprocal benefit for the advancement of BAL approaches. For example, small-scale in vitro bioreactor systems have been utilized to systematically examine the effects of shear stress and oxygen tension on hepatocyte function (76, 77). BAL device development is in desperate need of predictive biomarkers that point to the degree of hepatic function that is represented by a device. For example, the ability to monitor a single protein or metabolite in the perfusate as a broader indicator of reduced accumulation of systemic toxic metabolites, and hence the neuroprotective effects of a device, would be invaluable. Applying ‘omics’ methods in an effort to evaluate the state of these devices might provide clues towards the development of appropriate biomarker readouts.
In vitro strategies for improving hepatocyte viability and function
A major research area is the development of improved in vitro culture models, which would improve BAL design as well as provide platforms for the study of human hepatic function. Although systems employing single enzymes, liver slices or liver cell lines have found utility for addressing focused questions, each has limitations. For example, liver slices have limited viability and are not amenable to high-throughput screening; cell-free microsomes lack the dynamic gene expression and intact cellular machinery required for cellular responses (i.e. cytotoxicity); and carcinoma-derived cell lines and immortalized hepatocytes display an abnormal repertoire of liver proteins and limited liver-specific functions (5). For these reasons, isolated primary hepatocytes are considered to be the most suitable platform for a range of in vitro applications (3, 6). As a result, extensive research over several decades has been focused on identifying specific culture configurations and molecular stimuli that can maintain the phenotypic functions of hepatocytes (Figure 5). In general, the phenotype of an isolated hepatocyte is quite plastic and is exquisitely sensitive to its microenvironment. Many different manipulations regulate aspects of the differentiation program, although not necessarily in equivalent ways (the plethora of hepatocyte functions and the absence of a clinical biomarker of rescue means that many parameters must be measured). Such manipulations include culture medium, extracellular matrix, and interactions with non-parenchymal cells. Engineers and biologists have also tried manipulations that are not explicitly ‘biomimetic’ or obviously physiological but still manage to influence hepatocyte fate and function, presumably through some of the existing adult or developmental signaling pathways. For example, additives such as hormones, amino acids, corticosteroids, growth factors, as well as non-physiological small molecules, have been demonstrated to affect hepatocyte functions (78–81). Additionally, both extracellular matrix composition and topology can modulate hepatocyte morphology and phenotype. In the classic “double gel” culture format, hepatocytes are sandwiched between two layers of collagen gel (82). In this configuration, hepatocytes exhibit improved morphological features such as polarized bile canaliculi, and stabilized functions, including albumin and transferrin secretion, for weeks. However, key detoxification pathways such as oxidation and conjugation reactions have been shown to become imbalanced over time in this system (83, 84). Studies investigating the potential added benefits of complex mixtures of extracellular matrix components on hepatocyte function have utilized strategies including extracellular matrix preparations from native liver tissue (85, 86), or conversely, screening methods to systematically investigate defined extracellular matrix combinations (40, 87). Synthetic surface modifications, such as polyelectrolyte chemistries, also influence hepatocyte function in vitro (88, 89), and a polyurethane matrix identified by polymer library screening was shown to support hepatocellular differentiation and function (90). In addition to strictly 2D systems, hepatocyte culture under conditions that promote aggregation into 3D spheroids can affect functionality, with spheroid configurations displaying superior hepatocyte function compared to standard collagen monolayer cultures (91, 92). Potential mechanisms underlying these effects include the increased number of homotypic cell-cell contacts between hepatocytes, the retention of a 3D cytoarchitecture, and the asymmetric presentation of extracellular matrix and other signals surrounding the spheroids (93). Numerous technologies including arrays, rotational cultures, and encapsulation methods have been developed for the optimization and scale-up of hepatocyte spheroid culture (94–96).
Figure 5. In vitro culture systems for hepatocytes.
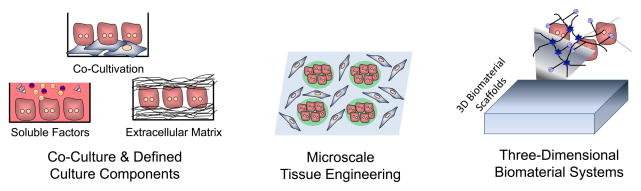
Improved in vitro systems have been developed for culturing primary human hepatocytes. Elucidating the roles of many microenvironmental signals in governing hepatocellular processes has enabled optimization of in vitro culture systems. These optimized systems include co-cultures to provide specific cell-cell interactions as well as culture conditions with defined concentrations of soluble factors and extracellular matrix molecules (left). The application of microfabrication technologies to in vitro hepatic tissue engineering has facilitated the control of culture platforms down to the microscale, such as the patterning of co-culture configurations (middle). Further, natural and synthetic biomaterial systems have been applied towards the optimization of three-dimensional in vitro culture platforms (right). Collectively, these engineering approaches have further advanced the understanding of how combinations of microenvironmental cues influence cell functions, and provided important insights into the temporal and spatial dynamics of hepatic cell and tissue function.
In both 2D and 3D formats, the addition of secondary supportive cells for heterotypic interactions is one of the most robust approaches for preserving the viability, morphology, and function of cultured hepatocytes. In addition to modulating cell fate through cell-cell sensing pathways such as cadherins, stromal cells additionally modify the extracellular matrix and the paracrine signals in the microenvironment. Specifically, beginning with the initial experiments by Guguen-Guillouzo and colleagues (97), substantial research efforts have demonstrated that both liver-derived cell types including liver biliary epithelial cells and non-parenchymal cells (e.g. stellate cells, sinusoidal endothelial cells), as well as numerous non-liver-derived cells (e.g. embryonic fibroblasts), are capable of supporting hepatocyte function in co-culture contexts (98). Although varied culture conditions have been explored, typically, primary hepatocyte functions can be rescued if co-cultures are initiated within 3–7 days following isolation. Transcriptional and post-transcriptional mechanisms have been implicated in the co-culture stabilization of hepatocyte-specific genes including albumin, transferrin, pyruvate kinase, and glutathione S-transferase (99, 100). Furthermore, cytochrome P450 (CYP) detoxification enzymes are elevated in co-cultures compared to hepatocytes cultured alone (101).
Co-culture systems have formed the basis for investigations into a broad range of hepatocellular processes such as the acute phase response, oxidative stress, mutagenesis, lipid and drug metabolism, and xenobiotic toxicity (98). For example, co-cultures of hepatocytes and Kupffer cells (the liver’s resident macrophages) have been used to examine mechanisms of hepatocellular damage (102, 103). Meanwhile co-cultures with liver sinusoidal endothelial cells have highlighted the importance of hepatocyte-endothelial cell interactions in the bidirectional stabilization of these cell types (104–109). Research efforts continue to employ co-culture platforms as in vitro models aimed at dissecting physiological cell-cell interactions in the liver, and as a tool for optimizing engineered liver microenvironments. In particular, for certain tissue engineering applications, replacing supportive cell types with acellular components that do not consume nutrients and occupy limited space may be advantageous. Studies focused on the underlying mechanisms of hepatocyte stabilization in co-culture have identified several cell surface and secreted factors that play a role including T-cadherin, E-cadherin, decorin, TGF-β1, and liver-regulating protein (110–116). Although such factors are currently incapable of supporting hepatocyte function at a level comparable to supportive stromal cell types, they represent a proof-of-concept for highly functional hepatocyte-only culture platforms.
Collectively, a set of in vitro strategies is emerging that may aid in BAL design as well as inform the development of in vitro liver models for discovery. Because the hepatocyte phenotype is quite plastic once the cell is isolated from its native environment, much of the focus has been in rescuing the hepatocyte phenotype ex vivo. There seems to be a shift in the field from strategies that are solely biomimetic (e.g. culturing hepatocytes in sandwich extracellular matrix, or under gradients of oxygen tension, or in co-culture with non-parenchymal hepatic cells) to culture models that do not necessarily mimic the native hepatic microenvironment (e.g. spheroids and co-cultures with embryonic fibroblasts). Strategies that optimize hepatic function, duration of rescue, accessibility to perfusate, and amenability to scale-up may or may not ultimately resemble native liver architecture. A systems-level picture of molecular signals that influence phenotypic stability is emerging and will aid these efforts (117).
Stem cells as sources of hepatocytes for cellular therapies
An independent approach to generating hepatocytes for therapeutics is to use stem and/or progenitor cells, which by definition have a high capacity for expansion (Figure 6), and may be sourced from a variety of tissues. Theoretically, such cells could be amplified, induced to differentiate, and used in diverse applications. Whereas progress has been made in genetically or immunologically identifying tissue-resident stem cell-like populations that arise in chemically damaged livers (42, 118–122), further work is needed to understand the role of these cells in normal liver physiology and repair (123) and to assess whether these populations represent a clinically-relevant source of hepatocytes. Other cell sources, including mesenchymal stem cells have been discussed recently in this context (124). Another current interest is on the potential of pluripotent stem cells, including human embryonic stem cell (hESC) and induced pluripotent stem cell (iPSC) lines, which have a high proliferative capacity and can differentiate into diverse lineages in vitro and in vivo (125). Various directed differentiation strategies have been applied to hESC and iPSC cultures, and have yielded populations that exhibit many phenotypic and some functional traits of mature hepatocytes, earning the derived cells the label of ‘hepatocyte-like cells” (11, 126–136). Yet hepatocyte-like cells exhibit numerous characteristics, such as distinctive CYP activities as well as expression of alpha-fetoprotein, that more closely resemble fetal-stage hepatocytes rather than mature adult cells (137). To further characterize these cells, some studies have tested the maturation and repopulation potential of both mouse and hESC-derived hepatic cells in rodent transplantation models, such as immune-deficient recipient mice that harbor genetic defects in resident hepatocytes (13, 138, 139). This form of inquiry helps to support the conclusion that hepatocyte-like cell populations can support at least some liver function in a replacement setting, but the best choice of animal models for these studies is still under debate (140). Attempts to identify small molecules that can induce the maturation of hepatocyte-like cells have suggested that ex vivo maturation may be possible (41).
Figure 6. Sources of Hepatocytes.

Obtaining appropriate sources of hepatocytes is a major limitation for developing cell-based therapies for treating liver disease. Many different approaches are under investigation including methods for improving the expansion of primary human hepatocytes in vitro, the directed differentiation of pluripotent stem cells (both embryonic stem cells and induced pluripotent stem cells), the differentiation of either intrahepatic or extrahepatic adult progenitor cells, as well as new methods for the direct reprogramming of hepatic cells from adult somatic cells.
One study utilized partially differentiated human iPSCs and admixed supportive stromal and endothelial cells to mimic aspects of early liver development. The resulting liver ‘buds’ were able to vascularize upon ectopic transplantation and provide functional rescue of mouse models of liver injury (141). Furthermore, recent progress suggests that hepatocyte-like cells derived by directed differentiation of fibroblasts also exhibit more adult hepatic traits, and, like iPSCs, might provide an autologous stem cell source that would bypass a need for clinical immune suppression (142–146). In light of this progress, the strategy of utilizing pluripotent cells as a source material for generating hepatocytes---either for clinical transplantation, as a cellular component in BAL devices, or in various model platforms to study disease and drug development---holds promise. Considerable experimental energy continues to be applied to find methods that improve the efficiency and extent of differentiation of these expansion-ready precursor cells (Figure 6). More needs to be done to obtain expansion of human stem cell- derived hepatocytes that have been implanted into mouse livers and evaluate their performance and safety. Regardless, the capacity to populate mouse model livers with human hepatocyte-like cells both from normal individuals and patients offers an experimental system of “humanized” mouse livers for use in toxicological studies on human hepatocytes (141, 147–150).
Translating existing technology into pre-clinical and therapeutic applications
Implantable therapeutic constructs for preclinical testing
Delivery of hepatocytes by cell transplantation requires cells to home to the liver from the portal blood stream and extravasate across the space of Disse into the hepatic lobular compartment. Early ‘tissue engineers’ sought to support these adhesion-dependent cells with biomaterials that could alleviate the need for homing and attachment. These seminal studies used available biomaterials (such as poly-lactic co-glycolic acid, PLGA) and demonstrated an improvement in the survival of the transplanted cells. Nonetheless, animal studies suggested that the persistence of hepatocyte phenotype and survival were dependent on the site of implantation, at least in this delivery context. For example, some studies demonstrated an improvement in the function of transplanted hepatocytes when bathed in the so-called hepatotrophic factors draining from the gut to the portal vein (151). Because the portal vein is an unattractive site for transplantation in chronic liver failure patients with elevated portal pressures, investigators have begun to explore ectopic sites (spleen, subcutaneous, renal capsule, intraperitoneal) (64, 152, 153). In these ectopic sites, manipulating the cellular microenvironment using biomaterials, peptides, or even other cells has led to a diminished dependence on the portal circulation, setting the stage for ectopic transplantation of engineered hepatic tissues (as is currently the practice for renal transplantation). Under strong regenerative stimulation such as that found in genetic mouse models of tyrosinemia, the transplanted cells can even repopulate host lymph nodes ectopically, suggesting that either the cells in these sites receive some hepatotrophic stimuli or the reliance on portal hepatotrophic factors may be mitigated in some regenerative contexts (26, 154). These findings are consistent with the classic parabiosis experiments of Bucher and colleagues, suggesting that regenerating liver produces bloodborne factors that stimulate hepatocyte expansion in a conjoined animal (155).
Since the early experiments with PLGA, a synthetic degradable polyester found in suture material, the community has explored many porous scaffold materials ranging from both natural (e.g. collagen, alginate) and synthetic (e.g. PLGA, PLLA) sources. Scaffold traits including porosity, material and chemical characteristics, and 3D architecture are among the parameters that are customizable in 3D platforms, and these properties play important roles in dictating cellular function and facilitating the transport of nutrients and secreted therapeutic factors. In addition, synthetic hydrogel systems based on poly(ethylene glycol) (PEG), which have been extensively utilized for tissue engineering studies (156), have found recent utility in 3D liver platforms (157–159). Hydrogel platforms offer the advantage of polymerization in the presence of cells, thereby enabling the fabrication of 3D networks with uniform cellular distribution without the need for cell migration or expansion in situ. Modifications in polymer chain length and the conjugation of bioactive factors such as adhesive peptides improve the survival and function of PEG hydrogel-encapsulated hepatic cells (157, 158, 160). Moreover, to generate scaffolds with a highly defined architecture that provide better control over the 3D environment at the microscale, a range of rapid prototyping and patterning strategies have been developed and tested for liver applications (161–164). For hydrogel systems, these include photolithography-based techniques for dictating the size and shape of constructs and for building multilayer geometries (157, 159, 165–167), and dielectrophoresis methods for controlling cellular positioning (168). Notably, cell-cell interactions (both homotypic and heterotypic) have been suggested to substantially affect hepatocellular survival and function in 3D biomaterial scaffolds (158, 167, 169, 170). As a result, similar to 2D culture models, the controlled integration of additional cell types or acellular factors that mimic key cell-cell interactions will be critical for the advancement of 3D liver platforms.
Early studies also uncovered a scale-up issue that paralleled that found with BAL devices: transplanting large numbers of cells in biomaterials without a sufficient nutrient supply rapidly leads to necrosis of the engineered tissue. Hepatocytes are highly metabolic and are normally in close contact with an extensive sinusoidal vasculature. As a result, significant efforts must be made in the design of implantable liver systems to avoid transport limitations that can greatly diminish tissue function. In particular, the site of implantation is a critical parameter in liver tissue engineering studies, with more highly vascularized sites such as the peritoneum or renal capsule generally promoting improved engraftment and function (158, 171, 172). Thus, another area of focus is the establishment of a vascular network that supports a large number of hepatocytes. Priming of the implantation site through pre-vascularization is a particularly effective strategy due to the minimization of early transport restrictions that occur prior to the establishment of functional vasculature. Microfabrication approaches, such as polymer molding with etched silicon, dissolvable sugar lattices, and microtissue molding of aligned endothelial cords have been employed as strategies to preform capillary-sized channel networks (163, 173, 174). Additionally, the integration of angiogenic growth factors into the implantable scaffolds has been shown to promote the recruitment of host vessels (175–178). Alternative strategies to achieve this goal include multilayer microfluidic networks, prevascularization through release of angiogenic growth factors, and inclusion of non-parenchymal cells that promote angiogenesis and vascular stabilization with pericytes. For instance, preceding hepatocyte delivery with the implantation of scaffolds that release angiogenic vascular endothelial growth factor (VEGF) enhanced capillary density and improved engraftment in rat liver lobules (179). Similarly, fibroblast growth factor 2 (FGF2) coated scaffolds served as a supportive environment for mouse ESC-derived hepatocyte inoculation in an in vivo hepatic failure mouse model (64). Furthermore, several recent studies aimed at engineering skeletal or cardiac muscle tissue have illustrated that significant improvements in survival and host vasculature connections can be achieved following in vitro formation of vessel structures within optimized tri-culture systems (muscle, endothelial and mesenchymal cells) (173, 180–182).
In addition to vascular integration, an improved understanding of multicellular organization and morphogenesis in the liver could also aid in the formation of functional biliary transport systems. Various in vitro models have been developed that exhibit organized bile canaliculi (183–185) or artificial duct structures (186), but their incorporation into implantable systems has yet to be fully explored. Although early work demonstrated engrafted bile ducts in ectopic sites (187), the degree to which the biliary tree must be reconstructed has not yet been established; in ectopic cell transplantation experiments the hepatocytes do not appear cholestatic, and biliary products do appear to find their way to the digestive tract. One hypothesis is that the biliary products are redirected or ‘leak’ into the bloodstream where they circulate and are processed by the remnant liver into bile. This scenario would argue against removal of the diseased liver in the setting of transplantable tissue engineered constructs, and is consistent with the functional outcome achieved in peritoneal transplantation of mature hepatocytes and hepatocyte-like cells that lacked biliary networks (26, 141). The need for reconstruction of the nervous and lymphatic systems is likewise even less well understood, although, if clinical transplantation is a guide, these are likely to be less critical.
Similar to whole organ transplantation, the host immune response following the introduction of tissue engineered constructs is a critical determinant of successful engraftment. Accordingly, the immune isolation capabilities of polymer scaffolds is a highly active research area (188), and represents the foundation of numerous tissue engineering approaches such as pancreatic islet transplantation (189–191). Recent liver tissue engineering studies demonstrate that hydrogel encapsulation enabled detectable human hepatic function in transplanted immunocompetent mouse strains for greater than one week (158), suggesting that aspects of immune isolation may find utility in various liver applications. In general, approaches to tune the degree of inflammatory cell recruitment to engineered implants could substantially aid in preventing foreign body responses that severely impair engraftment, while potentially maintaining the positive remodeling effects that inflammatory cells provide in numerous normal tissue regeneration contexts (192). For example, liver regeneration proceeds with a sequence of remodeling processes including protease expression and extracellular matrix deposition (193–195). The incorporation of protease-sensitive domains into hepatic hydrogel systems could allow for the degradation of the constructs following implantation and subsequent changes in ligand presentation and gel mechanics (196). Tailoring the degradation properties of constructs based on the kinetics of cell proliferation, inflammation, and angiogenesis could provide a means for efficient integration. Overall, the development of new biomaterials platforms that can collectively modulate host cell recruitment and material-resident cell function, as well as engraftment and release of implanted cell types (197), is a major focus and should advance liver tissue engineering approaches.
Animal models for pre-clinical testing of candidate interventions and barriers to overcome in clinical translation
Transplantation of liver cells has been shown to improve the survival of animals with chemically and surgically induced acute liver failure (198–207), end-stage liver failure secondary to cirrhosis (208), and to correct metabolic deficiencies and prolong survival in numerous models of liver-based metabolic diseases (199, 209). Whereas animal models have provided a foundation for clinical translation, the results of clinical trials using liver cell transplantation have been disappointing. The most promising results have come from the treatment of children with liver-based metabolic diseases, where evidence of functional replacement of the deficient enzyme has been documented and post-transplant biopsies have shown engrafted cells expressing a corrected form of the deficient enzyme (10).
Most disappointing has been the failure to successfully reverse acute hepatic failure in patients by hepatocyte transplantation. As the normal hepatic architecture remains intact in most cases of acute liver failure, transplanted hepatocytes, which in animals are infused through the portal vein or directly into the spleen, would be expected to translocate to the liver, engraft, and provide life-saving metabolic support while residual host hepatocytes regenerate. Yet, the field lacks an animal model that adequately recapitulates clinical hepatic failure such that the potential clinical effectiveness of cell transplantation or a liver assist device may be predicted. In the most used chemical and surgically-induced models of acute liver failure, animals develop severe histologic injury to the liver and elevated serum levels of ALT, AST, and bilirubin; however, the majority of animals do not die as a result of intracranial hypertension, which is the most common cause of death in acute liver failure. In addition, if the animals can be kept alive for as little as 72 hours after the injury, regeneration is rapid enough to completely correct the liver injury and animal survival approaches one hundred percent. In contrast, experience with auxiliary liver transplant in patients with acute liver failure indicates that native liver recovery could take weeks to months (210–212). Thus, the dramatic results obtained in animal studies may result from the short-term metabolic support provided by the transplanted hepatocytes rather than from stable replacement of hepatic function. Finally, it has not been possible to determine how many of each of the immediately available sources of functioning hepatocytes, which include cryopreserved cells, xenografts, or stem cell-derived hepatocytes, need to be transplanted to reverse clinical liver failure.
In addition to acute hepatic failure, intrasplenic hepatocyte transplantation to treat mouse models of chronic hepatic failure secondary to cirrhosis has been effective but transient, and the clinical experience has produced only anecdotal reports of improvement in some aspects of hepatic function. This inability to successfully reverse human liver failure associated with cirrhosis is not unexpected, as the cause of hepatic failure in cirrhosis is not completely understood (18, 195, 213). Furthermore, changes in liver architecture inhibit entry of transplanted hepatocytes into the abnormal cirrhotic environment, thus supporting the exploration of ectopic transplantation sites (195, 214). Cirrhosis represents a final phase of liver disease characterized by advanced liver fibrosis and nodular architecture. The two most commonly used animal models of experimental fibrosis are toxic damage (using the chemical CCl4) and bile duct ligation (18). Other mouse models that mimic specific liver diseases, including non-alcoholic steatohepatitis, may require special diets to induce injury (215) or they are genetic knockout models, such as Mdr2-null mice, that spontaneously develop fibrosis (216). Notably, the degree of reversibility of liver fibrosis in rodents following the discontinuation of the toxic agent varies between experimental model systems (208, 217). In addition, changes in hepatic function may result from acute effects of the intoxicating agent rather than from chronic injury to the liver. Reversal of human cirrhosis and secondary hepatic failure is not so easily accomplished (18, 218, 219). In rats, treatment with CCl4 for 28–32 weeks can reliably recapitulate many physiologic abnormalities found in patients with advanced cirrhosis and liver failure (217). In this model, hepatocytes transplanted into the spleen, to bypass vascular changes in the liver including portal hypertension, minimized hepatic encephalopathy and supported survival for a period of months (208, 220, 221). Alternatively, rodents that receive portacaval shunts and ammonium chloride treatments exhibit neurobehavioral changes similar to those associated with cirrhosis, without other hallmarks of hepatic failure. In this setting, hepatocyte transplantation into the spleen markedly improves hyperammonemia-induced hepatic encephalopathy and amino acid imbalances, and prevents development of hepatic coma (222, 223).
The failure of isolated hepatocyte transplantation to affect liver function and survival in patients with cirrhosis may be attributed to their infusion through the splenic artery, rather than by direct injection through the splenic capsule, which is the route of engraftment successfully used in rodents (224). Given that it appears that cells need to engraft in an extrahepatic site, treatment of chronic liver failure might also benefit from work in tissue engineering and whole organ liver bioengineering. Decellularized human or animal livers could serve as a biological scaffold for transplanted cells forming engineered internal auxiliary liver grafts (225–229).
Interestingly, transplantation of mesenchymal stem cells (MSCs) derived from bone marrow, adipose tissue, amniotic fluid, and other tissues has been associated in clinical trials with correction of portal hypertension and decompensated hepatic function. Evidence that this effect is mediated by replacement of diseased hepatocytes by MSC-derived cells, however, is lacking. [The complex role of MSCs in the treatment of liver disease is reviewed in (230, 231)]. Of course, any therapy that allows the native cirrhotic liver to remain in place will leave unresolved the management of coexisting portal hypertension and the risk of developing hepatocellular carcinoma unless the rescue is sufficient to ultimately allow explant of the cirrhotic liver.
Finally, beneficial improvement in liver function has been reported after hepatocyte transplantation in several models of life-threatening liver-based metabolic disease. Multiple animal models faithfully represent this class of diseases in man. These include the Gunn rat, a model of Crigler-Najjar syndrome type 1 (209); the fumarylacetoacetate-hydrolase (FAH)-deficient mouse, a model of tyrosinemia type I (232); the Long Evans cinnamon rat, a model of Wilson’s disease (233); the mdr2 null mouse, a model of progressive familial intrahepatic cholestasis type 3 (216); an arginosuccinate synthetase deficient mouse (234), the spf-ash mouse, a model of ornithine transcarbamylase (OTC) deficiency (235, 236); a liver arginase null mouse (237), the Agxt−/− mouse, a model for primary hyperoxaluria-1 (238); the Watanabe heritable hyperlipidemic (WHHL) rabbit, a model of familial hypercholesterolemia (239); the hyperuricemic Dalmatian dog (240) and the PiZ transgenic mouse, models of α-1-antitrypsin deficiency (241). Unfortunately, transplantation of liver cells has resulted in only partial correction of the genetic abnormality in most of these animal models, and the experience in humans has mirrored these results. In most cases of liver-based inherited diseases, the life-span and regenerative capacity of the host hepatocytes are normal, and transplanted hepatocytes do not compete successfully for survival in the host liver.
Transplantation studies in animal models of liver-based metabolic disease have also shown that it is not possible to engraft enough hepatocytes in a 24–48 hour time period to completely correct an enzyme deficiency even with multiple cell infusions. The number of donor cells that can be safely transplanted into the liver at any one time via the portal vein is usually less than 5% of the liver mass, or approximately 2 × 108 cells/ kg, as transplantation of greater numbers leads to either portal hypertension or translocation of cells out of the liver into the systemic circulation and embolization of cells in the lungs. Whereas replacement of 5% enzyme activity should be adequate to correct most metabolic liver diseases, it is thought that only 10 to 20% of transplanted cells engraft. Thus, improving engraftment rates with biomaterials, use of ectopic sites that do not harbor ongoing injurious stimuli, and the need for expansion of engrafted cells in response to regenerative cues are all strategies worth exploring further.
Experience in a selected group of animal models of human metabolic disease, however, has highlighted a strategy that could improve the outcome of hepatocyte transplantation in patients. In the FAH-deficient mouse model of hereditary tyrosinemia (242), the albumin-uPA transgenic mouse (243–245), and the transgenic mouse model of human α1-antitrypsin deficiency (241), host hepatocytes exhibit markedly reduced survival due to the inherited metabolic defect. In these cases, transplanted wild-type hepatocytes spontaneously replace the host cells over time, leading to near-complete replacement of host liver cells by donor hepatocytes. Recent advances in genome editing techniques suggest that in situ gene correction may even be beneficial in this model. In addition, another chimeric mouse model has been recently developed, which is based on thymidine kinase transgene expression in the liver of an immunodeficient mouse strain (TK-NOG) (246). In this model, brief exposure to ganciclovir causes a time-limited toxicity to host liver cells, which opens a window for chimerism in the absence of continuous injury during the repopulation phase. Partial repopulation by engrafted hepatocytes also occurs in the LEC rat model of Wilson’s disease and in the mdr2-deficient mouse, where the hepatocellular injury to native liver cells is more modest. Strategies have been developed to try to recapitulate this effect by exogenous means. For example, a selective growth advantage can be given to transplanted hepatocytes by preconditioning the liver using drugs that impair native liver regeneration, or that damage hepatocytes or sinusoidal endothelial cells, followed by a proliferation stimulus for the transplanted cells. Plant alkaloids, such as retrorsine, prevent hepatocellular proliferation (247), as does preparative irradiation of the liver; partial hepatectomy can provide a proliferative stimulus to the engrafted cells (248). Irradiating as little as 35% of the liver mass prior to allogeneic hepatocyte transplantation results in complete correction of hyperbilirubinemia in the Gunn rat model of Crigler-Najjar syndrome (249). Whereas this approach may resolve the problem concerning the adequacy of engraftment to treat metabolic liver disease, an additional barrier must be resolved, and that is the inability to directly monitor the status of the graft. As a result, rejection or other potential sources of graft loss cannot be determined, and inappropriate immune suppression can lead to rejection of the graft. New approaches for monitoring disease progression in the liver may offer a path to the identification of biomarkers that improve clinical decision-making (250).
Because of the proliferative advantage of donor hepatocytes in FAH-deficient, albumin-uPA transgenic mice, and in thymidine kinase transgenic mice during ganciclovir treatment, these animals, when crossed onto an immune-deficient background, have become the most frequently used animal models to study engraftment and expansion of primary human hepatocytes (251). Nearly complete replacement of the native liver can be achieved when primary human hepatocytes are transplanted into these models. In addition, in immune deficient FAH-deficient mice, serial transplantation of human hepatocytes produces several generations of animals with humanized livers (242). Serum concentrations of human albumin or α1-antitrypsin correlate with the degree of engraftment in these animals, as determined by immunohistochemistry of tissue sections. Use of these serum measurements is necessary to confirm the extent of human cell repopulation because autofluorescence in the liver can lead to misinterpretation of immunofluorescence data. Thus far, stem cell-derived hepatocyte-like cells have been shown to engraft and expand to a small degree in albumin-uPA SCID mice (13, 252), and in PiZ SCID mice (253). Transplanted human iPSC-derived hepatocytes have also been shown to engraft and expand in the livers of Gunn rats (254). Transplantation leads to partial correction of hyperbilirubinemia when part of the host was irradiated; hepatocyte growth factor was provided to stimulate expansion of transplanted cells and Tacrolimus was used for immune suppression. Similar results have been obtained in the mouse model of oxalosis (255). As mentioned above, recent studies indicate growing success in not only differentiating human ESCs and iPSCs into hepatocytes, but also the direct reprogramming of human fibroblasts into hepatocytes (144–146). Collectively, these results indicate that it may be possible to achieve an expandable, patient-specific source of transplantable human hepatocytes, and also bypass the need to generate iPSCs, which carry a measure of clinical risk of teratoma. No matter which animal model is utilized, is it important to apply more than one assay to monitor engraftment, as models of liver disease and techniques for cell delivery and transplantation are not yet standardized across most laboratories, and can generate results that have been confusing to investigators.
Clinical translation and scale-up
Clinical translation of candidate cell-based therapeutics that exhibit promise in faithful, robust preclinical models will require solutions to a range of practical challenges such as appropriate patient diagnosis and selection, detailed trial design, overcoming potential immune barriers, accounting for the impact of immune suppression regimens, as well as establishing ‘Good Manufacturing Practices’ or GMP conditions for the generation of any allogeneic cellular materials. The success of cell-based therapy will depend on the ability to scale the approach to a level that provides clinical effectiveness. BAL devices tested clinically have used between 0.5 × 109 and 1 × 1011 hepatoblastoma cells or porcine hepatocytes (69). The target capacity of most BAL designs is approximately 1 × 1010 hepatocytes corresponding to approximately 10% of total liver weight, the minimal mass estimated to be required to support vital metabolic functions such as gluconeogenesis. Hepatocyte transplantation experiments in rodent models and human subjects have demonstrated improvements in blood parameters following transplantation of cell numbers that are 1%–10% of total liver mass (256–258). Although distinct categories of liver diseases (acute liver failure, end-stage cirrhosis, inherited metabolic disorders) will have different scale requirements, it is expected that scale-up will represent a significant translational challenge for all cell-based liver therapies. To achieve clinical efficacy will require (i) efficient nutrient transport within the scaled-up systems and (ii) an expandable cell source. To address nutrient limitations in large-scale engineered tissues, efforts have focused on improving integration with the host vasculature as well as microfabrication approaches to develop constructs with 3D architectures and improved diffusion characteristics (173, 174). For extracorporeal approaches, increasing the number of cartridges (259) and fiber cartridge size (260), have been explored to increase the capacity of hollow fiber-based devices. Expanded configurations of flat plate or perfusion BAL device designs have been suggested, but these modifications may introduce heterogeneous flow distributions or large fluid volumes, respectively (261). Efforts exploring the utility of proliferative stem cell sources, as well as parallel methods for inducing substantial primary hepatocyte proliferation in vitro, are aimed at addressing the cell sourcing challenge (41). Notably, donor hepatocytes exhibit a proliferative advantage in the liver within various liver injury models (discussed above), which can result in near complete replacement of the native hepatocytes (245).
Modeling the healthy human liver
Progress has been made towards clinical application of cell-based therapeutic models, but it should not be overlooked that both animal models as well as in vitro liver platforms offer the potential to study normal liver function and biology (Figure 7). Insights gleaned about normal liver biology may be applied to tissue engineering and repair efforts, and may assist in assessing the pharmacokinetics (e.g. clearance), metabolism, and potential toxicity of new drugs. In vivo, the healthy liver is perfused and the hepatocyte phenotype is stabilized in its native microenvironment leading to two classes of models: (1) perfused hepatic cultures in bioreactors and (2) static monolayer cultures (2D) or aggregates such as spheroids (3D). Both types of model systems will be useful for understanding normal liver physiology and susceptibility to insult. Many of these studies were initially performed in rodents. Whereas establishing platforms using rodent hepatocytes can aid in the interpretation of pre-clinical in vivo rodent studies and serve as a test bed for optimizing formats for human cells, to achieve meaningful predictions of clinical outcomes it will be essential to incorporate human hepatocytes into these model systems (262, 263).
Figure 7. Liver cell and tissue engineering.

Progress in the field of liver cell and tissue engineering serves a bidirectional role as both a means for establishing robust model systems for investigating the human liver in health and disease, as well as the foundation for the development of new cell-based therapies. Consequently, applications exist on a continuum ranging from fundamental in vitro studies (left), to engineered approaches for interfacing with animal models (middle), and finally to translational clinical applications (right). In order to further understand human liver function and disease processes, engineered culture platforms can serve to complement animal models. Concurrently, the foundation of novel cell-based therapies is based on advances in cell and tissue engineering and the progression of these technologies from relevant animal disease models to clinical settings.
With perfused systems, a wide range of in vitro hepatocyte-based bioreactor platforms have been developed. These platforms offer the potential to examine flow-dependent phenomena such as the clearance of xenobiotics from the circulation over time or the bioactivation of a drug to a toxic metabolite that can cause damage downstream (264–267). Perfusion systems may contain hepatocellular aggregates to stabilize liver-specific functions (268–270) and recently have been used to examine the hepatic differentiation of hESCs (271, 272). The incorporation of multiple compartments in parallel may be valuable for increasing perfusion and throughput (273, 274), and integration of multiple reactors in series could be useful for investigating organ-organ interactions (275, 276). There has been much interest in building a ‘human on a chip’ to develop predictive models of human physiology and toxicology (277), and the liver module will be a vital part of this effort. In order to promote oxygen delivery while protecting hepatocytes from the deleterious effects of shear stress, gas-permeable membranes have been integrated into several bioreactor configurations (278, 279). Perfused systems also allow the capture of some of the physiological heterogeneity of hepatocyte gene expression along the liver sinusoids. This ‘zonal’ distribution is thought to arise from gradients in oxygen, hormones, and extracellular matrix molecules. A parallel-plate bioreactor revealed that a steady-state oxygen gradient contributed to the heterogeneous expression of drug metabolizing enzymes, which mimicked the expression gradients present in the liver as well as the regional susceptibility to acetaminophen in regions of low oxygen and high CYP activity (280).
Static systems that stabilize the hepatocyte phenotype in the absence of flow have provided insights into drug metabolism and drug-induced liver injury. These model systems have manipulated the composition and geometry of the extracellular matrix and perturbed the cytokine environment. They also include 3D aggregates formed on engineered surfaces or in droplets, or co-culture. Overall, co-culture of hepatocytes with non-parenchymal cell types appears to be most effective in providing maintenance of long-term function in vitro, likely due to the concordant impact on the local extracellular matrix and soluble microenvironment as well as cell-cell interactions (98). The utility of these platforms for drug development depends critically on their reproducibility and potential for miniaturization in order to minimize the amount of candidate drug needed. The vast number of species-specific metabolic properties of hepatocytes (281) underscores the importance of incorporating human hepatocytes in an effective and affordable manner.
Microtechnology offers the potential to readily miniaturize some of these culture environments. For instance, micropatterned co-cultures of hepatocytes and fibroblasts, fabricated with either photolithography or soft lithography techniques, have been used to precisely regulate homotypic and heterotypic interactions and have been optimized to support hepatocyte phenotypic functions for several weeks, including the maintenance of gene expression profiles, canalicular transport, phase I/II metabolism, and the secretion of liver-specific products (98, 282). Furthermore, drug-mediated modulation of CYP expression and activity was observed, illustrating the utility of the platform for ADME/Tox (absorption, distribution, metabolism, excretion, and toxicity) applications. Consistent with clinical studies and our current mechanistic understanding of drug-drug interactions, acetaminophen-induced hepatocyte toxicity was increased by a chemical inducer of CYP expression (phenobarbital) or an inhibitor of glucuronidation (probenecid), and species-specific differences in CYP1A induction (283) also were demonstrated. Subsequent studies have shown improved potential to predict generation of human metabolites and human hepatotoxicity compared to unpatterned or monoculture models (84, 284).
Microcontact printing and robotic spotting techniques have been used to fabricate microarrays of hepatic cells and investigate functional stabilization, differentiation, and injury responses (40, 285–287). Finally, microtechnology tools have also been applied to the analysis of dynamic processes. These tools include microfluidic devices for the study of drug clearance, toxicity, and inflammation-mediated gene expression changes (265, 288, 289), as well as mechanically actuated microfabricated substrates for deconstructing the role of contact and short-range paracrine signals in hepatocyte-stromal cell interactions (290). Overall, the fine spatial and temporal control of molecular signals provided by microtechnology approaches continues to reveal important mechanisms in liver biology and accelerate the development of therapeutic strategies.
In addition to in vitro approaches, humanized mouse models that exhibit human chimerism in the liver (discussed above) have been increasingly applied towards studies of human liver function and disease. In particular, for drug development, humanized mice could aid in investigations of drug-drug interactions and chronic toxicity within a framework of relevant in vivo pharmacokinetic profiles, and potentially also could detect risks that arise when a human-metabolized drug generates a toxic response in another organ. Recently, tissue engineering has been used to generate a humanized mouse rapidly with high yield and in an immunocompetent non-liver-injury context (158), thereby circumventing the limitations of current transgenic and transplantation approaches. In this study, human hepatocytes were transplanted into an ectopic (intraperitoneal) site within an optimized hydrogel scaffold, which served as a supportive microenvironment and barrier that delayed immune rejection. The transplanted constructs synthesized human liver proteins and exhibited human drug metabolism, drug-drug interactions, and drug-induced hepatocellular injury (158).
Modeling human disease processes
Engineered in vitro liver models facilitate studies into the behavior of pathogens that target human hepatocytes, including hepatitis C virus (HCV) and malaria. Initial in vitro experiments examining HCV replication used Huh7 liver carcinoma cells stably transfected with a subgenomic replicon (291). These studies provided important information about the HCV replication process and potential small molecule inhibitors of replicative enzymes, although the entire viral life cycle could not be completed due to the absence of structural proteins. Prior to 2005, there was no known viral genotype that could complete the viral life cycle in vitro. Following the identification of a genotype-2a strain of HCV responsible for fulminant hepatitis in a Japanese patient, termed JFH-1 (292), it was demonstrated that JFH-1 and a chimeric variant could complete a full viral life cycle in the Huh7 carcinoma cell line (293–295). Recent approaches have made it possible to examine HCV infection in primary human hepatocytes (296–298). In particular, stabilization of hepatocytes within a micropatterned co-culture environment enabled recapitulation of the full viral life cycle in vitro (298). With this platform, human hepatocytes expressed all known entry factors for HCV including scavenger receptor class B type 1 (SR-BI), claudin-1, CD81, and occludin (299). These stabilized hepatocytes also supported viral replication for several weeks, illustrating the potential of such in vitro systems for screening drug candidates that suppress HCV replication, with the goal of identifying non-hepatotoxic anti-HCV compounds that could be added to the recently expanding repertoire of FDA-approved protease and polymerase inhibitors (300–304). Furthermore, the demonstration that HCV infects human iPSC-derived hepatocytes will enable the role of host factors in HCV infection to be examined (305, 306). A similar approach can be taken to modeling hepatitis B virus as well as other hepatotropic viruses.
In vitro culture platforms have been explored to study infection of hepatocytes by Plasmodium sporozoites to simulate the liver stage of malaria. 2D collagen monolayer culture models have revealed that SR-BI and CD81 are entry receptors for the malaria parasite mediating hepatocyte invasion (307), and may yield insights into potential targets for attenuating parasite growth (308). However, analogous to HCV studies, engineered hepatocyte culture platforms that support long-term functional maintenance and enhance the efficiency and duration of Plasmodium infection in vitro, could clarify mechanisms of parasite dormancy and activation, in addition to serving as a testbed for candidate drugs and vaccines (309).
Adding to the potential value of both in vitro and in vivo models of diseased liver processes are current efforts to incorporate stem cell-derived hepatocyte lineage cells. Specifically, the capacity to generate iPSC lines from individuals bearing diseased genotypes, host-specific risk factors, or even from healthy donors whose resulting pluripotent cells could be subsequently altered genetically to harbor mutations of interest, opens the door for patient-specific assessment of liver disease, susceptibility to pathogens, and efficacy of candidate therapeutics. Although much attention has been paid to the potential application of iPSC-derived progeny in clinical transplantation settings, there are still major hurdles to overcome. For example, we need to determine how readily iPSCs can be amplified to produce sufficient numbers of terminally differentiated hepatocytes. In principle, hepatocyte populations generated from hESCs or iPSCs could be amplified in large animals that are made immune-deficient, such as pigs (310), but more studies are needed to assess the risks from potential infectious agents associated with hepatocytes that have passed through a xenogeneic model. In addition, prior to transplanting patients with pluripotent cell-derived material, it will be necessary to eliminate any lingering nondifferentiated cells that have the capacity to generate tumors (311, 312). This concern could be less of an issue if the differentiated cell populations are constrained in a BAL device that could be removed if tumor antigens were detected in the bloodstream. Alternatively, recent efforts to convert human fibroblasts to hepatic lineage cells have seemingly achieved this goal without requiring a pluripotent cell transition. Finally, these conversion protocols, as well as most of those used to derive iPSCs, require some form of gene transduction that can result in insertional mutagenesis. Techniques are being developed to transiently expose the target cells to the genes or proteins that promote reprogramming via plasmid-based or mRNA delivery-based methods, or with excisable constructs or viral carriers such as Sendai virus. Ultimately, it should be possible to generate autologous iPSCs from the dermal fibroblasts of an individual with a genetic liver disease, correct the genetic deficiency, and differentiate the iPSCs into hepatocytes for transplantation into the patient’s liver. A more near-term application of iPSC technology is to study interesting genotypes. Studies of this nature may offer insight into the fitness of different cell populations (e.g. cells from patients with different diseases), improved capacity to predict the effectiveness of candidate drugs or vaccine strategies, as well as permit the study of immunological issues and host-pathogen interactions by banking cell lines bearing certain HLA haplotypes. Indeed, the broader impact of iPSCs may be to model human diseases rather than be used therapeutically.
Although many challenges remain for the development of liver cell-based therapies and tissue engineering, tremendous progress that has been made in several key areas. These include cell sourcing approaches such as stem cell differentiation, the establishment of robust in vitro hepatocellular culture platforms, and the development of implantable therapeutic devices. Such innovations have advanced our overall understanding of liver function, disease, and regeneration, and have positioned the field to leverage parallel advances in transplant medicine and clinical diagnostics towards the realization of clinically effective cell-based treatments for liver disease and liver failure.
Acknowledgments
A mountain of gratitude for Heather Fleming for heroic editorial revision and without whom this State of the Art Review would have never come together. We also thank Robert Schwartz and Salman Khetani for insightful comments. K.S.Z. is supported by NIH R37 GM36477. S.N.B is an HHMI Investigator, and is supported by NIH R01 EB008396, UH2 EB017103, and R01 DK85713. I.J.F is supported by NIH RO1 DK48794, R01 DK099320, and PO1 DK096990, and by DOD W81XWH-09-1-0658 and W81XWH-11-1-0803.
References
- 1.Mann RE, Smart RG, Govoni R. The epidemiology of alcoholic liver disease. Alcohol research & health: the journal of the National Institute on Alcohol Abuse and Alcoholism. 2003;27:209–219. [PMC free article] [PubMed] [Google Scholar]
- 2.Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Kim WR, Brown RS, Jr, Terrault NA, El-Serag H. Burden of liver disease in the United States: summary of a workshop. Hepatology. 2002;36:227–242. doi: 10.1053/jhep.2002.34734. [DOI] [PubMed] [Google Scholar]
- 4.Brown KA. Liver transplantation. Curr Opin Gastroenterol. 2005;21:331–336. doi: 10.1097/01.mog.0000159830.36793.2b. [DOI] [PubMed] [Google Scholar]
- 5.Taub R. Liver regeneration: from myth to mechanism. Nature reviews Molecular cell biology. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 6.Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 7.Hassanein TI, Schade RR, Hepburn IS. Acute-on-chronic liver failure: extracorporeal liver assist devices. Current opinion in critical care. 2011;17:195–203. doi: 10.1097/MCC.0b013e328344b3aa. [DOI] [PubMed] [Google Scholar]
- 8.Rademacher S, Oppert M, Jorres A. Artificial extracorporeal liver support therapy in patients with severe liver failure. Expert review of gastroenterology & hepatology. 2011;5:591–599. doi: 10.1586/egh.11.59. [DOI] [PubMed] [Google Scholar]
- 9.Phua J, Lee KH. Liver support devices. Current opinion in critical care. 2008;14:208–215. doi: 10.1097/MCC.0b013e3282f70057. [DOI] [PubMed] [Google Scholar]
- 10.Dhawan A, Puppi J, Hughes RD, Mitry RR. Human hepatocyte transplantation: current experience and future challenges. Nat Rev Gastroenterol Hepatol. 2010;7:288–298. doi: 10.1038/nrgastro.2010.44. [DOI] [PubMed] [Google Scholar]
- 11.Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sullivan GJ, Hay DC, Park IH, Fletcher J, Hannoun Z, Payne CM, Dalgetty D, Black JR, Ross JA, Samuel K, Wang G, Daley GQ, Lee JH, Church GM, Forbes SJ, Iredale JP, Wilmut I. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010;51:329–335. doi: 10.1002/hep.23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basma H, Soto-Gutierrez A, Yannam GR, Liu L, Ito R, Yamamoto T, Ellis E, Carson SD, Sato S, Chen Y, Muirhead D, Navarro-Alvarez N, Wong RJ, Roy-Chowdhury J, Platt JL, Mercer DF, Miller JD, Strom SC, Kobayashi N, Fox IJ. Differentiation and transplantation of human embryonic stem cell-derived hepatocytes. Gastroenterology. 2009;136:990–999. doi: 10.1053/j.gastro.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding Q, Lee YK, Schaefer EA, Peters DT, Veres A, Kim K, Kuperwasser N, Motola DL, Meissner TB, Hendriks WT, Trevisan M, Gupta RM, Moisan A, Banks E, Friesen M, Schinzel RT, Xia F, Tang A, Xia Y, Figueroa E, Wann A, Ahfeldt T, Daheron L, Zhang F, Rubin LL, Peng LF, Chung RT, Musunuru K, Cowan CA. A TALEN Genome-Editing System for Generating Human Stem Cell-Based Disease Models. Cell stem cell. 2012 doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monga SPS. Springer, editor. Molecular Pathology of Liver Diseases. In: Cagle PT, editor. Molecular Pathology Library. 1. Vol. 5. 2011. pp. 7–25. [Google Scholar]
- 16.Lee WM, Squires RH, Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: Summary of a workshop. Hepatology. 2008;47:1401–1415. doi: 10.1002/hep.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O’Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez-Ruiz F, Perico N, Phillips D, Pierce K, Pope CA, 3rd, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De Leon FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui-Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berry MN, Friend DS. High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J Cell Biol. 1969;43:506–520. doi: 10.1083/jcb.43.3.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta S, Rajvanshi P, Lee CD. Integration of transplanted hepatocytes into host liver plates demonstrated with dipeptidyl peptidase IV-deficient rats. Proc Natl Acad Sci U S A. 1995;92:5860–5864. doi: 10.1073/pnas.92.13.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta S, Rajvanshi P, Sokhi R, Slehria S, Yam A, Kerr A, Novikoff PM. Entry and integration of transplanted hepatocytes in rat liver plates occur by disruption of hepatic sinusoidal endothelium. Hepatology. 1999;29:509–519. doi: 10.1002/hep.510290213. [DOI] [PubMed] [Google Scholar]
- 22.Ponder KP, Gupta S, Leland F, Darlington G, Finegold M, DeMayo J, Ledley FD, Chowdhury JR, Woo SL. Mouse hepatocytes migrate to liver parenchyma and function indefinitely after intrasplenic transplantation. Proc Natl Acad Sci U S A. 1991;88:1217–1221. doi: 10.1073/pnas.88.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strom SC, Chowdhury JR, Fox IJ. Hepatocyte transplantation for the treatment of human disease. Seminars in liver disease. 1999;19:39–48. doi: 10.1055/s-2007-1007096. [DOI] [PubMed] [Google Scholar]
- 24.Habibullah CM, Syed IH, Qamar A, Taher-Uz Z. Human fetal hepatocyte transplantation in patients with fulminant hepatic failure. Transplantation. 1994;58:951–952. doi: 10.1097/00007890-199410270-00016. [DOI] [PubMed] [Google Scholar]
- 25.Ohashi K, Waugh JM, Dake MD, Yokoyama T, Kuge H, Nakajima Y, Yamanouchi M, Naka H, Yoshioka A, Kay MA. Liver tissue engineering at extrahepatic sites in mice as a potential new therapy for genetic liver diseases. Hepatology. 2005;41:132–140. doi: 10.1002/hep.20484. [DOI] [PubMed] [Google Scholar]
- 26.Komori J, Boone L, DeWard A, Hoppo T, Lagasse E. The mouse lymph node as an ectopic transplantation site for multiple tissues. Nature biotechnology. 2012;30:976–983. doi: 10.1038/nbt.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LeCluyse EL. Human hepatocyte culture systems for the in vitro evaluation of cytochrome P450 expression and regulation. European journal of pharmaceutical sciences: official journal of the European Federation for Pharmaceutical Sciences. 2001;13:343–368. doi: 10.1016/s0928-0987(01)00135-x. [DOI] [PubMed] [Google Scholar]
- 28.Soltys KA, Soto-Gutierrez A, Nagaya M, Baskin KM, Deutsch M, Ito R, Shneider BL, Squires R, Vockley J, Guha C, Roy-Chowdhury J, Strom SC, Platt JL, Fox IJ. Barriers to the successful treatment of liver disease by hepatocyte transplantation. Journal of hepatology. 2010;53:769–774. doi: 10.1016/j.jhep.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fox IJ, Chowdhury JR, Kaufman SS, Goertzen TC, Chowdhury NR, Warkentin PI, Dorko K, Sauter BV, Strom SC. Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation. N Engl J Med. 1998;338:1422–1426. doi: 10.1056/NEJM199805143382004. [DOI] [PubMed] [Google Scholar]
- 30.Puppi J, Tan N, Mitry RR, Hughes RD, Lehec S, Mieli-Vergani G, Karani J, Champion MP, Heaton N, Mohamed R, Dhawan A. Hepatocyte transplantation followed by auxiliary liver transplantation--a novel treatment for ornithine transcarbamylase deficiency. Am J Transplant. 2008;8:452–457. doi: 10.1111/j.1600-6143.2007.02058.x. [DOI] [PubMed] [Google Scholar]
- 31.Dhawan A, Mitry RR, Hughes RD, Lehec S, Terry C, Bansal S, Arya R, Wade JJ, Verma A, Heaton ND, Rela M, Mieli-Vergani G. Hepatocyte transplantation for inherited factor VII deficiency. Transplantation. 2004;78:1812–1814. doi: 10.1097/01.tp.0000146386.77076.47. [DOI] [PubMed] [Google Scholar]
- 32.Fox IJ, Chowdhury JR, Kaufman SS, Goertzen TC, Chowdhury NR, Warkentin PI, Dorko K, Sauter BV, Strom SC. Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation. N Engl J Med. 1998;338:1422–1426. doi: 10.1056/NEJM199805143382004. [DOI] [PubMed] [Google Scholar]
- 33.Horslen SP, McCowan TC, Goertzen TC, Warkentin PI, Cai HB, Strom SC, Fox IJ. Isolated hepatocyte transplantation in an infant with a severe urea cycle disorder. Pediatrics. 2003;111:1262–1267. doi: 10.1542/peds.111.6.1262. [DOI] [PubMed] [Google Scholar]
- 34.Mito M, Kusano M, Kawaura Y. Hepatocyte transplantation in man. Transplantation proceedings. 1992;24:3052–3053. [PubMed] [Google Scholar]
- 35.Muraca M, Gerunda G, Neri D, Vilei MT, Granato A, Feltracco P, Meroni M, Giron G, Burlina AB. Hepatocyte transplantation as a treatment for glycogen storage disease type 1a. Lancet. 2002;359:317–318. doi: 10.1016/S0140-6736(02)07529-3. [DOI] [PubMed] [Google Scholar]
- 36.Sokal EM, Smets F, Bourgois A, Van Maldergem L, Buts JP, Reding R, Bernard Otte J, Evrard V, Latinne D, Vincent MF, Moser A, Soriano HE. Hepatocyte transplantation in a 4-year-old girl with peroxisomal biogenesis disease: technique, safety, and metabolic follow-up. Transplantation. 2003;76:735–738. doi: 10.1097/01.TP.0000077420.81365.53. [DOI] [PubMed] [Google Scholar]
- 37.Overturf K, al-Dhalimy M, Ou CN, Finegold M, Grompe M. Serial transplantation reveals the stem-cell-like regenerative potential of adult mouse hepatocytes. The American journal of pathology. 1997;151:1273–1280. [PMC free article] [PubMed] [Google Scholar]
- 38.Cho CH, Berthiaume F, Tilles AW, Yarmush ML. A new technique for primary hepatocyte expansion in vitro. Biotechnology and bioengineering. 2008;101:345–356. doi: 10.1002/bit.21911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michalopoulos GK, DeFrances MC. Liver Regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 40.Flaim CJ, Chien S, Bhatia SN. An extracellular matrix microarray for probing cellular differentiation. Nature methods. 2005;2:119–125. doi: 10.1038/nmeth736. [DOI] [PubMed] [Google Scholar]
- 41.Shan J, Schwartz RE, Ross NT, Logan DJ, Thomas D, Duncan SA, North TE, Goessling W, Carpenter AE, Bhatia SN. Identification of small molecules for human hepatocyte expansion and iPS differentiation. Nature chemical biology. 2013 doi: 10.1038/nchembio.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riehle KJ, Dan YY, Campbell JS, Fausto N. New concepts in liver regeneration. Journal of gastroenterology and hepatology. 2011;26(Suppl 1):203–212. doi: 10.1111/j.1440-1746.2010.06539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurinna S, Barton MC. Cascades of transcription regulation during liver regeneration. The international journal of biochemistry & cell biology. 2011;43:189–197. doi: 10.1016/j.biocel.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Kiyokawa H, Dennewitz MB, Costa RH. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc Natl Acad Sci U S A. 2002;99:16881–16886. doi: 10.1073/pnas.252570299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wirth KG, Ricci R, Gimenez-Abian JF, Taghybeeglu S, Kudo NR, Jochum W, Vasseur-Cognet M, Nasmyth K. Loss of the anaphase-promoting complex in quiescent cells causes unscheduled hepatocyte proliferation. Genes & development. 2004;18:88–98. doi: 10.1101/gad.285404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wuestefeld T, Pesic M, Rudalska R, Dauch D, Longerich T, Kang TW, Yevsa T, Heinzmann F, Hoenicke L, Hohmeyer A, Potapova A, Rittelmeier I, Jarek M, Geffers R, Scharfe M, Klawonn F, Schirmacher P, Malek NP, Ott M, Nordheim A, Vogel A, Manns MP, Zender L. A Direct in vivo RNAi screen identifies MKK4 as a key regulator of liver regeneration. Cell. 2013;153:389–401. doi: 10.1016/j.cell.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 48.Carpentier B, Gautier A, Legallais C. Artificial and bioartificial liver devices: present and future. Gut. 2009;58:1690–1702. doi: 10.1136/gut.2008.175380. [DOI] [PubMed] [Google Scholar]
- 49.Stange J, Ramlow W, Mitzner S, Schmidt R, Klinkmann H. Dialysis against a recycled albumin solution enables the removal of albumin-bound toxins. Artificial organs. 1993;17:809–813. doi: 10.1111/j.1525-1594.1993.tb00635.x. [DOI] [PubMed] [Google Scholar]
- 50.Rifai K, Ernst T, Kretschmer U, Bahr MJ, Schneider A, Hafer C, Haller H, Manns MP, Fliser D. Prometheus--a new extracorporeal system for the treatment of liver failure. Journal of hepatology. 2003;39:984–990. doi: 10.1016/s0168-8278(03)00468-9. [DOI] [PubMed] [Google Scholar]
- 51.Wolff B, Machill K, Schumacher D, Schulzki I. MARS dialysis in decompensated alcoholic liver disease: a single-center experience. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2007;13:1189–1192. doi: 10.1002/lt.21235. [DOI] [PubMed] [Google Scholar]
- 52.Evenepoel P, Laleman W, Wilmer A, Claes K, Kuypers D, Bammens B, Nevens F, Vanrenterghem Y. Prometheus versus molecular adsorbents recirculating system: comparison of efficiency in two different liver detoxification devices. Artificial organs. 2006;30:276–284. doi: 10.1111/j.1525-1594.2006.00215.x. [DOI] [PubMed] [Google Scholar]
- 53.Skwarek A, Grodzicki M, Nyckowski P, Kotulski M, Zieniewicz K, Michalowicz B, Patkowski W, Grzelak I, Paczkowska A, Giercuszkiewicz D, Sanko-Resmer J, Paczek L, Krawczyk M. The use Prometheus FPSA system in the treatment of acute liver failure: preliminary results. Transplantation proceedings. 2006;38:209–211. doi: 10.1016/j.transproceed.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 54.Wauters J, Wilmer A. Albumin dialysis: current practice and future options. Liver international: official journal of the International Association for the Study of the Liver. 2011;31(Suppl 3):9–12. doi: 10.1111/j.1478-3231.2011.02589.x. [DOI] [PubMed] [Google Scholar]
- 55.Banares R, Nevens F, Larsen FS, Jalan R, Albillos A, Dollinger M, Saliba F, Sauerbruch T, Klammt S, Ockenga J, Pares A, Wendon J, Brunnler T, Kramer L, Mathurin P, de la Mata M, Gasbarrini A, Mullhaupt B, Wilmer A, Laleman W, Eefsen M, Sen S, Zipprich A, Tenorio T, Pavesi M, Schmidt HH, Mitzner S, Williams R, Arroyo V Rs group. Extracorporeal albumin dialysis with the molecular adsorbent recirculating system in acute-on-chronic liver failure: the RELIEF trial. Hepatology. 2013;57:1153–1162. doi: 10.1002/hep.26185. [DOI] [PubMed] [Google Scholar]
- 56.Kribben A, Gerken G, Haag S, Herget-Rosenthal S, Treichel U, Betz C, Sarrazin C, Hoste E, Van Vlierberghe H, Escorsell A, Hafer C, Schreiner O, Galle PR, Mancini E, Caraceni P, Karvellas CJ, Salmhofer H, Knotek M, Gines P, Kozik-Jaromin J, Rifai K, Group HS. Effects of fractionated plasma separation and adsorption on survival in patients with acute-on-chronic liver failure. Gastroenterology. 2012;142:782–789. e783. doi: 10.1053/j.gastro.2011.12.056. [DOI] [PubMed] [Google Scholar]
- 57.Matsumura KN, Guevara GR, Huston H, Hamilton WL, Rikimaru M, Yamasaki G, Matsumura MS. Hybrid bioartificial liver in hepatic failure: preliminary clinical report. Surgery. 1987;101:99–103. [PubMed] [Google Scholar]
- 58.Millis JM, Cronin DC, Johnson R, Conjeevaram H, Conlin C, Trevino S, Maguire P. Initial experience with the modified extracorporeal liver-assist device for patients with fulminant hepatic failure: system modifications and clinical impact. Transplantation. 2002;74:1735–1746. doi: 10.1097/00007890-200212270-00016. [DOI] [PubMed] [Google Scholar]
- 59.van de Kerkhove MP, Di Florio E, Scuderi V, Mancini A, Belli A, Bracco A, Dauri M, Tisone G, Di Nicuolo G, Amoroso P, Spadari A, Lombardi G, Hoekstra R, Calise F, Chamuleau RA. Phase I clinical trial with the AMC-bioartificial liver. The International journal of artificial organs. 2002;25:950–959. doi: 10.1177/039139880202501009. [DOI] [PubMed] [Google Scholar]
- 60.Gerlach JC, Mutig K, Sauer IM, Schrade P, Efimova E, Mieder T, Naumann G, Grunwald A, Pless G, Mas A, Bachmann S, Neuhaus P, Zeilinger K. Use of primary human liver cells originating from discarded grafts in a bioreactor for liver support therapy and the prospects of culturing adult liver stem cells in bioreactors: a morphologic study. Transplantation. 2003;76:781–786. doi: 10.1097/01.TP.0000083319.36931.32. [DOI] [PubMed] [Google Scholar]
- 61.Demetriou AA, Brown RS, Jr, Busuttil RW, Fair J, McGuire BM, Rosenthal P, Am Esch JS, 2nd, Lerut J, Nyberg SL, Salizzoni M, Fagan EA, de Hemptinne B, Broelsch CE, Muraca M, Salmeron JM, Rabkin JM, Metselaar HJ, Pratt D, De La Mata M, McChesney LP, Everson GT, Lavin PT, Stevens AC, Pitkin Z, Solomon BA. Prospective, randomized, multicenter, controlled trial of a bioartificial liver in treating acute liver failure. Annals of surgery. 2004;239:660–667. doi: 10.1097/01.sla.0000124298.74199.e5. discussion 667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nyberg SL, Remmel RP, Mann HJ, Peshwa MV, Hu WS, Cerra FB. Primary hepatocytes outperform Hep G2 cells as the source of biotransformation functions in a bioartificial liver. Annals of surgery. 1994;220:59–67. [PMC free article] [PubMed] [Google Scholar]
- 63.Baccarani U, Donini A, Sanna A, Risaliti A, Cariani A, Nardo B, Cavallari A, Martinelli G, Ridolfi L, Bellini G, Scalamogna M, Bresadola F. First report of cryopreserved human hepatocytes based bioartificial liver successfully used as a bridge to liver transplantation. Am J Transplant. 2004;4:286–289. doi: 10.1046/j.1600-6143.2003.00310.x. [DOI] [PubMed] [Google Scholar]
- 64.Soto-Gutierrez A, Kobayashi N, Rivas-Carrillo JD, Navarro-Alvarez N, Zhao D, Okitsu T, Noguchi H, Basma H, Tabata Y, Chen Y, Tanaka K, Narushima M, Miki A, Ueda T, Jun HS, Yoon JW, Lebkowski J, Tanaka N, Fox IJ. Reversal of mouse hepatic failure using an implanted liver-assist device containing ES cell-derived hepatocytes. Nature biotechnology. 2006;24:1412–1419. doi: 10.1038/nbt1257. [DOI] [PubMed] [Google Scholar]
- 65.Katenz E, Vondran FW, Schwartlander R, Pless G, Gong X, Cheng X, Neuhaus P, Sauer IM. Cryopreservation of primary human hepatocytes: the benefit of trehalose as an additional cryoprotective agent. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2007;13:38–45. doi: 10.1002/lt.20921. [DOI] [PubMed] [Google Scholar]
- 66.Poyck PP, Hoekstra R, van Wijk AC, Attanasio C, Calise F, Chamuleau RA, van Gulik TM. Functional and morphological comparison of three primary liver cell types cultured in the AMC bioartificial liver. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2007;13:589–598. doi: 10.1002/lt.21090. [DOI] [PubMed] [Google Scholar]
- 67.Park JK, Lee DH. Bioartificial liver systems: current status and future perspective. Journal of bioscience and bioengineering. 2005;99:311–319. doi: 10.1263/jbb.99.311. [DOI] [PubMed] [Google Scholar]
- 68.Rozga J, Holzman MD, Ro MS, Griffin DW, Neuzil DF, Giorgio T, Moscioni AD, Demetriou AA. Development of a hybrid bioartificial liver. Annals of surgery. 1993;217:502–509. doi: 10.1097/00000658-199305010-00010. discussion 509–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van de Kerkhove MP, Hoekstra R, Chamuleau RA, van Gulik TM. Clinical application of bioartificial liver support systems. Annals of surgery. 2004;240:216–230. doi: 10.1097/01.sla.0000132986.75257.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coward SM, Legallais C, David B, Thomas M, Foo Y, Mavri-Damelin D, Hodgson HJ, Selden C. Alginate-encapsulated HepG2 cells in a fluidized bed bioreactor maintain function in human liver failure plasma. Artificial organs. 2009;33:1117–1126. doi: 10.1111/j.1525-1594.2009.00821.x. [DOI] [PubMed] [Google Scholar]
- 71.Rahman TM, Selden C, Khalil M, Diakanov I, Hodgson HJ. Alginate-encapsulated human hepatoblastoma cells in an extracorporeal perfusion system improve some systemic parameters of liver failure in a xenogeneic model. Artificial organs. 2004;28:476–482. doi: 10.1111/j.1525-1594.2004.07259.x. [DOI] [PubMed] [Google Scholar]
- 72.Sauer IM, Neuhaus P, Gerlach JC. Concept for modular extracorporeal liver support for the treatment of acute hepatic failure. Metab Brain Dis. 2002;17:477–484. doi: 10.1023/a:1021938708670. [DOI] [PubMed] [Google Scholar]
- 73.Washizu J, Berthiaume F, Chan C, Tompkins RG, Toner M, Yarmush ML. Optimization of rat hepatocyte culture in citrated human plasma. J Surg Res. 2000;93:237–246. doi: 10.1006/jsre.2000.5986. [DOI] [PubMed] [Google Scholar]
- 74.Chan C, Berthiaume F, Lee K, Yarmush ML. Metabolic flux analysis of hepatocyte function in hormone- and amino acid-supplemented plasma. Metab Eng. 2003;5:1–15. doi: 10.1016/s1096-7176(02)00011-3. [DOI] [PubMed] [Google Scholar]
- 75.Wang Y, Susando T, Lei X, Anene-Nzelu C, Zhou H, Liang LH, Yu H. Current development of bioreactors for extracorporeal bioartificial liver (Review) Biointerphases. 2010;5:FA116–131. doi: 10.1116/1.3521520. [DOI] [PubMed] [Google Scholar]
- 76.Park J, Berthiaume F, Toner M, Yarmush ML, Tilles AW. Microfabricated grooved substrates as platforms for bioartificial liver reactors. Biotechnology and bioengineering. 2005;90:632–644. doi: 10.1002/bit.20463. [DOI] [PubMed] [Google Scholar]
- 77.Tilles AW, Baskaran H, Roy P, Yarmush ML, Toner M. Effects of oxygenation and flow on the viability and function of rat hepatocytes cocultured in a microchannel flat-plate bioreactor. Biotechnology and bioengineering. 2001;73:379–389. doi: 10.1002/bit.1071. [DOI] [PubMed] [Google Scholar]
- 78.Guillouzo A. Liver cell models in in vitro toxicology. Environ Health Perspect. 1998;106(Suppl 2):511–532. doi: 10.1289/ehp.98106511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laishes BA, Williams GM. Conditions affecting primary cell cultures of functional adult rat hepatocytes. II. Dexamethasone enhanced longevity and maintenance of morphology. In Vitro. 1976;12:821–832. doi: 10.1007/BF02796367. [DOI] [PubMed] [Google Scholar]
- 80.Isom HC, Secott T, Georgoff I, Woodworth C, Mummaw J. Maintenance of differentiated rat hepatocytes in primary culture. Proc Natl Acad Sci U S A. 1985;82:3252–3256. doi: 10.1073/pnas.82.10.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miyazaki M, Handa Y, Oda M, Yabe T, Miyano K, Sato J. Long-term survival of functional hepatocytes from adult rat in the presence of phenobarbital in primary culture. Exp Cell Res. 1985;159:176–190. doi: 10.1016/s0014-4827(85)80047-1. [DOI] [PubMed] [Google Scholar]
- 82.Dunn JC, Tompkins RG, Yarmush ML. Hepatocytes in collagen sandwich: evidence for transcriptional and translational regulation. J Cell Biol. 1992;116:1043–1053. doi: 10.1083/jcb.116.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Richert L, Binda D, Hamilton G, Viollon-Abadie C, Alexandre E, Bigot-Lasserre D, Bars R, Coassolo P, LeCluyse E. Evaluation of the effect of culture configuration on morphology, survival time, antioxidant status and metabolic capacities of cultured rat hepatocytes. Toxicol In Vitro. 2002;16:89–99. doi: 10.1016/s0887-2333(01)00099-6. [DOI] [PubMed] [Google Scholar]
- 84.Khetani SR, Kanchagar C, Ukairo O, Krzyzewski S, Moore A, Shi J, Aoyama S, Aleo M, Will Y. The Use of Micropatterned Co-cultures to Detect Compounds that Cause Drug induced Liver Injury in Humans. Toxicological sciences: an official journal of the Society of Toxicology. 2012 doi: 10.1093/toxsci/kfs326. [DOI] [PubMed] [Google Scholar]
- 85.Lin P, Chan WC, Badylak SF, Bhatia SN. Assessing porcine liver-derived biomatrix for hepatic tissue engineering. Tissue engineering. 2004;10:1046–1053. doi: 10.1089/ten.2004.10.1046. [DOI] [PubMed] [Google Scholar]
- 86.Sellaro TL, Ranade A, Faulk DM, McCabe GP, Dorko K, Badylak SF, Strom SC. Maintenance of human hepatocyte function in vitro by liver-derived extracellular matrix gels. Tissue engineering Part A. 2010;16:1075–1082. doi: 10.1089/ten.tea.2008.0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reticker-Flynn NE, Malta DF, Winslow MM, Lamar JM, Xu MJ, Underhill GH, Hynes RO, Jacks TE, Bhatia SN. A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nature communications. 2012;3:1122. doi: 10.1038/ncomms2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen AA, Khetani SR, Lee S, Bhatia SN, Van Vliet KJ. Modulation of hepatocyte phenotype in vitro via chemomechanical tuning of polyelectrolyte multilayers. Biomaterials. 2009;30:1113–1120. doi: 10.1016/j.biomaterials.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Janorkar AV, Rajagopalan P, Yarmush ML, Megeed Z. The use of elastin-like polypeptide-polyelectrolyte complexes to control hepatocyte morphology and function in vitro. Biomaterials. 2008;29:625–632. doi: 10.1016/j.biomaterials.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 90.Hay DC, Pernagallo S, Diaz-Mochon JJ, Medine CN, Greenhough S, Hannoun Z, Schrader J, Black JR, Fletcher J, Dalgetty D, Thompson AI, Newsome PN, Forbes SJ, Ross JA, Bradley M, Iredale JP. Unbiased screening of polymer libraries to define novel substrates for functional hepatocytes with inducible drug metabolism. Stem cell research. 2011;6:92–102. doi: 10.1016/j.scr.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 91.Wu FJ, Friend JR, Remmel RP, Cerra FB, Hu WS. Enhanced cytochrome P450 IA1 activity of self-assembled rat hepatocyte spheroids. Cell transplantation. 1999;8:233–246. doi: 10.1177/096368979900800304. [DOI] [PubMed] [Google Scholar]
- 92.Koide N, Sakaguchi K, Koide Y, Asano K, Kawaguchi M, Matsushima H, Takenami T, Shinji T, Mori M, Tsuji T. Formation of multicellular spheroids composed of adult rat hepatocytes in dishes with positively charged surfaces and under other nonadherent environments. Exp Cell Res. 1990;186:227–235. doi: 10.1016/0014-4827(90)90300-y. [DOI] [PubMed] [Google Scholar]
- 93.Yuasa C, Tomita Y, Shono M, Ishimura K, Ichihara A. Importance of cell aggregation for expression of liver functions and regeneration demonstrated with primary cultured hepatocytes. J Cell Physiol. 1993;156:522–530. doi: 10.1002/jcp.1041560311. [DOI] [PubMed] [Google Scholar]
- 94.Kojima R, Yoshimoto K, Takahashi E, Ichino M, Miyoshi H, Nagasaki Y. Spheroid array of fetal mouse liver cells constructed on a PEG-gel micropatterned surface: upregulation of hepatic functions by co-culture with nonparenchymal liver cells. Lab on a chip. 2009;9:1991–1993. doi: 10.1039/b903388b. [DOI] [PubMed] [Google Scholar]
- 95.Brophy CM, Luebke-Wheeler JL, Amiot BP, Khan H, Remmel RP, Rinaldo P, Nyberg SL. Rat hepatocyte spheroids formed by rocked technique maintain differentiated hepatocyte gene expression and function. Hepatology. 2009;49:578–586. doi: 10.1002/hep.22674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Drewitz M, Helbling M, Fried N, Bieri M, Moritz W, Lichtenberg J, Kelm JM. Towards automated production and drug sensitivity testing using scaffold-free spherical tumor microtissues. Biotechnology journal. 2011;6:1488–1496. doi: 10.1002/biot.201100290. [DOI] [PubMed] [Google Scholar]
- 97.Guguenguillouzo C, Clement B, Baffet G, Beaumont C, Morelchany E, Glaise D, Guillouzo A. MAINTENANCE AND REVERSIBILITY OF ACTIVE ALBUMIN SECRETION BY ADULT-RAT HEPATOCYTES CO-CULTURED WITH ANOTHER LIVER EPITHELIAL-CELL TYPE. Exp Cell Res. 1983;143:47–54. doi: 10.1016/0014-4827(83)90107-6. [DOI] [PubMed] [Google Scholar]
- 98.Bhatia SN, Balis UJ, Yarmush ML, Toner M. Effect of cell-cell interactions in preservation of cellular phenotype: cocultivation of hepatocytes and nonparenchymal cells. The FASEB journal. 1999;13:1883–1900. doi: 10.1096/fasebj.13.14.1883. [DOI] [PubMed] [Google Scholar]
- 99.Fraslin JM, Kneip B, Vaulont S, Glaise D, Munnich A, Guguen-Guillouzo C. Dependence of hepatocyte-specific gene expression on cell-cell interactions in primary culture. EMBO J. 1985;4:2487–2491. doi: 10.1002/j.1460-2075.1985.tb03960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vandenberghe Y, Tee L, Rogiers V, Yeoh G. Transcriptional- and post-transcriptional-dependent regulation of glutathione S-transferase expression in rat hepatocytes as a function of culture conditions. FEBS Lett. 1992;313:155–159. doi: 10.1016/0014-5793(92)81434-n. [DOI] [PubMed] [Google Scholar]
- 101.Akrawi M, Rogiers V, Vandenberghe Y, Palmer CN, Vercruysse A, Shephard EA, Phillips IR. Maintenance and induction in co-cultured rat hepatocytes of components of the cytochrome P450-mediated mono-oxygenase. Biochemical pharmacology. 1993;45:1583–1591. doi: 10.1016/0006-2952(93)90298-b. [DOI] [PubMed] [Google Scholar]
- 102.Milosevic N, Schawalder H, Maier P. Kupffer cell-mediated differential down-regulation of cytochrome P450 metabolism in rat hepatocytes. European journal of pharmacology. 1999;368:75–87. doi: 10.1016/s0014-2999(98)00988-1. [DOI] [PubMed] [Google Scholar]
- 103.Hoebe KH, Witkamp RF, Fink-Gremmels J, Van Miert AS, Monshouwer M. Direct cell-to-cell contact between Kupffer cells and hepatocytes augments endotoxin-induced hepatic injury. American journal of physiology Gastrointestinal and liver physiology. 2001;280:G720–728. doi: 10.1152/ajpgi.2001.280.4.G720. [DOI] [PubMed] [Google Scholar]
- 104.Morin O, Normand C. Long-term maintenance of hepatocyte functional activity in co-culture: requirements for sinusoidal endothelial cells and dexamethasone. J Cell Physiol. 1986;129:103–110. doi: 10.1002/jcp.1041290115. [DOI] [PubMed] [Google Scholar]
- 105.Krause P, Markus PM, Schwartz P, Unthan-Fechner K, Pestel S, Fandrey J, Probst I. Hepatocyte-supported serum-free culture of rat liver sinusoidal endothelial cells. Journal of hepatology. 2000;32:718–726. doi: 10.1016/s0168-8278(00)80239-1. [DOI] [PubMed] [Google Scholar]
- 106.Nahmias Y, Casali M, Barbe L, Berthiaume F, Yarmush ML. Liver endothelial cells promote LDL-R expression and the uptake of HCV-like particles in primary rat and human hepatocytes. Hepatology. 2006;43:257–265. doi: 10.1002/hep.21016. [DOI] [PubMed] [Google Scholar]
- 107.Hwa AJ, Fry RC, Sivaraman A, So PT, Samson LD, Stolz DB, Griffith LG. Rat liver sinusoidal endothelial cells survive without exogenous VEGF in 3D perfused co-cultures with hepatocytes. The FASEB journal. 2007;21:2564–2579. doi: 10.1096/fj.06-7473com. [DOI] [PubMed] [Google Scholar]
- 108.March S, Hui EE, Underhill GH, Khetani S, Bhatia SN. Microenvironmental regulation of the sinusoidal endothelial cell phenotype in vitro. Hepatology. 2009;50:920–928. doi: 10.1002/hep.23085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim Y, Rajagopalan P. 3D hepatic cultures simultaneously maintain primary hepatocyte and liver sinusoidal endothelial cell phenotypes. PloS one. 2010;5:e15456. doi: 10.1371/journal.pone.0015456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goulet F, Normand C, Morin O. Cellular interactions promote tissue-specific function, biomatrix deposition and junctional communication of primary cultured hepatocytes. Hepatology. 1988;8:1010–1018. doi: 10.1002/hep.1840080506. [DOI] [PubMed] [Google Scholar]
- 111.Corlu A, Kneip B, Lhadi C, Leray G, Glaise D, Baffet G, Bourel D, Guguen-Guillouzo C. A plasma membrane protein is involved in cell contact-mediated regulation of tissue-specific genes in adult hepatocytes. J Cell Biol. 1991;115:505–515. doi: 10.1083/jcb.115.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rojkind M, Novikoff PM, Greenwel P, Rubin J, Rojas-Valencia L, de Carvalho AC, Stockert R, Spray D, Hertzberg EL, Wolkoff AW. Characterization and functional studies on rat liver fat-storing cell line and freshly isolated hepatocyte coculture system. The American journal of pathology. 1995;146:1508–1520. [PMC free article] [PubMed] [Google Scholar]
- 113.Hamilton GA, Jolley SL, Gilbert D, Coon DJ, Barros S, LeCluyse EL. Regulation of cell morphology and cytochrome P450 expression in human hepatocytes by extracellular matrix and cell-cell interactions. Cell and tissue research. 2001;306:85–99. doi: 10.1007/s004410100429. [DOI] [PubMed] [Google Scholar]
- 114.Brieva TA, Moghe PV. Functional engineering of hepatocytes via heterocellular presentation of a homoadhesive molecule, E-cadherin. Biotechnology and bioengineering. 2001;76:295–302. doi: 10.1002/bit.10041. [DOI] [PubMed] [Google Scholar]
- 115.Chia SM, Lin PC, Yu H. TGF-beta1 regulation in hepatocyte-NIH3T3 co-culture is important for the enhanced hepatocyte function in 3D microenvironment. Biotechnology and bioengineering. 2005;89:565–573. doi: 10.1002/bit.20372. [DOI] [PubMed] [Google Scholar]
- 116.Khetani SR, Chen AA, Ranscht B, Bhatia SN. T-cadherin modulates hepatocyte functions in vitro. The FASEB journal. 2008;22:3768–3775. doi: 10.1096/fj.07-105155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Odom DT, Dowell RD, Jacobsen ES, Nekludova L, Rolfe PA, Danford TW, Gifford DK, Fraenkel E, Bell GI, Young RA. Core transcriptional regulatory circuitry in human hepatocytes. Molecular systems biology. 2006;2:2006 0017. doi: 10.1038/msb4100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dorrell C, Erker L, Schug J, Kopp JL, Canaday PS, Fox AJ, Smirnova O, Duncan AW, Finegold MJ, Sander M, Kaestner KH, Grompe M. Prospective isolation of a bipotential clonogenic liver progenitor cell in adult mice. Genes & development. 2011;25:1193–1203. doi: 10.1101/gad.2029411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shin S, Walton G, Aoki R, Brondell K, Schug J, Fox A, Smirnova O, Dorrell C, Erker L, Chu AS, Wells RG, Grompe M, Greenbaum LE, Kaestner KH. Foxl1-Cre-marked adult hepatic progenitors have clonogenic and bilineage differentiation potential. Genes & development. 2011;25:1185–1192. doi: 10.1101/gad.2027811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cardinale V, Wang Y, Carpino G, Mendel G, Alpini G, Gaudio E, Reid LM, Alvaro D. The biliary tree--a reservoir of multipotent stem cells. Nat Rev Gastroenterol Hepatol. 2012;9:231–240. doi: 10.1038/nrgastro.2012.23. [DOI] [PubMed] [Google Scholar]
- 121.Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, Sato T, Hamer K, Sasaki N, Finegold MJ, Haft A, Vries RG, Grompe M, Clevers H. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494:247–250. doi: 10.1038/nature11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Suzuki A, Zheng Y, Kondo R, Kusakabe M, Takada Y, Fukao K, Nakauchi H, Taniguchi H. Flow-cytometric separation and enrichment of hepatic progenitor cells in the developing mouse liver. Hepatology. 2000;32:1230–1239. doi: 10.1053/jhep.2000.20349. [DOI] [PubMed] [Google Scholar]
- 123.Diehl AM, Chute J. Underlying potential: cellular and molecular determinants of adult liver repair. J Clin Invest. 2013;123:1858–1860. doi: 10.1172/JCI69966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Meier RP, Muller YD, Morel P, Gonelle-Gispert C, Buhler LH. Transplantation of mesenchymal stem cells for the treatment of liver diseases, is there enough evidence? Stem cell research. 2013;11:1348–1364. doi: 10.1016/j.scr.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 125.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 126.Rambhatla L, Chiu CP, Kundu P, Peng Y, Carpenter MK. Generation of hepatocyte-like cells from human embryonic stem cells. Cell transplantation. 2003;12:1–11. doi: 10.3727/000000003783985179. [DOI] [PubMed] [Google Scholar]
- 127.Zaret KS. Genetic programming of liver and pancreas progenitors: lessons for stem-cell differentiation. Nature reviews Genetics. 2008;9:329–340. doi: 10.1038/nrg2318. [DOI] [PubMed] [Google Scholar]
- 128.Gouon-Evans V, Boussemart L, Gadue P, Nierhoff D, Koehler CI, Kubo A, Shafritz DA, Keller G. BMP-4 is required for hepatic specification of mouse embryonic stem cell-derived definitive endoderm. Nature biotechnology. 2006;24:1402–1411. doi: 10.1038/nbt1258. [DOI] [PubMed] [Google Scholar]
- 129.Agarwal S, Holton KL, Lanza R. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem cells. 2008;26:1117–1127. doi: 10.1634/stemcells.2007-1102. [DOI] [PubMed] [Google Scholar]
- 130.Cai J, Zhao Y, Liu Y, Ye F, Song Z, Qin H, Meng S, Chen Y, Zhou R, Song X, Guo Y, Ding M, Deng H. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology. 2007;45:1229–1239. doi: 10.1002/hep.21582. [DOI] [PubMed] [Google Scholar]
- 131.Hay DC, Zhao D, Ross A, Mandalam R, Lebkowski J, Cui W. Direct differentiation of human embryonic stem cells to hepatocyte-like cells exhibiting functional activities. Cloning and stem cells. 2007;9:51–62. doi: 10.1089/clo.2006.0045. [DOI] [PubMed] [Google Scholar]
- 132.Lavon N, Yanuka O, Benvenisty N. Differentiation and isolation of hepatic-like cells from human embryonic stem cells. Differentiation; research in biological diversity. 2004;72:230–238. doi: 10.1111/j.1432-0436.2004.07205002.x. [DOI] [PubMed] [Google Scholar]
- 133.Song Z, Cai J, Liu Y, Zhao D, Yong J, Duo S, Song X, Guo Y, Zhao Y, Qin H, Yin X, Wu C, Che J, Lu S, Ding M, Deng H. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell research. 2009;19:1233–1242. doi: 10.1038/cr.2009.107. [DOI] [PubMed] [Google Scholar]
- 134.Touboul T, Hannan NR, Corbineau S, Martinez A, Martinet C, Branchereau S, Mainot S, Strick-Marchand H, Pedersen R, Di Santo J, Weber A, Vallier L. Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology. 2010;51:1754–1765. doi: 10.1002/hep.23506. [DOI] [PubMed] [Google Scholar]
- 135.Vosough M, Omidinia E, Kadivar M, Shokrgozar MA, Pournasr B, Aghdami N, Baharvand H. Generation of functional hepatocyte-like cells from human pluripotent stem cells in a scalable suspension culture. Stem cells and development. 2013;22:2693–2705. doi: 10.1089/scd.2013.0088. [DOI] [PubMed] [Google Scholar]
- 136.Ma X, Duan Y, Tschudy-Seney B, Roll G, Behbahan IS, Ahuja TP, Tolstikov V, Wang C, McGee J, Khoobyari S, Nolta JA, Willenbring H, Zern MA. Highly efficient differentiation of functional hepatocytes from human induced pluripotent stem cells. Stem cells translational medicine. 2013;2:409–419. doi: 10.5966/sctm.2012-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schwartz RE, Fleming HE, Khetani SR, Bhatia SN. Pluripotent stem cell-derived hepatocyte-like cells. Biotechnology advances. 2014;32:504–513. doi: 10.1016/j.biotechadv.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Heo J, Factor VM, Uren T, Takahama Y, Lee JS, Major M, Feinstone SM, Thorgeirsson SS. Hepatic precursors derived from murine embryonic stem cells contribute to regeneration of injured liver. Hepatology. 2006;44:1478–1486. doi: 10.1002/hep.21441. [DOI] [PubMed] [Google Scholar]
- 139.Li F, Liu P, Liu C, Xiang D, Deng L, Li W, Wangensteen K, Song J, Ma Y, Hui L, Wei L, Li L, Ding X, Hu Y, He Z, Wang X. Hepatoblast-like progenitor cells derived from embryonic stem cells can repopulate livers of mice. Gastroenterology. 2010;139:2158–2169. e2158. doi: 10.1053/j.gastro.2010.08.042. [DOI] [PubMed] [Google Scholar]
- 140.Liu H, Kim Y, Sharkis S, Marchionni L, Jang YY. In vivo liver regeneration potential of human induced pluripotent stem cells from diverse origins. Sci Transl Med. 2011;3:82ra39. doi: 10.1126/scitranslmed.3002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang RR, Ueno Y, Zheng YW, Koike N, Aoyama S, Adachi Y, Taniguchi H. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013 doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- 142.Huang P, He Z, Ji S, Sun H, Xiang D, Liu C, Hu Y, Wang X, Hui L. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature. 2011;475:386–389. doi: 10.1038/nature10116. [DOI] [PubMed] [Google Scholar]
- 143.Sekiya S, Suzuki A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature. 2011;475:390–393. doi: 10.1038/nature10263. [DOI] [PubMed] [Google Scholar]
- 144.Zhu S, Rezvani M, Harbell J, Mattis AN, Wolfe AR, Benet LZ, Willenbring H, Ding S. Mouse liver repopulation with hepatocytes generated from human fibroblasts. Nature. 2014;508:93–97. doi: 10.1038/nature13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Huang P, Zhang L, Gao Y, He Z, Yao D, Wu Z, Cen J, Chen X, Liu C, Hu Y, Lai D, Hu Z, Chen L, Zhang Y, Cheng X, Ma X, Pan G, Wang X, Hui L. Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell stem cell. 2014;14:370–384. doi: 10.1016/j.stem.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 146.Du Y, Wang J, Jia J, Song N, Xiang C, Xu J, Hou Z, Su X, Liu B, Jiang T, Zhao D, Sun Y, Shu J, Guo Q, Yin M, Sun D, Lu S, Shi Y, Deng H. Human hepatocytes with drug metabolic function induced from fibroblasts by lineage reprogramming. Cell stem cell. 2014;14:394–403. doi: 10.1016/j.stem.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 147.Katoh M, Yokoi T. Application of chimeric mice with humanized liver for predictive ADME. Drug metabolism reviews. 2007;39:145–157. doi: 10.1080/03602530601021340. [DOI] [PubMed] [Google Scholar]
- 148.Legrand N, Ploss A, Balling R, Becker PD, Borsotti C, Brezillon N, Debarry J, de Jong Y, Deng H, Di Santo JP, Eisenbarth S, Eynon E, Flavell RA, Guzman CA, Huntington ND, Kremsdorf D, Manns MP, Manz MG, Mention JJ, Ott M, Rathinam C, Rice CM, Rongvaux A, Stevens S, Spits H, Strick-Marchand H, Takizawa H, van Lent AU, Wang C, Weijer K, Willinger T, Ziegler P. Humanized mice for modeling human infectious disease: challenges, progress, and outlook. Cell host & microbe. 2009;6:5–9. doi: 10.1016/j.chom.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dorner M, Ploss A. Deconstructing hepatitis C virus infection in humanized mice. Annals of the New York Academy of Sciences. 2011;1245:59–62. doi: 10.1111/j.1749-6632.2011.06317.x. [DOI] [PubMed] [Google Scholar]
- 150.Medine CN, Lucendo-Villarin B, Storck C, Wang F, Szkolnicka D, Khan F, Pernagallo S, Black JR, Marriage HM, Ross JA, Bradley M, Iredale JP, Flint O, Hay DC. Developing high-fidelity hepatotoxicity models from pluripotent stem cells. Stem cells translational medicine. 2013;2:505–509. doi: 10.5966/sctm.2012-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim SS, Kaihara S, Benvenuto MS, Kim BS, Mooney DJ, Vacanti JP. Small intestinal submucosa as a small-caliber venous graft: a novel model for hepatocyte transplantation on synthetic biodegradable polymer scaffolds with direct access to the portal venous system. Journal of pediatric surgery. 1999;34:124–128. doi: 10.1016/s0022-3468(99)90241-5. [DOI] [PubMed] [Google Scholar]
- 152.Ohashi K, Koyama F, Tatsumi K, Shima M, Park F, Nakajima Y, Okano T. Functional life-long maintenance of engineered liver tissue in mice following transplantation under the kidney capsule. Journal of tissue engineering and regenerative medicine. 2010;4:141–148. doi: 10.1002/term.225. [DOI] [PubMed] [Google Scholar]
- 153.Hamazaki K, Doi Y, Koide N. Microencapsulated multicellular spheroid of rat hepatocytes transplanted intraperitoneally after 90% hepatectomy. Hepato-gastroenterology. 2002;49:1514–1516. [PubMed] [Google Scholar]
- 154.Hoppo T, Komori J, Manohar R, Stolz DB, Lagasse E. Rescue of lethal hepatic failure by hepatized lymph nodes in mice. Gastroenterology. 2011;140:656–666. e652. doi: 10.1053/j.gastro.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bucher NL, Scott JF, Aub JC. Regeneration of the liver in parabiotic rats. Cancer research. 1951;11:457–465. [PubMed] [Google Scholar]
- 156.Hoffman AS. Hydrogels for biomedical applications. Advanced drug delivery reviews. 2002;54:3–12. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 157.Underhill GH, Chen AA, Albrecht DR, Bhatia SN. Assessment of hepatocellular function within PEG hydrogels. Biomaterials. 2007;28:256–270. doi: 10.1016/j.biomaterials.2006.08.043. [DOI] [PubMed] [Google Scholar]
- 158.Chen AA, Thomas DK, Ong LL, Schwartz RE, Golub TR, Bhatia SN. Humanized mice with ectopic artificial liver tissues. Proc Natl Acad Sci U S A. 2011;108:11842–11847. doi: 10.1073/pnas.1101791108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu Tsang V, Chen AA, Cho LM, Jadin KD, Sah RL, DeLong S, West JL, Bhatia SN. Fabrication of 3D hepatic tissues by additive photopatterning of cellular hydrogels. The FASEB journal. 2007;21:790–801. doi: 10.1096/fj.06-7117com. [DOI] [PubMed] [Google Scholar]
- 160.Itle LJ, Koh WG, Pishko MV. Hepatocyte viability and protein expression within hydrogel microstructures. Biotechnol Prog. 2005;21:926–932. doi: 10.1021/bp049681i. [DOI] [PubMed] [Google Scholar]
- 161.Tan W, Desai TA. Microfluidic patterning of cells in extracellular matrix biopolymers: effects of channel size, cell type, and matrix composition on pattern integrity. Tissue engineering. 2003;9:255–267. doi: 10.1089/107632703764664729. [DOI] [PubMed] [Google Scholar]
- 162.Vozzi G, Flaim C, Ahluwalia A, Bhatia SN. Fabrication of PLGA scaffolds using soft lithography and microsyringe deposition. Biomaterials. 2003;24:2533–2540. doi: 10.1016/s0142-9612(03)00052-8. [DOI] [PubMed] [Google Scholar]
- 163.Kaihara S, Borenstein J, Koka R, Lalan S, Ochoa ER, Ravens M, Pien H, Cunningham B, Vacanti JP. Silicon micromachining to tissue engineer branched vascular channels for liver fabrication. Tissue engineering. 2000;6:105–117. doi: 10.1089/107632700320739. [DOI] [PubMed] [Google Scholar]
- 164.Kim SS, Utsunomiya H, Koski JA, Wu BM, Cima MJ, Sohn J, Mukai K, Griffith LG, Vacanti JP. Survival and function of hepatocytes on a novel three-dimensional synthetic biodegradable polymer scaffold with an intrinsic network of channels[see comments] Annals of surgery. 1998;228:8–13. doi: 10.1097/00000658-199807000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Revzin A, Russell RJ, Yadavalli VK, Koh WG, Deister C, Hile DD, Mellott MB, Pishko MV. Fabrication of poly(ethylene glycol) hydrogel microstructures using photolithography. Langmuir. 2001;17:5440–5447. doi: 10.1021/la010075w. [DOI] [PubMed] [Google Scholar]
- 166.Hahn MS, Taite LJ, Moon JJ, Rowland MC, Ruffino KA, West JL. Photolithographic patterning of polyethylene glycol hydrogels. Biomaterials. 2006;27:2519–2524. doi: 10.1016/j.biomaterials.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 167.Stevens KR, Ungrin MD, Schwartz RE, Ng S, Carvalho B, Christine KS, Chaturvedi RR, Li CY, Zandstra PW, Chen CS, Bhatia SN. InVERT molding for scalable control of tissue microarchitecture. Nature communications. 2013;4:1847. doi: 10.1038/ncomms2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Albrecht DR, Underhill GH, Wassermann TB, Sah RL, Bhatia SN. Probing the role of multicellular organization in three-dimensional microenvironments. Nature methods. 2006;3:369–375. doi: 10.1038/nmeth873. [DOI] [PubMed] [Google Scholar]
- 169.Moghe PV, Coger RN, Toner M, Yarmush ML. Cell-cell interactions are essential for maintenance of hepatocyte function in collagen gel but not on matrigel. Biotechnology and bioengineering. 1997;56:706–711. doi: 10.1002/(SICI)1097-0290(19971220)56:6<706::AID-BIT14>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 170.Thomas RJ, Bennett A, Thomson B, Shakesheff KM. Hepatic stellate cells on poly(DL-lactic acid) surfaces control the formation of 3D hepatocyte co-culture aggregates in vitro. European cells & materials. 2006;11:16–26. doi: 10.22203/ecm.v011a03. discussion 26. [DOI] [PubMed] [Google Scholar]
- 171.Ohashi K, Park F, Kay MA. Hepatocyte transplantation: clinical and experimental application. J Mol Med (Berl) 2001;79:617–630. doi: 10.1007/s001090100260. [DOI] [PubMed] [Google Scholar]
- 172.Lee H, Cusick RA, Utsunomiya H, Ma PX, Langer R, Vacanti JP. Effect of implantation site on hepatocytes heterotopically transplanted on biodegradable polymer scaffolds. Tissue engineering. 2003;9:1227–1232. doi: 10.1089/10763270360728134. [DOI] [PubMed] [Google Scholar]
- 173.Baranski JD, Chaturvedi RR, Stevens KR, Eyckmans J, Carvalho B, Solorzano RD, Yang MT, Miller JS, Bhatia SN, Chen CS. Geometric control of vascular networks to enhance engineered tissue integration and function. Proc Natl Acad Sci U S A. 2013;110:7586–7591. doi: 10.1073/pnas.1217796110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Miller JS, Stevens KR, Yang MT, Baker BM, Nguyen DH, Cohen DM, Toro E, Chen AA, Galie PA, Yu X, Chaturvedi R, Bhatia SN, Chen CS. Rapid casting of patterned vascular networks for perfusable engineered three-dimensional tissues. Nature materials. 2012;11:768–774. doi: 10.1038/nmat3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Richardson TP, Peters MC, Ennett AB, Mooney DJ. Polymeric system for dual growth factor delivery. Nature biotechnology. 2001;19:1029–1034. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- 176.Lee H, Cusick RA, Browne F, Ho Kim T, Ma PX, Utsunomiya H, Langer R, Vacanti JP. Local delivery of basic fibroblast growth factor increases both angiogenesis and engraftment of hepatocytes in tissue-engineered polymer devices. Transplantation. 2002;73:1589–1593. doi: 10.1097/00007890-200205270-00011. [DOI] [PubMed] [Google Scholar]
- 177.Smith MK, Peters MC, Richardson TP, Garbern JC, Mooney DJ. Locally enhanced angiogenesis promotes transplanted cell survival. Tissue engineering. 2004;10:63–71. doi: 10.1089/107632704322791709. [DOI] [PubMed] [Google Scholar]
- 178.Peattie RA, Nayate AP, Firpo MA, Shelby J, Fisher RJ, Prestwich GD. Stimulation of in vivo angiogenesis by cytokine-loaded hyaluronic acid hydrogel implants. Biomaterials. 2004;25:2789–2798. doi: 10.1016/j.biomaterials.2003.09.054. [DOI] [PubMed] [Google Scholar]
- 179.Kedem A, Perets A, Gamlieli-Bonshtein I, Dvir-Ginzberg M, Mizrahi S, Cohen S. Vascular endothelial growth factor-releasing scaffolds enhance vascularization and engraftment of hepatocytes transplanted on liver lobes. Tissue engineering. 2005;11:715–722. doi: 10.1089/ten.2005.11.715. [DOI] [PubMed] [Google Scholar]
- 180.Lesman A, Habib M, Caspi O, Gepstein A, Arbel G, Levenberg S, Gepstein L. Transplantation of a Tissue-Engineered Human Vascularized Cardiac Muscle. Tissue engineering. Part A. 2009 doi: 10.1089/ten.TEA.2009.0130. [DOI] [PubMed] [Google Scholar]
- 181.Levenberg S, Rouwkema J, Macdonald M, Garfein ES, Kohane DS, Darland DC, Marini R, van Blitterswijk CA, Mulligan RC, D’Amore PA, Langer R. Engineering vascularized skeletal muscle tissue. Nature biotechnology. 2005;23:879–884. doi: 10.1038/nbt1109. [DOI] [PubMed] [Google Scholar]
- 182.Stevens KR, Kreutziger KL, Dupras SK, Korte FS, Regnier M, Muskheli V, Nourse MB, Bendixen K, Reinecke H, Murry CE. Physiological function and transplantation of scaffold-free and vascularized human cardiac muscle tissue. Proc Natl Acad Sci U S A. 2009;106:16568–16573. doi: 10.1073/pnas.0908381106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sudo R, Mitaka T, Ikeda M, Tanishita K. Reconstruction of 3D stacked-up structures by rat small hepatocytes on microporous membranes. The FASEB journal. 2005;19:1695–1697. doi: 10.1096/fj.04-3269fje. [DOI] [PubMed] [Google Scholar]
- 184.Ishida Y, Smith S, Wallace L, Sadamoto T, Okamoto M, Auth M, Strazzabosco M, Fabris L, Medina J, Prieto J, Strain A, Neuberger J, Joplin R. Ductular morphogenesis and functional polarization of normal human biliary epithelial cells in three-dimensional culture. Journal of hepatology. 2001;35:2–9. doi: 10.1016/s0168-8278(01)00078-2. [DOI] [PubMed] [Google Scholar]
- 185.Auth MK, Joplin RE, Okamoto M, Ishida Y, McMaster P, Neuberger JM, Blaheta RA, Voit T, Strain AJ. Morphogenesis of primary human biliary epithelial cells: induction in high-density culture or by coculture with autologous human hepatocytes. Hepatology. 2001;33:519–529. doi: 10.1053/jhep.2001.22703. [DOI] [PubMed] [Google Scholar]
- 186.Miyazawa M, Torii T, Toshimitsu Y, Okada K, Koyama I, Ikada Y. A tissue-engineered artificial bile duct grown to resemble the native bile duct. Am J Transplant. 2005;5:1541–1547. doi: 10.1111/j.1600-6143.2005.00845.x. [DOI] [PubMed] [Google Scholar]
- 187.Ebata H, Onodera K, Sawa M, Mito M. A study of liver regeneration using fetal rat liver tissue transplanted into the spleen. The Japanese journal of surgery. 1988;18:540–547. doi: 10.1007/BF02471488. [DOI] [PubMed] [Google Scholar]
- 188.Wilson JT, Chaikof EL. Challenges and emerging technologies in the immunoisolation of cells and tissues. Advanced drug delivery reviews. 2008;60:124–145. doi: 10.1016/j.addr.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sawhney AS, Pathak CP, Hubbell JA. Modification of islet of langerhans surfaces with immunoprotective poly(ethylene glycol) coatings via interfacial photopolymerization. Biotechnology and bioengineering. 1994;44:383–386. doi: 10.1002/bit.260440317. [DOI] [PubMed] [Google Scholar]
- 190.Hill RS, Cruise GM, Hager SR, Lamberti FV, Yu X, Garufis CL, Yu Y, Mundwiler KE, Cole JF, Hubbell JA, Hegre OD, Scharp DW. Immunoisolation of adult porcine islets for the treatment of diabetes mellitus. The use of photopolymerizable polyethylene glycol in the conformal coating of mass-isolated porcine islets. Annals of the New York Academy of Sciences. 1997;831:332–343. doi: 10.1111/j.1749-6632.1997.tb52208.x. [DOI] [PubMed] [Google Scholar]
- 191.Cheung CY, Anseth KS. Synthesis of immunoisolation barriers that provide localized immunosuppression for encapsulated pancreatic islets. Bioconjugate chemistry. 2006;17:1036–1042. doi: 10.1021/bc060023o. [DOI] [PubMed] [Google Scholar]
- 192.Bryers JD, Giachelli CM, Ratner BD. Engineering biomaterials to integrate and heal: The biocompatibility paradigm shifts. Biotechnology and bioengineering. 2012;109:1898–1911. doi: 10.1002/bit.24559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kim TH, Mars WM, Stolz DB, Michalopoulos GK. Expression and activation of pro-MMP-2 and pro-MMP-9 during rat liver regeneration. Hepatology. 2000;31:75–82. doi: 10.1002/hep.510310114. [DOI] [PubMed] [Google Scholar]
- 194.Knittel T, Mehde M, Grundmann A, Saile B, Scharf JG, Ramadori G. Expression of matrix metalloproteinases and their inhibitors during hepatic tissue repair in the rat. Histochem Cell Biol. 2000;113:443–453. doi: 10.1007/s004180000150. [DOI] [PubMed] [Google Scholar]
- 195.Liu LY, GR, Nishikawa T, Yamamoto T, Basma H, Ito R, Nagaya M, Dutta-Moscato J, Stolz DB, Duan F, Kaestner KH, Vodovotz Y, Soto-Gutierrez A, Fox IJ. The microenvirontment in hepatocyte regeneration and function in rats with advance cirrhosis. Hepatology. 2011 doi: 10.1002/hep.24815. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kloxin AM, Kloxin CJ, Bowman CN, Anseth KS. Mechanical properties of cellularly responsive hydrogels and their experimental determination. Advanced Materials. 2010;22:3484–3494. doi: 10.1002/adma.200904179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Arany PR, Mooney DJ. At the edge of translation - materials to program cells for directed differentiation. Oral diseases. 2011;17:241–251. doi: 10.1111/j.1601-0825.2010.01735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kobayashi N, Fujiwara T, Westerman KA, Inoue Y, Sakaguchi M, Noguchi H, Miyazaki M, Cai J, Tanaka N, Fox IJ, Leboulch P. Prevention of acute liver failure in rats with reversibly immortalized human hepatocytes. Science. 2000;287:1258–1262. doi: 10.1126/science.287.5456.1258. [DOI] [PubMed] [Google Scholar]
- 199.Lee SW, Wang X, Chowdhury NR, Roy-Chowdhury J. Hepatocyte transplantation: state of the art and strategies for overcoming existing hurdles. Ann Hepatol. 2004;3:48–53. [PubMed] [Google Scholar]
- 200.Myren JVT. The effect of carbon tetrachloride on liver and liver transplants in mice. Acta Pathol Microbiol Scand (Suppl) 1952;93:173–179. [PubMed] [Google Scholar]
- 201.Kawai Y, Price JB, Hardy MA. Reversal of liver failure in rats by ultraviolet-irradiated hepatocyte transplantation. Transplantation proceedings. 1987;19:989–991. [PubMed] [Google Scholar]
- 202.Sutherland DE, Numata M, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation in acute liver failure. Surgery. 1977;82:124–132. [PubMed] [Google Scholar]
- 203.Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplantation proceedings. 1979;11:578–584. [PubMed] [Google Scholar]
- 204.Makowka L, Falk RE, Rotstein LE, Falk JA, Nossal N, Langer B, Blendis LM, Phillips MJ. Reversal of experimental acute hepatic failure in the rat. Journal of Surgical Research. 1980;29:479–487. doi: 10.1016/0022-4804(80)90016-5. [DOI] [PubMed] [Google Scholar]
- 205.Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology. 1988;8:1006–1009. doi: 10.1002/hep.1840080505. [DOI] [PubMed] [Google Scholar]
- 206.Bernardi M, Tacconi C, Somaroli M, Gasbarrini G, Mazziotti A. Hyperammonemic, ammonia-independent coma in experimental acute liver failure induced in the pig. Gastroenterology. 1981;81:191–192. [PubMed] [Google Scholar]
- 207.Sommer BG, Sutherland DE, Simmons RL, Najarian JS. Hepatocellular transplantation for experimental ischemic acute liver failure in dogs. Journal of Surgical Research. 1980;29:319–325. doi: 10.1016/0022-4804(80)90064-5. [DOI] [PubMed] [Google Scholar]
- 208.Kobayashi N, Ito M, Nakamura J, Cai J, Gao C, Hammel JM, Fox IJ. Hepatocyte transplantation in rats with decompensated cirrhosis. Hepatology. 2000;31:851–857. doi: 10.1053/he.2000.5636. [DOI] [PubMed] [Google Scholar]
- 209.Matas AJ, Sutherland DE, Steffes MW, Mauer SM, Sowe A, Simmons RL, Najarian JS. Hepatocellular transplantation for metabolic deficiencies: decrease of plasms bilirubin in Gunn rats. Science. 1976;192:892–894. doi: 10.1126/science.818706. [DOI] [PubMed] [Google Scholar]
- 210.Fujita M, Furukawa H, Hattori M, Todo S, Ishida Y, Nagashima K. Sequential observation of liver cell regeneration after massive hepatic necrosis in auxiliary partial orthotopic liver transplantation. Mod Pathol. 2000;13:152–157. doi: 10.1038/modpathol.3880029. [DOI] [PubMed] [Google Scholar]
- 211.Quaglia A, Portmann BC, Knisely AS, Srinivasan P, Muiesan P, Wendon J, Heneghan MA, O’Grady JG, Samyn M, Hadzic D, Dhawan A, Mieli-Vergani G, Heaton N, Rela M. Auxiliary transplantation for acute liver failure: Histopathological study of native liver regeneration. Liver Transplantation. 2008;14:1437–1448. doi: 10.1002/lt.21568. [DOI] [PubMed] [Google Scholar]
- 212.Katoonizadeh A, Nevens F, Verslype C, Pirenne J, Roskams T. Liver regeneration in acute severe liver impairment: a clinicopathological correlation study. Liver International. 2006;26:1225–1233. doi: 10.1111/j.1478-3231.2006.01377.x. [DOI] [PubMed] [Google Scholar]
- 213.Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–436. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 214.Gagandeep S, Rajvanshi P, Sokhi RP, Slehria S, Palestro CJ, Bhargava KK, Gupta S. Transplanted hepatocytes engraft, survive, and proliferate in the liver of rats with carbon tetrachloride-induced cirrhosis. J Pathol. 2000;191:78–85. doi: 10.1002/(SICI)1096-9896(200005)191:1<78::AID-PATH587>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 215.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. Journal of hepatology. 2005;43:1045–1054. doi: 10.1016/j.jhep.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 217.Quinn PS, Higginson J. Reversible and Irreversible Changes in Experimental Cirrhosis. American Journal of Pathology. 1965;47:353–369. [PMC free article] [PubMed] [Google Scholar]
- 218.Iredale JP. Hepatic stellate cell behavior during resolution of liver injury. Seminars in liver disease. 2001;21:427–436. doi: 10.1055/s-2001-17557. [DOI] [PubMed] [Google Scholar]
- 219.Dufour JF, DeLellis R, Kaplan MM. Reversibility of hepatic fibrosis in autoimmune hepatitis. Ann Intern Med. 1997;127:981–985. doi: 10.7326/0003-4819-127-11-199712010-00006. [DOI] [PubMed] [Google Scholar]
- 220.Cai J, Ito M, Nagata H, Westerman KA, Lafleur D, Chowdhury JR, Leboulch P, Fox IJ. Treatment of liver failure in rats with end-stage cirrhosis by transplantation of immortalized hepatocytes. Hepatology. 2002;36:386–394. doi: 10.1053/jhep.2002.34614. [DOI] [PubMed] [Google Scholar]
- 221.Nagata H, Ito M, Cai J, Edge AS, Platt JL, Fox IJ. Treatment of cirrhosis and liver failure in rats by hepatocyte xenotransplantation. Gastroenterology. 2003;124:422–431. doi: 10.1053/gast.2003.50065. [DOI] [PubMed] [Google Scholar]
- 222.Ribeiro J, Nordlinger B, Ballet F, Cynober L, Coudray-Lucas C, Baudrimont M, Legendre C, Delelo R, Panis Y. Intrasplenic hepatocellular transplantation corrects hepatic encephalopathy in portacaval-shunted rats. Hepatology. 1992;15:12–18. doi: 10.1002/hep.1840150104. [DOI] [PubMed] [Google Scholar]
- 223.Schumacher IK, Okamoto T, Kim BH, Chowdhury NR, Chowdhury JR, Fox IJ. Transplantation of conditionally immortalized hepatocytes to treat hepatic encephalopathy. Hepatology. 1996;24:337–343. doi: 10.1002/hep.510240209. [DOI] [PubMed] [Google Scholar]
- 224.Nagata H, Ito M, Shirota C, Edge A, McCowan TC, Fox IJ. Route of hepatocyte delivery affects hepatocyte engraftment in the spleen. Transplantation. 2003;76:732–734. doi: 10.1097/01.TP.0000081560.16039.67. [DOI] [PubMed] [Google Scholar]
- 225.Soto-Gutierrez A, Zhang L, Medberry C, Fukumitsu K, Faulk D, Jiang H, Reing J, Gramignoli R, Komori J, Ross M, Nagaya M, Lagasse E, Stolz D, Strom SC, Fox IJ, Badylak SF. A whole-organ regenerative medicine approach for liver replacement. Tissue Eng Part C Methods. 2011;17:677–686. doi: 10.1089/ten.tec.2010.0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Uygun BE, Soto-Gutierrez A, Yagi H, Izamis ML, Guzzardi MA, Shulman C, Milwid J, Kobayashi N, Tilles A, Berthiaume F, Hertl M, Nahmias Y, Yarmush ML, Uygun K. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat Med. 2010;16:814–820. doi: 10.1038/nm.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Soto-Gutierrez A, Kobayashi N, Rivas-Carrillo JD, Navarro-Alvarez N, Zhao D, Okitsu T, Noguchi H, Basma H, Tabata Y, Chen Y, Tanaka K, Narushima M, Miki A, Ueda T, Jun HS, Yoon JW, Lebkowski J, Tanaka N, Fox IJ. Reversal of mouse hepatic failure using an implanted liver-assist device containing ES cell-derived hepatocytes. Nature Biotechnology. 2006;24:1412–1419. doi: 10.1038/nbt1257. [DOI] [PubMed] [Google Scholar]
- 228.Baptista PM, Siddiqui MM, Lozier G, Rodriguez SR, Atala A, Soker S. The use of whole organ decellularization for the generation of a vascularized liver organoid. Hepatology. 2011;53:604–617. doi: 10.1002/hep.24067. [DOI] [PubMed] [Google Scholar]
- 229.Barakat O, Abbasi S, Rodriguez G, Rios J, Wood RP, Ozaki C, Holley LS, Gauthier PK. Use of decellularized porcine liver for engineering humanized liver organ. J Surg Res. 2012;173:e11–25. doi: 10.1016/j.jss.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 230.Caplan AI, Correa D. The MSC: an injury drugstore. Cell stem cell. 2011;9:11–15. doi: 10.1016/j.stem.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ren G, Chen X, Dong F, Li W, Ren X, Zhang Y, Shi Y. Concise review: mesenchymal stem cells and translational medicine: emerging issues. Stem cells translational medicine. 2012;1:51–58. doi: 10.5966/sctm.2011-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Overturf K, Al-Dhalimy M, Tanguay R, Brantly M, Ou CN, Finegold M, Grompe M. Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1996;12:266–273. doi: 10.1038/ng0396-266. [DOI] [PubMed] [Google Scholar]
- 233.Malhi H, Joseph B, Schilsky ML, Gupta S. Development of cell therapy strategies to overcome copper toxicity in the LEC rat model of Wilson disease. Regen Med. 2008;3:165–173. doi: 10.2217/17460751.3.2.165. [DOI] [PubMed] [Google Scholar]
- 234.Patejunas G, Bradley A, Beaudet AL, O’Brien WE. Generation of a mouse model for citrullinemia by targeted disruption of the argininosuccinate synthetase gene. Somatic cell and molecular genetics. 1994;20:55–60. doi: 10.1007/BF02257486. [DOI] [PubMed] [Google Scholar]
- 235.Grompe M, Jones SN, Loulseged H, Caskey CT. Retroviral-mediated gene transfer of human ornithine transcarbamylase into primary hepatocytes of spf and spf-ash mice. Hum Gene Ther. 1992;3:35–44. doi: 10.1089/hum.1992.3.1-35. [DOI] [PubMed] [Google Scholar]
- 236.Ohtake A, Takayanagi M, Yamamoto S, Kakinuma H, Nakajima H, Tatibana M, Mori M. Molecular basis of ornithine transcarbamylase deficiency in spf and spf-ash mutant mice. J Inherit Metab Dis. 1986;9:289–291. doi: 10.1007/BF01799667. [DOI] [PubMed] [Google Scholar]
- 237.Iyer RK, Yoo PK, Kern RM, Rozengurt N, Tsoa R, O’Brien WE, Yu H, Grody WW, Cederbaum SD. Mouse model for human arginase deficiency. Molecular and cellular biology. 2002;22:4491–4498. doi: 10.1128/MCB.22.13.4491-4498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Jiang J, Salido EC, Guha C, Wang X, Moitra R, Liu L, Roy-Chowdhury J, Roy-Chowdhury N. Correction of hyperoxaluria by liver repopulation with hepatocytes in a mouse model of primary hyperoxaluria type-1. Transplantation. 2008;85:1253–1260. doi: 10.1097/TP.0b013e31816de49e. [DOI] [PubMed] [Google Scholar]
- 239.Wiederkehr JC, Kondos GT, Pollak R. Hepatocyte transplantation for the low-density lipoprotein receptor-deficient state. A study in the Watanabe rabbit. Transplantation. 1990;50:466–471. doi: 10.1097/00007890-199009000-00021. [DOI] [PubMed] [Google Scholar]
- 240.Kocken JM, Borel Rinkes IH, Bijma AM, de Roos WK, Bouwman E, Terpstra OT, Sinaasappel M. Correction of an inborn error of metabolism by intraportal hepatocyte transplantation in a dog model. Transplantation. 1996;62:358–364. doi: 10.1097/00007890-199608150-00010. [DOI] [PubMed] [Google Scholar]
- 241.Ding J, Yannam GR, Roy-Chowdhury N, Hidvegi T, Basma H, Rennard SI, Wong RJ, Avsar Y, Guha C, Perlmutter DH, Fox IJ, Roy-Chowdhury J. Spontaneous hepatic repopulation in transgenic mice expressing mutant human alpha1-antitrypsin by wild-type donor hepatocytes. J Clin Invest. 2011;121:1930–1934. doi: 10.1172/JCI45260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, Strom S, Kay MA, Finegold M, Grompe M. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nature biotechnology. 2007;25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Sandgren EP, Palmiter RD, Heckel JL, Daugherty CC, Brinster RL, Degen JL. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell. 1991;66:245–256. doi: 10.1016/0092-8674(91)90615-6. [DOI] [PubMed] [Google Scholar]
- 244.Rhim JA, Sandgren EP, Palmiter RD, Brinster RL. Complete reconstitution of mouse liver with xenogeneic hepatocytes. Proc Natl Acad Sci U S A. 1995;92:4942–4946. doi: 10.1073/pnas.92.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Rhim JA, Sandgren EP, Degen JL, Palmiter RD, Brinster RL. Replacement of diseased mouse liver by hepatic cell transplantation. Science. 1994;263:1149–1152. doi: 10.1126/science.8108734. [DOI] [PubMed] [Google Scholar]
- 246.Hasegawa M, Kawai K, Mitsui T, Taniguchi K, Monnai M, Wakui M, Ito M, Suematsu M, Peltz G, Nakamura M, Suemizu H. The reconstituted ‘humanized liver’ in TK-NOG mice is mature and functional. Biochemical and biophysical research communications. 2011;405:405–410. doi: 10.1016/j.bbrc.2011.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Laconi S, Curreli F, Diana S, Pasciu D, De Filippo G, Sarma DS, Pani P, Laconi E. Liver regeneration in response to partial hepatectomy in rats treated with retrorsine: a kinetic study. Journal of hepatology. 1999;31:1069–1074. doi: 10.1016/s0168-8278(99)80320-1. [DOI] [PubMed] [Google Scholar]
- 248.Yamanouchi K, Zhou H, Roy-Chowdhury N, Macaluso F, Liu L, Yamamoto T, Yannam GR, Enke C, Solberg TD, Adelson AB, Platt JL, Fox IJ, Roy-Chowdhury J, Guha C. Hepatic irradiation augments engraftment of donor cells following hepatocyte transplantation. Hepatology. 2009;49:258–267. doi: 10.1002/hep.22573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Guha C, Parashar B, Deb NJ, Garg M, Gorla GR, Singh A, Roy-Chowdhury N, Vikram B, Roy-Chowdhury J. Normal hepatocytes correct serum bilirubin after repopulation of Gunn rat liver subjected to irradiation/partial resection. Hepatology. 2002;36:354–362. doi: 10.1053/jhep.2002.34516. [DOI] [PubMed] [Google Scholar]
- 250.Kwong GA, von Maltzahn G, Murugappan G, Abudayyeh O, Mo S, Papayannopoulos IA, Sverdlov DY, Liu SB, Warren AD, Popov Y, Schuppan D, Bhatia SN. Mass-encoded synthetic biomarkers for multiplexed urinary monitoring of disease. Nature biotechnology. 2013;31:63–70. doi: 10.1038/nbt.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Grompe M, Strom S. Mice with human livers. Gastroenterology. 2013;145:1209–1214. doi: 10.1053/j.gastro.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 252.Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, Miranda E, Ordonez A, Hannan NR, Rouhani FJ, Darche S, Alexander G, Marciniak SJ, Fusaki N, Hasegawa M, Holmes MC, Di Santo JP, Lomas DA, Bradley A, Vallier L. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ding J, Roy-Chowdhury N, Wang X, Neufeld DS, Hansel MC, Strom SC, Fox IJ, Guha C, Roy-Chowdhury J. SCID/Piz mice: a novel animal model for evaluating engraftment and proliferation of human stem cell-derived hepatocytes. Hepatology. 2011;54:957A. [Google Scholar]
- 254.Roy-Chowdhury N, Chen Y, Atienza K, Chang C, Wang X, Guha C, Fox IJ, Bouhassira EE, Roy-Chowdhury J. Amelioration of hyperbilirubinemia in Gunn rats after transplantation of human hepatocytes derived from induced pluripotent stem cells. Hepatology. 2010;52:426A. [Google Scholar]
- 255.Atienza KV, Ding J, Chang C-J, Bouhassira EE, Chen Y, Wang X, Avsar Y, Laibin L, Guha C, Fox IJ, Salido E, Roy-Chowdhury J, Roy-Chowdhury N. Reduction of urinary oxalate excretion after transplantation of human induced pluripotent stem cell-derived hepatocytes in a mouse model of primary hyperoxaluria-1. Hepatology. 2011;54:412A. [Google Scholar]
- 256.Bilir BM, Guinette D, Karrer F, Kumpe DA, Krysl J, Stephens J, McGavran L, Ostrowska A, Durham J. Hepatocyte transplantation in acute liver failure. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2000;6:32–40. doi: 10.1002/lt.500060113. [DOI] [PubMed] [Google Scholar]
- 257.Strom SC, Chowdhury JR, Fox IJ. Hepatocyte transplantation for the treatment of human disease. Semin Liver Dis. 1999;19:39–48. doi: 10.1055/s-2007-1007096. [DOI] [PubMed] [Google Scholar]
- 258.Gupta S, Chowdhury JR. Therapeutic potential of hepatocyte transplantation. Semin Cell Dev Biol. 2002;13:439–446. doi: 10.1016/s1084952102001325. [DOI] [PubMed] [Google Scholar]
- 259.Maguire PJ, Stevens C, Humes HD, Shander A, Halpern NA, Pastores SM. Bioartificial organ support for hepatic, renal, and hematologic failure. Critical care clinics. 2000;16:681–694. doi: 10.1016/s0749-0704(05)70140-8. [DOI] [PubMed] [Google Scholar]
- 260.Gerlach JC, Encke J, Hole O, Müller C, Ryan CJ, Neuhaus P. Bioreactor for a larger scale hepatocyte in vitro perfusion. Transplantation. 1994;58:984–988. doi: 10.1097/00007890-199411150-00002. [DOI] [PubMed] [Google Scholar]
- 261.Allen JW, Hassanein T, Bhatia SN. Advances in bioartificial liver devices. Hepatology. 2001;34:447–455. doi: 10.1053/jhep.2001.26753. [DOI] [PubMed] [Google Scholar]
- 262.Olson H, Betton G, Robinson D, Thomas K, Monro A, Kolaja G, Lilly P, Sanders J, Sipes G, Bracken W, Dorato M, Van Deun K, Smith P, Berger B, Heller A. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regulatory toxicology and pharmacology: RTP. 2000;32:56–67. doi: 10.1006/rtph.2000.1399. [DOI] [PubMed] [Google Scholar]
- 263.Shih H, Pickwell GV, Guenette DK, Bilir B, Quattrochi LC. Species differences in hepatocyte induction of CYP1A1 and CYP1A2 by omeprazole. Human & experimental toxicology. 1999;18:95–105. doi: 10.1177/096032719901800206. [DOI] [PubMed] [Google Scholar]
- 264.Lee PJ, Hung PJ, Lee LP. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnology and bioengineering. 2007;97:1340–1346. doi: 10.1002/bit.21360. [DOI] [PubMed] [Google Scholar]
- 265.Chao P, Maguire T, Novik E, Cheng KC, Yarmush ML. Evaluation of a microfluidic based cell culture platform with primary human hepatocytes for the prediction of hepatic clearance in human. Biochemical pharmacology. 2009;78:625–632. doi: 10.1016/j.bcp.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Novik E, Maguire TJ, Chao P, Cheng KC, Yarmush ML. A microfluidic hepatic coculture platform for cell-based drug metabolism studies. Biochemical pharmacology. 2010;79:1036–1044. doi: 10.1016/j.bcp.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Zhang S, Tong W, Zheng B, Susanto TA, Xia L, Zhang C, Ananthanarayanan A, Tuo X, Sakban RB, Jia R, Iliescu C, Chai KH, McMillian M, Shen S, Leo H, Yu H. A robust high-throughput sandwich cell-based drug screening platform. Biomaterials. 2011;32:1229–1241. doi: 10.1016/j.biomaterials.2010.09.064. [DOI] [PubMed] [Google Scholar]
- 268.Eschbach E, Chatterjee SS, Noldner M, Gottwald E, Dertinger H, Weibezahn KF, Knedlitschek G. Microstructured scaffolds for liver tissue cultures of high cell density: morphological and biochemical characterization of tissue aggregates. Journal of cellular biochemistry. 2005;95:243–255. doi: 10.1002/jcb.20360. [DOI] [PubMed] [Google Scholar]
- 269.Knedlitschek G, Schneider F, Gottwald E, Schaller T, Eschbach E, Weibezahn KF. A tissue-like culture system using microstructures: influence of extracellular matrix material on cell adhesion and aggregation. J Biomech Eng. 1999;121:35–39. doi: 10.1115/1.2798040. [DOI] [PubMed] [Google Scholar]
- 270.Sivaraman A, Leach JK, Townsend S, Iida T, Hogan BJ, Stolz DB, Fry R, Samson LD, Tannenbaum SR, Griffith LG. A microscale in vitro physiological model of the liver: predictive screens for drug metabolism and enzyme induction. Curr Drug Metab. 2005;6:569–591. doi: 10.2174/138920005774832632. [DOI] [PubMed] [Google Scholar]
- 271.Miki T, Ring A, Gerlach J. Hepatic differentiation of human embryonic stem cells is promoted by three-dimensional dynamic perfusion culture conditions. Tissue Eng Part C Methods. 2011;17:557–568. doi: 10.1089/ten.TEC.2010.0437. [DOI] [PubMed] [Google Scholar]
- 272.Sivertsson L, Synnergren J, Jensen J, Bjorquist P, Ingelman-Sundberg M. Hepatic differentiation and maturation of human embryonic stem cells cultured in a perfused three-dimensional bioreactor. Stem cells and development. 2013;22:581–594. doi: 10.1089/scd.2012.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Gerlach JC, Schnoy N, Encke J, Smith MD, Muller C, Neuhaus P. Improved hepatocyte in vitro maintenance in a culture model with woven multicompartment capillary systems: electron microscopy studies. Hepatology. 1995;22:546–552. [PubMed] [Google Scholar]
- 274.Domansky K, Inman W, Serdy J, Dash A, Lim MH, Griffith LG. Perfused multiwell plate for 3D liver tissue engineering. Lab on a chip. 2010;10:51–58. doi: 10.1039/b913221j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Sung JH, Kam C, Shuler ML. A microfluidic device for a pharmacokinetic-pharmacodynamic (PK-PD) model on a chip. Lab on a chip. 2010;10:446–455. doi: 10.1039/b917763a. [DOI] [PubMed] [Google Scholar]
- 276.Vunjak-Novakovic G, Bhatia S, Chen C, Hirschi K. HeLiVa platform: integrated heart-liver-vascular systems for drug testing in human health and disease. Stem cell research & therapy. 2013;4(Suppl 1):S8. doi: 10.1186/scrt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends in cell biology. 2011;21:745–754. doi: 10.1016/j.tcb.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Gebhardt R, Hengstler JG, Muller D, Glockner R, Buenning P, Laube B, Schmelzer E, Ullrich M, Utesch D, Hewitt N, Ringel M, Hilz BR, Bader A, Langsch A, Koose T, Burger HJ, Maas J, Oesch F. New hepatocyte in vitro systems for drug metabolism: metabolic capacity and recommendations for application in basic research and drug development, standard operation procedures. Drug metabolism reviews. 2003;35:145–213. doi: 10.1081/dmr-120023684. [DOI] [PubMed] [Google Scholar]
- 279.Roy P, Baskaran H, Tilles AW, Yarmush ML, Toner M. Analysis of oxygen transport to hepatocytes in a flat-plate microchannel bioreactor. Ann Biomed Eng. 2001;29:947–955. doi: 10.1114/1.1415524. [DOI] [PubMed] [Google Scholar]
- 280.Allen JW, Khetani SR, Bhatia SN. In vitro zonation and toxicity in a hepatocyte bioreactor. Toxicological sciences: an official journal of the Society of Toxicology. 2005;84:110–119. doi: 10.1093/toxsci/kfi052. [DOI] [PubMed] [Google Scholar]
- 281.Pritchard JF, Jurima-Romet M, Reimer ML, Mortimer E, Rolfe B, Cayen MN. Making better drugs: Decision gates in non-clinical drug development. Nature reviews Drug discovery. 2003;2:542–553. doi: 10.1038/nrd1131. [DOI] [PubMed] [Google Scholar]
- 282.Khetani SR, Bhatia SN. Microscale culture of human liver cells for drug development. Nature biotechnology. 2008;26:120–126. doi: 10.1038/nbt1361. [DOI] [PubMed] [Google Scholar]
- 283.Hewitt NJ, Lechon MJ, Houston JB, Hallifax D, Brown HS, Maurel P, Kenna JG, Gustavsson L, Lohmann C, Skonberg C, Guillouzo A, Tuschl G, Li AP, LeCluyse E, Groothuis GM, Hengstler JG. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug metabolism reviews. 2007;39:159–234. doi: 10.1080/03602530601093489. [DOI] [PubMed] [Google Scholar]
- 284.Wang WW, Khetani SR, Krzyzewski S, Duignan DB, Obach RS. Assessment of a micropatterned hepatocyte coculture system to generate major human excretory and circulating drug metabolites. Drug metabolism and disposition: the biological fate of chemicals. 2010;38:1900–1905. doi: 10.1124/dmd.110.034876. [DOI] [PubMed] [Google Scholar]
- 285.Fukuda J, Sakai Y, Nakazawa K. Novel hepatocyte culture system developed using microfabrication and collagen/polyethylene glycol microcontact printing. Biomaterials. 2006;27:1061–1070. doi: 10.1016/j.biomaterials.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 286.Jones CN, Tuleuova N, Lee JY, Ramanculov E, Reddi AH, Zern MA, Revzin A. Cultivating hepatocytes on printed arrays of HGF and BMP7 to characterize protective effects of these growth factors during in vitro alcohol injury. Biomaterials. 2010;31:5936–5944. doi: 10.1016/j.biomaterials.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Lee MY, Kumar RA, Sukumaran SM, Hogg MG, Clark DS, Dordick JS. Three-dimensional cellular microarray for high-throughput toxicology assays. Proc Natl Acad Sci U S A. 2008;105:59–63. doi: 10.1073/pnas.0708756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.King KR, Wang S, Irimia D, Jayaraman A, Toner M, Yarmush ML. A high-throughput microfluidic real-time gene expression living cell array. Lab on a chip. 2007;7:77–85. doi: 10.1039/b612516f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zhang C, Chia SM, Ong SM, Zhang S, Toh YC, van Noort D, Yu H. The controlled presentation of TGF-beta1 to hepatocytes in a 3D-microfluidic cell culture system. Biomaterials. 2009;30:3847–3853. doi: 10.1016/j.biomaterials.2009.03.052. [DOI] [PubMed] [Google Scholar]
- 290.Hui EE, Bhatia SN. Micromechanical control of cell-cell interactions. Proc Natl Acad Sci U S A. 2007;104:5722–5726. doi: 10.1073/pnas.0608660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Lohmann V, Korner F, Koch JO, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 292.Kato T, Furusaka A, Miyamoto M, Date T, Yasui K, Hiramoto J, Nagayama K, Tanaka T, Wakita T. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. Journal of Medical Virology. 2001;64:334–339. doi: 10.1002/jmv.1055. [DOI] [PubMed] [Google Scholar]
- 293.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 294.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao ZJ, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Zhong J, Gastaminza P, Cheng GF, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Buck M. Direct infection and replication of naturally occurring hepatitis C virus genotypes 1, 2, 3 and 4 in normal human hepatocyte cultures. PloS one. 2008;3:e2660. doi: 10.1371/journal.pone.0002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Molina S, Castet V, Pichard-Garcia L, Wychowski C, Meurs E, Pascussi JM, Sureau C, Fabre JM, SaCunha A, Larrey D, Dubuisson J, Coste J, McKeating J, Maurel P, Fournier-Wirth C. Serum-derived hepatitis C virus infection of primary human hepatocytes is tetraspanin CD81 dependent. Journal of Virology. 2008;82:569–574. doi: 10.1128/JVI.01443-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Ploss A, Khetani SR, Jones CT, Syder AJ, Trehan K, Gaysinskaya VA, Mu K, Ritola K, Rice CM, Bhatia SN. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc Natl Acad Sci U S A. 2010;107:3141–3145. doi: 10.1073/pnas.0915130107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Lindenbach BD, Rice CM. The ins and outs of hepatitis C virus entry and assembly. Nature reviews Microbiology. 2013;11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Kwong AD, Kauffman RS, Hurter P, Mueller P. Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nature biotechnology. 2011;29:993–1003. doi: 10.1038/nbt.2020. [DOI] [PubMed] [Google Scholar]
- 301.Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Fried MW, Buti M, Dore GJ, Flisiak R, Ferenci P, Jacobson I, Marcellin P, Manns M, Nikitin I, Poordad F, Sherman M, Zeuzem S, Scott J, Gilles L, Lenz O, Peeters M, Sekar V, De Smedt G, Beumont-Mauviel M. Once-daily simeprevir (TMC435) with pegylated interferon and ribavirin in treatment-naive genotype 1 hepatitis C: the randomized PILLAR study. Hepatology. 2013;58:1918–1929. doi: 10.1002/hep.26641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, Jacobson IM, Kowdley KV, Nyberg L, Subramanian GM, Hyland RH, Arterburn S, Jiang D, McNally J, Brainard D, Symonds WT, McHutchison JG, Sheikh AM, Younossi Z, Gane EJ. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368:1878–1887. doi: 10.1056/NEJMoa1214853. [DOI] [PubMed] [Google Scholar]
- 304.Jacobson IM, Gordon SC, Kowdley KV, Yoshida EM, Rodriguez-Torres M, Sulkowski MS, Shiffman ML, Lawitz E, Everson G, Bennett M, Schiff E, Al-Assi MT, Subramanian GM, An D, Lin M, McNally J, Brainard D, Symonds WT, McHutchison JG, Patel K, Feld J, Pianko S, Nelson DR. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med. 2013;368:1867–1877. doi: 10.1056/NEJMoa1214854. [DOI] [PubMed] [Google Scholar]
- 305.Schwartz RE, Trehan K, Andrus L, Sheahan TP, Ploss A, Duncan SA, Rice CM, Bhatia SN. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1121400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Wu X, Robotham JM, Lee E, Dalton S, Kneteman NM, Gilbert DM, Tang H. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS pathogens. 2012;8:e1002617. doi: 10.1371/journal.ppat.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Yalaoui S, Huby T, Franetich JF, Gego A, Rametti A, Moreau M, Collet X, Siau A, van Gemert GJ, Sauerwein RW, Luty AJF, Vaillant JC, Hannoun L, Chapman J, Mazier D, Froissard P. Scavenger receptor BI boosts hepatocyte permissiveness to Plasmodium infection. Cell host & microbe. 2008;4:283–292. doi: 10.1016/j.chom.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 308.van Schaijk BCL, Janse CJ, van Gemert G-J, van Dijk MR, Gego A, Franetich J-F, van de Vegte-Bolmer M, Yalaoui S, Silvie O, Hoffman SL, Waters AP, Mazier D, Sauerwein RW, Khan SM. Gene disruption of Plasmodium falciparum p52 results in attenuation of malaria liver stage development in cultured primary human hepatocytes. PloS one. 2008;3:e3549. doi: 10.1371/journal.pone.0003549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.March S, Ng S, Velmurugan S, Galstian A, Shan J, Logan David J, Carpenter Anne E, Thomas D, Kim L, Sim B, Mota Maria M, Hoffman Stephen L, Bhatia Sangeeta N. A Microscale Human Liver Platform that Supports the Hepatic Stages of Plasmodium falciparum and vivax. Cell host & microbe. 2013;14:104–115. doi: 10.1016/j.chom.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Hickey RD, Lillegard JB, Fisher JE, McKenzie TJ, Hofherr SE, Finegold MJ, Nyberg SL, Grompe M. Efficient production of Fah-null heterozygote pigs by chimeric adeno-associated virus-mediated gene knockout and somatic cell nuclear transfer. Hepatology. 2011;54:1351–1359. doi: 10.1002/hep.24490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Schuldiner M, Itskovitz-Eldor J, Benvenisty N. Selective ablation of human embryonic stem cells expressing a “suicide” gene. Stem cells. 2003;21:257–265. doi: 10.1634/stemcells.21-3-257. [DOI] [PubMed] [Google Scholar]
- 312.Kiuru M, Boyer JL, O’Connor TP, Crystal RG. Genetic control of wayward pluripotent stem cells and their progeny after transplantation. Cell stem cell. 2009;4:289–300. doi: 10.1016/j.stem.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]