

Abstract
Chronic, low-grade inflammation in osteoarthritis (OA) contributes to symptoms and disease progression. Effective disease-modifying medical OA therapies are lacking, but better understanding inflammatory pathophysiology in OA could lead to transformative therapy. Networks of diverse innate inflammatory danger signals, including complement and alarmins, are activated in OA. Through inflammatory mediators, biomechanical cartilage injury and oxidative stress compromise chondrocyte viability and reprogram viable chondrocytes to hypertrophic differentiation and proinflammatory, and procatabolic responses in mechanistically similar ways. Integral to this reprogramming are certain ‘switching’ pathways in transcriptional signals, other than the well-characterized effects of NFκB and mitogen-activated protein kinase signalling. HIF-2α transcriptional signalling and ZIP8-mediated Zn2+ uptake, with downstream MTF1 transcriptional signalling, have been implicated in chondrocyte reprogramming, but further validation is required. Permissive factors in procatabolic reprogramming of OA chondrocytes by inflammatory mediators also have come to light, including impaired bioenergetics, such as altered mitochondrial function and decreased activity of the bioenergy sensors AMPK and SIRT1. These factors interact with molecular inflammatory responses and proteostasis mechanisms that normally resolve cell stress, such as the unfolded protein response and autophagy. Bioenergy-sensing by AMPK and SIRT1 modulates proteostasis and provides ‘stop signals’ for oxidative stress, inflammatory, and matrix catabolic processes in chondrocytes. The complexity of molecular inflammatory processes in OA, and the involvement of multiple inflammatory mediators in tissue repair responses, raises daunting questions about how to therapeutically target inflammatory processes and macroscopic inflammation in OA. Bioenergy sensing might provide a pragmatic ‘entry point’ for novel strategies to limit OA progression.
Introduction
Osteoarthritis (OA) is both the most common cause of arthritis, and a major public health problem. The prevalence of OA is growing in developed countries owing to ageing of the population and other, acquired factors including biomechanical injury and obesity. Currently, we lack effective disease-modifying medical therapy for OA. To help discuss translation in this area, this review applies a vocabulary to a variety of processes that regulate OA progression (Box 1). The central rationale of this Review is that chronic, low-grade inflammatory processes in OA not only promote disease symptomatology, but also accelerate disease progression. Better understanding of the inflammatory pathophysiology of OA could lead to novel approaches to slow destructive changes in the joint and prevent permanent functional impairment. Therefore, this Review moves from discussing networks of innate inflammatory danger signals to how they are related to permissive and ‘stop signals’ for chondrocyte procatabolic reprogramming. We discuss linkages in impairment of certain chondrocyte proteostasis responses and of chondrocyte bioenergetics, such as altered mitochondrial function and decreased activity of the bioenergy sensors AMPK and SIRT1, and how these processes regulate both inflammatory and matrix catabolic processes in chondrocytes in OA. Last, we evaluate and compare the relevance for potential clinical translation of the inflammatory and bioenergetics processes discussed.
Box 1. Definitions and qualifying statements for major concepts discussed in this Review.
Inflammation: Terminology used in this review to denote overt, macroscopic inflammation in OA, mainly but not solely, as synovitis and synovial effusion.
Inflammatory: Describes changes exerted at the molecular and cellular level, employing mediators, such as cytokines and DAMPs, that promote cell stress and jeopardize joint tissue homeostasis.
Proteostasis: Protein homeostasis carried out by multiple interconnected pathways and systems, which include the unfolded protein response, autophagy, ubiquitin-proteasome system, and lysosomes. Proteostasis regulates protein fate, from biogenesis to folding, subcellular trafficking, and degradation in cells.
Oxidative stress: A promoter of inflammatory and matrix catabolic responses, including biomechanical stress-induced inflammatory and matrix catabolic responses in mechanotransduction. Also, is a regulator of cell stress responses including proteostasis.
Bioenergetics and bioenergy sensors: We focus here on altered mitochondrial function and activity of the bioenergy sensors AMPK and SIRT1, which react to changes in nutrition, energy balance, cell stressors, and inflammatory processes.
Inflammatory manifestations in OA
Progressive articular cartilage degradation is central to OA, and is driven by well-understood mechanisms of cartilage matrix catabolic effects (including via metalloproteinases [MMPs], aggrecanases, and other enzymes) and anti-anabolic effects (via factors including increased nitric oxide generation) of chondrocytes (1–5). However, OA is a disease mediated by and affecting the entire ‘synovial joint organ’, which includes not only meniscal fibrocartilage and hyaline articular cartilage, but also subchondral bone and synovium (1–4). Changes in periarticular musculature, and in articular and periarticular tendons and ligaments, can induce substantial biomechanical stress, compounded by the loss of other joint homeostatic functions including lubricant production (6).
Increased attention is now being paid to the role in OA-associated inflammation of metabolic factors related to obesity, such as insulin resistance and the effects of adipokines (7,8). Modulation of OA inflammation by articular adipose tissue is exemplified by the infrapatellar fat pad of the knee, as addressed in detail elsewhere (8). We now recognize that certain mediators of OA (including MMPs, TNF, and Toll-like receptor [TLR] and p38 mitogen-activated protein kinase signalling) can promote neural mechanisms of chronic joint pain and, conversely, that pain itself, mediated in part by increased joint-fluid glutamate in OA (9), can modulate joint inflammation and cartilage degradation (10,11).
OA is a heterogeneous disorder (BOX 2), with inflammation and bone erosion playing more prominent roles in particular forms of disease, including a subset of small hand OA joint (12). Changes in subchondral bone, including transforming growth factor (TGF)-β-mediated angiogenesis, can be among the earliest pathologic changes in OA (13). Macroscopic synovitis clearly contributes to clinical signs and symptoms, becomes more prominent in the middle and late stages of OA as part of a proinflammatory interaction between joint tissues, and is linked to OA progression (1,2,3,14).
BOX 2. Synovium and subchondral bone in OA.
OA is a heterogeneous disorder, with inflammation and bone erosion playing more prominent roles in particular forms of disease, including a subset of small hand OA joint
Changes in subchondral bone, including TGFβ -mediated angiogenesis, may be among the earliest pathologic changes in OA
Synovitis and other manifestations of macroscopic inflammation become more prominent in middle and later stages of OA, and contributes to clinical signs and symptoms, and disease progression
Macroscopic synovitis has been detected in up to 75% of human knee OA by imaging, and synovial gene expression changes have marked regional variability in OA
Whether or not there is frank synovitis, dysregulated synovial function and eventual fibrotic changes in OA are likely important in OA pathophysiology
In cross-sectional imaging analyses, macroscopic synovitis has been detected in up to 75% of human knee OA (14–17), but such studies probably underestimate synovitis as macroscopic synovitis could precede ultimate synovial fibrosis in some circumstances. Gene expression patterns for mediators of not only inflammation, but also cartilage matrix catabolism, anabolism, and angiogenesis, are strikingly different in inflamed and non-inflamed areas of the synovium in individual patients with knee OA (18). Moreover, dysregulated synovial function without gross inflammation also is probably important in OA pathophysiology (19). A decrease in synovial production of lubricants such as hyaluronan and lubricin contributes to compromised protection of articular cartilage from wear in OA, and lubricin is a physiologic suppressor of synovial proliferation (6,20).
Molecular inflammatory processes in OA
‘Conventional’ inflammatory cytokines
As with many other organ diseases of ageing, basal inflammatory processes in OA are generally low-grade and ’molecular’, with many ‘usual suspects‘ implicated, including oxidative stress (1,21). So-called conventional inflammatory cytokines (such as TNF, IL-1β, IL-6, and multiple chemokines), released from various cell types, can promote OA progression by, for example, promoting synovitis and altering chondrocyte differentiation, function, and viability (22,23). Within this cytokine network, it is pertinent that autocrine activation of the NLRP3 inflammasome by chondrocytes and chondrocyte-derived IL-1β might not be functionally critical to OA progression (24). In this context, nitric oxide, which is generated in increased amounts by OA chondrocytes in situ, suppresses NLRP3 activation (25,26). A study of human and animal models of OA showed that the inflammasome constituents NLRP3, ASC, and caspase-1 were not robustly expressed in OA cartilage and, moreover, OA cartilage did not produce active IL-1β (24). In addition, cartilage explant catabolic responses to TNF, lipopolysaccharide, and biomechanical load were not inhibited by deficiency of either NLRP3 or IL-1 receptor (24). Hence, attention has turned to danger-associated molecular patterns (DAMPS) in OA inflammation pathophysiology.
Alarmins, DAMPs, and complement
Multiple alarmins (for example, high mobility group box protein 1 and the calgranulins S100A8 and S100A9), degradation products of cartilage extracellular matrix proteins (such as collagen and fibronectin) and of proteoglycans constituents (for example, low-molecular-weight hyaluronan), free fatty acids, and other DAMPs are increased in OA joints, essentially forming a network within a larger network of innate immunity (27–31)(Figure 1). These agents induce multiple ’conventional’ inflammatory cytokines, mediated in large part by signalling through various pattern recognition receptors (PRRs) expressed in OA cartilage and synovium, including TLR2, TLR4 and receptor for advanced glycation endproducts (RAGE) (1,27–31). In turn, the induced cytokines promote expression and release of many DAMPs (Figure 1). The consequent induction and amplification of synovial-cell proliferation and other inflammatory responses can directly and indirectly drive procatabolic responses of chondrocytes, and thus OA progression (1,27–31).
Figure 1. Relationships between inflammatory mediator networks in OA.
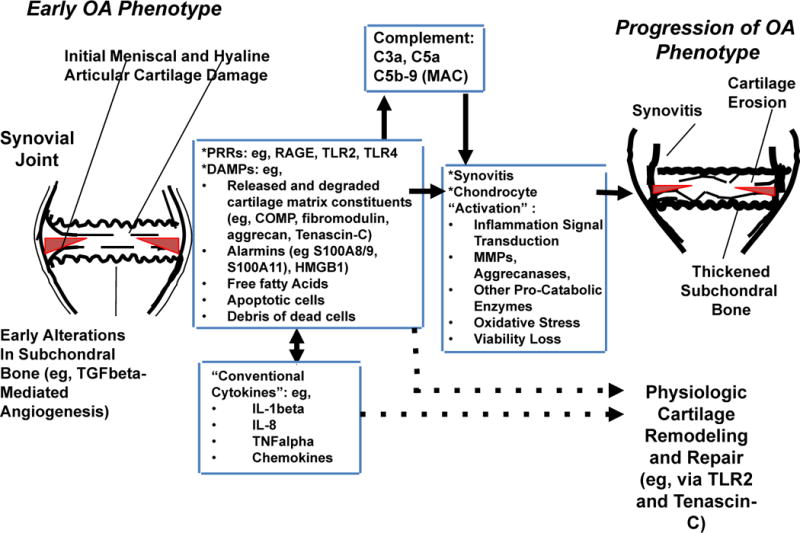
This schematic illustrates how several of the different classes of inflammatory mediators, including PRRs and their DAMP ligands, conventional inflammatory cytokines, and activated complement proteins C5a and C5b-9 network to augment meniscal fibrocartilage and articular cartilage damage in early and progressive OA. These mediators promote macroscopic inflammation, including synovitis, and can drive cartilage matrix catabolism, but some also promote cartilage remodelling and repair. The number and diversity of inflammatory mediators in OA joints, the paradoxical roles of some of these moieties in tissue damage and repair, and the physiological roles of some mediators in host defense, means targeting individual mediators for OA therapy is difficult. Abbreviations: COMP, cartilage oligomeric matrix protein; DAMP, danger associated molecular pattern; HMGB1, high mobility group box protein 1; LMW-HA, low molecular weight hyaluronan; MAC, membrane attack complex; OA, osteoarthritis; PRR, pattern recognition receptor; RAGE, receptor for advanced glycation endproducts; TGF-β, transforming growth factor β; TLR, Toll-like receptor.
Complement activation has emerged as a substantial factor in disease progression in experimental OA (32,33). Increased expression and activity of manifold effector molecules of the classical, alternative, and membrane attack complex (MAC) pathways of complement activation occurs in early human OA, and synovial expression of multiple complement inhibitors, including the C5a-inactivating enzyme carboxypeptidase B, is decreased in human knee OA (32,33). In OA joints, complement can be activated by DAMPs including cartilage matrix constituents (such as fibromodulin, aggrecan, and cartilage oligomeric matrix protein), by hydroxyapatite and calcium pyrophosphate dihydrate crystals, and also by apoptotic cells and the debris of dead cells. Generation of C5a, and of MAC (C5b-9), have been implicated in OA progression in elegant studies of knee OA mouse models (32). Activated complement components accumulate in cartilage, where C5b-9 can activate and kill chondrocytes (32). Although DAMPs activate various complement pathways that converge on C3, mouse knee OA was not substantially affected by C3 knockout, probably because coagulation factor-mediated compensatory mechanisms operate in C3−/− mice that enable C5 activation to proceed (32).
Networking of inflammatory mediators
The term hormesis describes the protective effects of low-level (subtoxic, sublethal) exposure to stressors, and can include adaptive responses that protect against deleterious responses to toxic levels of noxious stimuli, exemplified by low level physiologic mitochondrial reactive oxygen species (ROS) production potentially protecting against noxious levels of ROS (108). In the big picture, physiological hormetic processes within cartilage that are necessary for cartilage tissue remodelling and repair probably use inflammatory processes. One example seems to be the enzymatically stimulated release of latent chondrocyte growth factors such as fibroblast growth factor 2 and TGF-β stored within the extracellular matrix, which can have beneficial or detrimental effects depending on the target cells and timing. Another example is the capacity of inflammatory processes to have beneficial effects by influencing the recruitment, reorganization, and fate of mesenchymal precursor cells and chondrocytes within cartilage. Some mediators of innate inflammation, including TLR2, tenascin-C, and possibly RAGE, can have different effects in different phases of OA, and in the presence or absence of synovitis in OA (27). The chemokine receptor CXCR2, whose ligands include CXCL8 (IL-8), transduces not only pro-catabolic responses in vitro (42) but also promotes chondrocyte phenotypic stability and limits chondrocyte apoptosis; CXCR2 knockout in vivo increases OA progression (Sherwood J, Bertrand J, Nalesso G, Poulet B, Pitsillides A, Brandolini L, Karystinou A, De Bari C, Luyten FP, Pitzalis C, Pap T, Dell’Accio F. A homeostatic function of CXCR2 signalling in articular cartilage. Ann Rheum Dis. 2014 Aug 18. pii: annrheumdis-2014-205546. doi: 10.1136/annrheumdis-2014-205546. [Epub ahead of print] PubMed PMID: 25135253.)
Clearly, the number and diversity of inflammatory mediators in OA joints, the paradoxical roles of some of these moieties in tissue damage and repair, and the physiological roles of some mediators in host defense renders their individual targeting for OA therapy a daunting task. Although complement activation promotes OA progression, the safe and pragmatic use of complement inhibition in OA, including in patients of advanced age and debilitation, would need substantial technological innovation to ensure the preferential and durable targeting of complement activation in OA joints. Given the collective limitations in the ability to control inflammatory mediators in OA, there is great interest in identifying and targeting the major ‘go signals’ (that is, stimulatory and permissive factors) for the procatabolic activities of chondrocytes in OA.
Catabolic reprogramming of chondrocytes
Cytotoxic effects of inflammatory and oxidative stress can both compromise chondrocyte viability and activate inflammatory transcriptional signalling, exemplified by NFκB and certain mitogen activated protein kinases, in surviving chondrocytes (34). The effects of several inflammatory mediators in cartilage evidently are transduced, in part, by certain ’go signals’ that transcriptionally reprogram chondrocytes into extracellular-matrix-catabolizing cells (35–41). One manifestation of procatabolic reprogramming by inflammatory mediators is chondrocyte maturation to hypertrophic differentiation (30,43–45). Moreover, mediators from hypertrophic chondrocytes including vascular endothelial growth factor and bone sialoprotein can promote angiogenesis in joint tissues, including synovium (46).
A HIF-2α-mediated gene expression programme was discovered to promote chondrocyte hypertrophy and increased cartilage matrix catabolism in response to multiple inflammatory cytokines in vitro, and to have a major role in OA progression in vivo (BOX 3) (39,40). Additionally, influx of Zn2+ into chondrocytes via ZIP8, and Zn2+-dependent transcriptional signalling through MTF1, have been reported to mediate OA pathogenesis in response to inflammatory mediators such as IL-1β in vitro and to biomechanical injury in vivo (BOX 3) (41). Evidently, the HIF-2α and MTF1 transcriptional regulatory pathways induce MMPs including MMP-13, and their deficiency prevents cartilage loss and OA progression in mouse models of OA (39–41). Previous work showed that NFκB is upstream of HIF-2α, but whether it is also upstream of ZIP8-mediated Zn2+ influx has not yet been addressed, and NFκB does not have a defined role in ZIP8-mediated expression of MMPs (41). Moreover, the role in OA of the HIF-2α transcriptional signalling pathway remains controversial, given the anabolic effects of HIF-2α on chondrocytes (47–49). The ZIP8-mediated and MTF1 pathway mechanism would also benefit from further validation studies. Hence, we appreciate the limits of designating HIF-2α and the ZIP8–Zn2+–MTF1 axis as ‘go signals’. Furthermore, we need to learn if these pathways are induced and activated independently of each other and the other mechanisms discussed in this Review, or whether their regulation is coordinated upstream or downstream.
BOX 3. Transcriptional reprogramming of chondrocytes.
Several “go signals” transcriptionally reprogram chondrocytes into extracellular matrix catabolizing cells in response to inflammation
The HIF-2α -mediated gene expression program promotes chondrocyte hypertrophy and increased cartilage matrix catabolism in response to multiple inflammatory cytokines and drives OA progression in vivo
Zn2+ importation by ZIP8, and Zn2+-dependent transcriptional signalling through MTF1, mediate chondrocyte activation in response to inflammatory mediators, and OA progression following biomechanical injury in vivo
Impaired chondrocyte bioenergy
Mitochondrial damage and dysfunction
Inflammatory transcriptional ’go signals’ and chondrocyte procatabolic reprogramming are only part of the evolving story of how OA progression could be therapeutically targeted. Specifically, in several organ-degenerative diseases linked to both low-grade tissue inflammation and ageing, acquired mitochondrial dysfunction and damage is not only induced by oxidative stress, but also promotes oxidative stress and related inflammatory processes, such as redox-sensitive inflammatory transcriptional signalling. In this context, chondrocytes derive ATP for their bioenergy needs from high basal rates of glycolysis. In OA, inflammatory mediators drive increased generation of nitric oxide by chondrocytes, which suppresses mitochondrial oxidative phosphorylation and can promote calcification (50–52). Moreover, oxidative stress in OA damages the chondrocyte mitochondrial respiratory chain protein complex (52). Concordantly, there is decreased mitochondrial ATP generation and an associated loss in the chondrocyte ’energy reserve’, contributing to the impaired matrix synthetic function and viability of chondrocytes in acquired OA (51,52). Notably certain mitochondrial haplotypes predispose individuals to OA via potentially related mechanisms (53). In OA chondrocytes, the mitochondrially localized antioxidant SOD2 is depleted, which promotes mitochondrial dysfunction and increased production of reactive oxygen species (54).
Mitochondria also mediate tissue inflammation and damage by way of effects beyond oxidative stress (55). For example, some agents that activate the NLRP3 inflammasome induce inflammasome assembly by causing the depletion of nicotinamide adenine dinucleotide (NAD+), with mitochondrially mediated microtubule assembly (56). In chondrocytes, mitochondrial dysfunction amplifies manifold chondrocyte inflammatory and matrix catabolic responses to IL-1β and TNF, mediated by oxidative stress and NFκB activation (57,58). Evolving evidence suggests the need for increased attention to the change, in OA chondrocytes, from anti-inflammatory reliance on oxidative phosphorylation and the tricarboxylic acid cycle for energy reserve to proinflammatory reliance on glycolysis for energy (59). Moreover, mitochondrial dysfunction, and associated oxidative stress, mediate inflammation related to ER stress and impaired autophagy (60.61), as discussed below.
Bioenergy sensors AMPK and SIRT1
The serine/threonine kinase AMPK, a master regulator of cellular energy balance, enables cells to adjust to changes in energy demand (62,63). Metabolic stress, such as in the hypoxic state of normal chondrocytes, normally activates AMPK (62,63), which phosphorylates multiple downstream targets that promote the inhibition of ATP-consuming pathways and the activation of ATP-producing pathways (62,63). Dysregulation of AMPK has been linked to multiple age-related diseases associated with mitochondrial dysfunction and cellular energy imbalance, including diabetes mellitus, atherosclerosis, cardiovascular disease, cancer, neurodegenerative diseases (62,63), and, most recently, OA (64,65).
Our group discovered that AMPK activity is constitutively robust in normal articular chondrocytes and cartilage, but is decreased in human knee OA chondrocytes and cartilage (64), in mouse knee OA cartilage, in aged mouse knee cartilage, and in chondrocytes after biomechanical injury or treatment with IL-1β and TNF (64,65). Notably, chondrocytes with decreased AMPK activity exhibited increased catabolic responses to IL-1β and TNF, and to biomechanical injury; these responses were attenuated by pharmacological activation of AMPK (64,65). The homeostatic effects of sustained AMPK activity in articular chondrocytes could be particularly important in ageing, as responsiveness to AMPK activators declines during the ageing process (66), and low-grade molecular inflammation present in ageing tissue can further inhibit effects of AMPK (64).
Biochemical regulation of AMPK activity involves phosphorylation and dephosphorylation by certain kinases and phosphatases. The liver protein kinase B1 (LKB1) is the primary upstream kinase that promotes AMPK activity in chondrocytes (65). Chondrocyte catabolic responses to IL-1β and TNF are increased by loss of LKB1 activity in chondrocytes, (65) and we have observed the concomitant reduction of phosphorylation of both LKB1 and AMPKα (the marker for AMPK activity) in primary human knee OA chondrocytes, in mouse knee OA cartilage, in aged mouse knee cartilage, and in bovine chondrocytes after biomechanical injury (65). Hence, in cartilage affected by age or OA, dysregulation of LKB1 might have a major role in suppression of chondrocyte AMPK activity.
AMPK activity regulates energy metabolism via downstream mediators including the NAD+-dependent protein deacetylase SIRT1 (66) (Figure 2). In cartilage, SIRT1 promotes cartilage-specific gene expression (67–69), protects chondrocytes from radiation-induced senescence (70), and inhibits chondrocyte apoptosis (71–73). Cartilage expression of SIRT1 is decreased in human knee OA, mouse knee OA and knees of aged mice (69,74). Inhibition of SIRT1 in chondrocytes results in enhanced procatabolic responses to IL-1β and TNF (75,76). Moreover, adult heterozygous Sirt1 knockout mice and mice with a Sirt1 point mutation exhibit increased OA progression (68,73), and cartilage-specific Sirt1 knockout mice develop accelerated OA progression (74). A path to translation to new clinical therapy exists, as pharmacologic SIRT1 activation exerts chondroprotective effects (77–82). Moreover, AMPK and SIRT1 can interact or impinge on the same pathways in multiple ways (Figure 2), including in inflammation (83). For example, activation of AMPK can stimulate the activity of SIRT1 by increasing intracellular NAD+ (66), and SIRT1 is able to deacetylate LKB1, which increases LKB1 activity and leads to AMPK activation (66). This positive feedback loop between SIRT1 and AMPK potentiates AMPK activity (66).
Figure 2. Functions of AMPK and SIRT1 in chondrocyte resistance to cell stress and inflammatory processes that promote matrix catabolism.

AMPK, which has multiple endogenous and exogenous activators, promotes activation of SIRT1. LKB1 is the primary upstream kinase that promotes AMPK activity, and decreased LKB1 increases chondrocyte matrix procatabolic responses to IL-1β and TNF. Active LKB1 and AMPK are decreased in OA, injured, and ageing chondrocytes. AMPK and SIRT1 exert anti-inflammatory effects, including inhibition of NFκB activation via SIRT1, which deacetylates the p65 subunit of NFκB and thereby primes p65 for proteasomal degradation; these effects limit chondrocyte matrix procatabolic responses. In addition, AMPK and SIRT1 promote autophagy, with attendant repair of damaged organelles, such as mitochondria and ER, and AMPK and SIRT1 inhibit ER stress, providing further means to limit inflammation in OA cartilage.
Abbreviations: AMPK, 5′-AMP-activated protein kinase; ER, endoplasmic reticulum; LKB1, liver protein kinase B1; SIRT1, NAD-dependent protein deacetylase sirtuin-1.
The functions of AMPK and SIRT1 are not restricted to the maintenance of energy metabolism during increased energy consumption; they coordinate several ‘housekeeping’ mechanisms of resistance to cell stress and inflammation (66). First, activation of AMPK can limit oxidative stress, most likely by improving mitochondrial function (66,84). Second, AMPK and SIRT1 have anti-inflammatory effects and limit matrix catabolism by chondrocytes (Figure 2). For example, AMPK inhibits NFκB activation via SIRT1, which deacetylates the p65 subunit of NFκB and thereby primes p65 for proteasomal degradation (66). Activation of AMPK or SIRT1 inhibits chondrocyte procatabolic responses to proinflammatory cytokines IL-1β and TNF, via attenuation of NFκB activation (64,74,75). Finally, AMPK and SIRT1 promote autophagy, with attendant repair of damaged mitochondria (and ER), and limiting ER stress (Figure 2), as discussed below.
Bioenergy sensing and proteostasis
Protein homeostasis or ’proteostasis’ involves several thousand mediators, and is carried out by multiple interconnected pathways and systems, which include the unfolded protein response (UPR), autophagy, the ubiquitin–proteasome system, and lysosomes (84–86). Proteostasis regulates protein fate from biogenesis to conformation folding, subcellular trafficking, and degradation within cells. Normally, proteostasis maintains the health of cells in development, ageing, and resistance to environmental stressors, and prevents diseases mediated by excessive protein misfolding, aggregation, or degradation (84–86). In OA, however, not only changes in cell differentiation and fate, but also the proinflammatory effects of abnormal proteostasis (including increased UPR activation and dysfunction of autophagy in chondrocytes), seem to contribute to pathogenesis (BOX 4). Similar changes in other tissues are involved in both inflammatory diseases (87) and many metabolic and degenerative diseases of ageing (84–86,88). A prime example, in rheumatic disease, is ankylosing spondylitis, where abnormal proteostasis driven by protein misfolding amplifies TLR-mediated inflammatory signaling in response to DAMPs (87).
BOX 4. Abnormal chondrocyte proteostasis in OA.
Inflammatory effects via abnormal proteostasis in chondrocytes, including increased UPR activation and impaired autophagy, are involved in OA pathophysiology
Similar alterations in proteostasis are involved in multiple tissue degenerative disease of aging and in emerging paradigms in other rheumatic diseases involving inflammation, including ankylosing spondylitis
Autophagy is a cellular homeostasis process that functions in part by removing damaged organelles and protein aggregates; in so doing, autophagy generates energy that can assist cell survival under conditions of stress such as starvation, resistance to ongoing infection, and inflammation (85,89,90). Autophagy has substantial chrondroprotective effects, but is defective in ageing and OA chondrocytes (85). Disordered autophagy promotes inflammation in multiple tissues (85,89,90), and probably contributes to OA chondrocyte pathophysiology, at least in part, in this way. Autophagy in OA has been reviewed in detail, including in this journal (85,89,90). Here, we focus on the UPR, a response system activated by conditions including oxidative stress and inflammation, and one central to successful resolution of ER stress caused by protein misfolding (86,88,91,92).
The UPR lessens pressure on the stressed ER by limiting protein production, and by switching on a transcriptome replete with chaperones and protein-folding catalysts that boosts the protein-folding machinery (88,91,92) (Figure 3). Importantly, unsuccessful resolution of ER stress by the UPR promotes oxidative stress inflammation and apoptosis (88,91,92). The three UPR signalling cascades stem from functional activation of the ER transmembrane proteins PERK, IRE1, and ATF6, respectively, and are triggered by dissociation of the chaperone GRP78 (also termed BiP) from these ER transmembrane proteins (88,91,92)(Figure 3). The UPR cascades participate in normal skeletal development, where ER stress-induced chondrocyte dysfunction was first pinpointed in pathophysiology studies of hereditary chondrodysplasia triggered by misfolded transgenic mutant cartilage matrix proteins (93,94).
Figure 3. Modulation of inflammatory processes by the UPR in chondrocytes in OA.
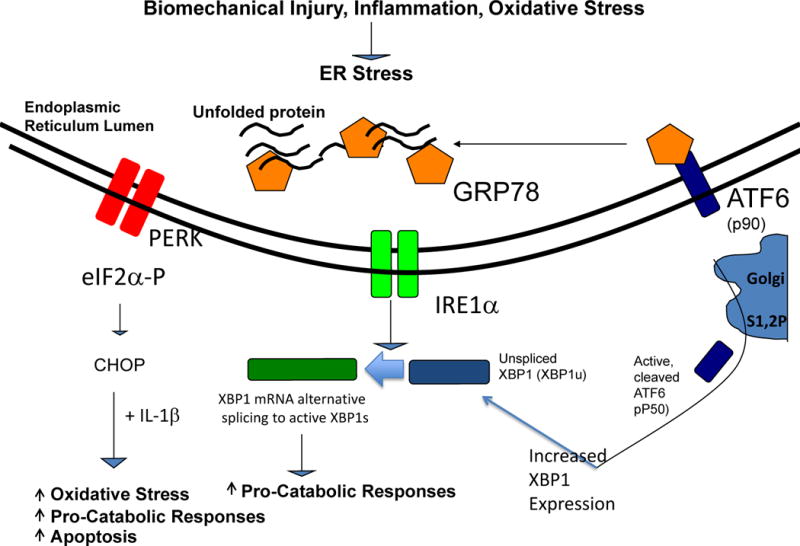
The UPR signalling cascades triggered by dissociation of the chaperone GRP78 from the ER transmembrane proteins PERK, IRE1, and ATF6, and activated by biomechanical injury, nitric oxide, and certain inflammatory mediators. Alternatively spliced, transcriptionally activated XBP1 (XBP1s) promotes chondrocyte maturation to procatabolic hypertrophic differentiation, mediated in part by cross-talk with the ATF6 pathway. XBP1s also drives matrix pro-catabolic responses in response to IL-1β, as does excess CHOP, another terminal UPR effector. CHOP also promotes chondrocyte superoxide production and apoptosis. Knock-out of CHOP is chondroprotective in mouse knee instability-induced OA in vivo.
Abbreviations: ATF6, activating transcription factor 6; CHOP, C/EBP homologous protein; ER, endoplasmic reticulum; GRP78, 78 kDa glucose-regulated protein; IRE1, inositol-requiring protein 1; PERK, protein kinase RNA-like ER kinase; UPR, unfolded protein response; XBP1, X-box-binding protein 1; XBP1s, X-box-binding protein 1 splicing form.
In vitro studies have elucidated that biomechanical injury, IL-1β and nitric oxide are among the factors in OA pathogenesis that can activate the UPR in cultured chondrocytes (86,95)(Figure 3). Moreover, findings predominately related to the terminal effectors of the PERK and IRE1 pathways (CHOP and XBP1, respectively) have demonstrated increased UPR activation in OA articular chondrocytes (86,95–97). Alternatively spliced, transcriptionally activated XBP1 (XBP1s), the generation of which is UPR-specific, promotes chondrocyte maturation to hypertrophic differentiation through its association with the transcription factor RUNX2, and by modulation of IHH (Indian hedgehog) and parathyroid hormone-related peptide signalling (98). Notably, XBP1 expression is induced by activated ATF6 (97)(Figure 3), one of the modes of cross-talk between UPR cascades in chondrocyte biology.
The generation of XBP1s is central to not only TLR-mediated inflammation signalling and expression of multiple inflammatory cytokines, but also ‘killing’ responses for intracellular pathogens in macrophages (99). XBP1s has a long half-life, whereas unspliced XBP1, as well as CHOP, normally has a short half-life. Our group demonstrated that biomechanical injury induces robust expression of CHOP in chondrocytes (86). CHOP constitutively works to resolve the UPR by restoring protein synthesis, but excessive CHOP loads the stressed ER with more protein, and promotes oxidative stress and apoptosis (87,88,91,92) (Figure 3).
XBP1s and CHOP potentiate the capacity of IL-1β to induce nitric oxide and MMP-3 release by cultured chondrocytes (86) (Figure 3). However, XBP1s also can have salutary effects on chondrocyte survival in some experimental conditions (97). In vivo analyses of a surgically induced instability model of knee OA in mice revealed that global knockout of Chop partially protected against increased chondrocyte apoptosis and cartilage degradation (mediated by anabolic and catabolic mechanisms), and these effects were not attributable to discernible differences in chondrocyte ER stress in situ (109).
Relevance for clinical translation
Many treatments that affect symptoms in OA, but are not truly disease-modifying, exert inhibitory effects on inflammatory processes. These treatments include glucocorticosteroids, NSAIDs and coxibs (100), and other FDA-approved medical treatments such as intra-articular hyaluronan (101,102). Moreover, certain nutraceuticals can modulate proteostasis or inflammatory processes, or both (102–104). Conceptually attractive, rational, inflammation-targeting therapeutic approaches for investigation include intra-articular injection of lubricin to decrease synovial proliferation (20), or inhibiting DAMPS and their signalling, for example by limiting intra-articular complement activation or by constraining the effects of S100 calgranulins and of TLR4 signalling. Biologic therapies targeting single cytokines that are increased in OA joint tissues (for example, IL-1β, IL-6, TNFα) have not yet been established as either effective or pragmatic systemic or local approaches in human OA. Moreover, there are major limitations in how pragmatic such approaches can be, and we will probably need innovations in delivery systems, such as nanotechnology, to selectively and safely target joints in a durable manner.
In this Review, we have attempted to capsulize emerging thinking on potential preventative and therapeutic strategies for OA that target inflammation in an integrative manner. Entry points for therapeutic interventions that promote ‘stop signals’ for inflammatory processes in chondrocytes start with exercise, targeted strength training, and achievement of ideal body weight for overweight and obese patients, which are of proven benefit in human knee OA (105). More-specific entry points for therapy can target proteostasis-related inflammatory and aging processes, such as delivery to the joint of compounds known to directly promote chondrocyte autophagy, or to improve protein folding and limit ER stress, or both (85,88–92,108). Direct activation of AMPK (BOX 5) is an alternative approach (Figure 4). AMPK can alleviate ER stress, and AMPK activation inhibits CHOP expression and catabolic responses induced by biomechanical injury and IL-1β in chondrocytes (86). AMPK and SIRT1 signalling also promote autophagy (66,106). Importantly, metformin, sodium salicylate, high-dose aspirin, and methotrexate are among drugs known to activate AMPK that are already employed in the clinic for arthritis and other conditions (59). Moreover, a randomized placebo-controlled trial of methotrexate in knee OA demonstrated salutary clinical benefits and associated reduction of synovitis (107). As such, strategies to inhibit inflammatory reprogramming of chondrocytes in OA by modulating bioenergy sensors might be well within reach for definitive testing in further clinical trials.
BOX 5. Can AMPK activation be developed for OA?
AMPK and SIRT1 signalling promote autophagy and can limit ER stress
Metformin, sodium salicylate, high-dose aspirin, and methotrexate are among drugs, known to activate AMPK, that are already employed in the clinic for arthritis and some other conditions
A recent randomized placebo-controlled trial of methotrexate in knee OA demonstrated salutary clinical benefits and associated reduction of synovitis
Figure 4. Therapeutic modulation of AMPK and SIRT1 as an Entry Point for “Stop Signals” for Chondrocyte Pro-Catabolic Reprogramming by Inflammation, Oxidative Stress, and Altered UPR and Autophagy.
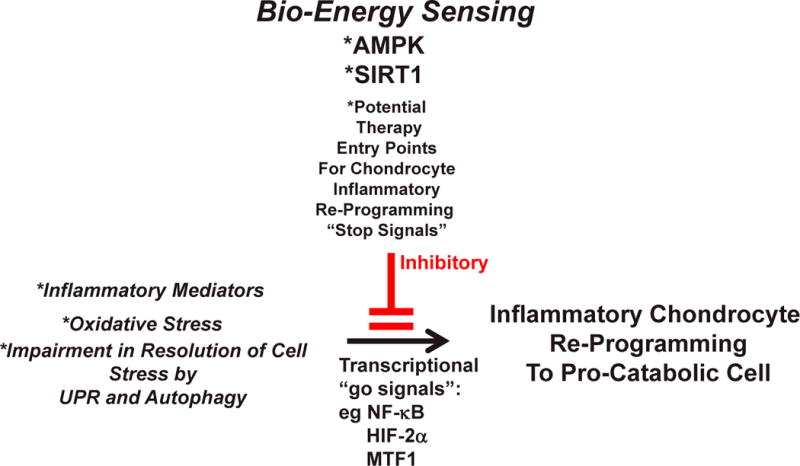
The Figure schematizes the proposal that the bio-energy sensors AMPK and SIRT1 can be an integrative entry point to therapeutic suppression (red lines) of the inflammatory process-mediated re-programming of chondrocytes to a pro-catabolic state. In this manner, there is potential to slow the progression of OA, but this question needs direct testing in vivo.
Conclusions
To paraphrase Confucius, the quest to pinpoint fundamental roles of specific inflammatory processes that promote OA progression has often been “like looking for a black cat in a dark room; there may or may not be a cat in the room”. We now see a clearer portrait, with a broad network of inflammatory mediators and their receptors, and novel ‘go signals’ for chondrocyte differentiation reprogramming. Moreover, we better appreciate how articular chondrocyte metabolic factors and certain bioenergy sensors can refine inflammatory responses, and how loss of quality controls in certain cell-responses to stress are involved in inflammatory processes in chondrocytes (Figure 4). Future investigation should lead to better understanding of how bioenergy sensors, modulated by body weight, nutrition, exercise, ageing, and comorbidities, link metabolism with inflammatory processes to regulate both joint physiology and OA clinical phenotypes. Such research should open up more integrative and effective preventative and therapeutic strategies for OA.
Key Points.
Multiple danger associated molecular patterns, including activators of complement, are increased within joints affected by osteoarthritis (OA), and complement activation is a major factor in progression of experimental knee OA
Biomechanical cartilage injury and joint inflammation compromise chondrocyte viability and reprogram viable chondrocytes to procatabolic differentiation using transcriptional ‘go signals’, including NFκB, and possibly HIF-2α, and MTF1
Oxidative stress and dysregulated chondrocyte mitochondrial function contribute not only to impaired matrix synthetic function and viability, but also to molecular inflammatory processes and matrix catabolism in OA
Biomechanical injury, oxidative stress, and inflammatory mediators modulate proteostasis responses, including autophagy and the unfolded protein response to endoplasmic reticulum (ER) stress
Chondrocyte bioenergy sensors including AMPK and SIRT1 can modulate deleterious chondrocyte responses to oxidative stress and inflammatory mediators, potentially providing therapeutic ‘entry points’ for limiting OA progression
Acknowledgments
The authors’ work is supported by funding from the VA Research Service, Arthritis Foundation, and NIH (PAG07996, AI81881).
Biographies
Ru Liu-Bryan PhD has a research laboratory based at the VA Medical Center in San Diego, where she is Associate Professor of Medicine, University of California, San Diego, USA. She received her PhD in molecular biology and biochemistry from Imperial College London, UK. Her research focuses on understanding molecular mechanisms of pathogenesis of innate immunity in diseases including osteoarthritis and crystal arthropathy.
Robert Terkeltaub MD is Chief of Rheumatology at the VA Medical Center and Professor of Medicine at University of California San Diego, CA, USA. Dr Terkeltaub received his MD at McGill University, Montreal, QC, Canada, and completed residency and training programs in Clinical Rheumatology and Internal Medicine at Montreal General Hospital. Dr Terkeltaub’s research focuses principally on interfaces between metabolism and inflammation in diseases including osteoarthritis, gout, and pathologic calcification. Dr Terkeltaub regularly serves on NIH and other arthritis research Study Sections and is Co-Editor of Arthritis & Rheumatology.
Footnotes
Competing interests: R.T. declares he serves as a Scientific Advisory Board Member for Cardax.
Author contributions: Both authors contributed equally to researching the data for the article, providing a substantial contribution to discussions of the content, writing the article and review and/or editing of the manuscript before submission.
Review Criteria:
We searched MEDLINE and PubMed databases for original articles focusing on osteoarthritis and on mechanisms of low-grade inflammation, published between 2006 and February 2014. The search terms, used in various combinations, were “osteoarthritis”, “inflammation”, “alarmin”, “danger associated molecular pattern”, “chondrocyte”, “mitochondria”, “inflammasome”, “cytokine”, “AMPK”, SIRT1”, and “unfolded protein response”. All papers identified were English-language and full-text. We also searched reference lists within the identified manuscripts for additional relevant papers. The reference list was last updated August 2014.
References
- 1.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–1707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6:625–635. doi: 10.1038/nrrheum.2010.159. [DOI] [PubMed] [Google Scholar]
- 3.Karsdal MA, et al. The coupling of bone and cartilage turnover in osteoarthritis: opportunities for bone antiresorptives and anabolics as potential treatments? Ann Rheum Dis. 2014;73:336–348. doi: 10.1136/annrheumdis-2013-204111. [DOI] [PubMed] [Google Scholar]
- 4.Goldring MB, Goldring SR. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann N Y Acad Sci. 2010;1192:230–237. doi: 10.1111/j.1749-6632.2009.05240.x. [DOI] [PubMed] [Google Scholar]
- 5.Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7:161–169. doi: 10.1038/nrrheum.2010.213. [DOI] [PubMed] [Google Scholar]
- 6.Bao JP, Chen WP, Wu LD. Lubricin: a novel potential biotherapeutic approaches for the treatment of osteoarthritis. Mol Biol Rep. 2011;38:2879–2885. doi: 10.1007/s11033-010-9949-9. [DOI] [PubMed] [Google Scholar]
- 7.Berenbaum F, Eymard F, Houard X. Osteoarthritis, inflammation and obesity. Curr Opin Rheumatol. 2013;25:114–118. doi: 10.1097/BOR.0b013e32835a9414. [DOI] [PubMed] [Google Scholar]
- 8.Issa RI, Griffin TM. Pathobiology of obesity and osteoarthritis: integrating biomechanics and inflammation. Pathobiol Aging Age Relat Dis. 2012;2:17470. doi: 10.3402/pba.v2i0.17470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonnet CS, et al. AMPA/kainate glutamate receptors contribute to inflammation, degeneration and pain related behaviour in inflammatory stages of arthritis. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2013-203670. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malfait AM, Schnitzer TJ. Towards a mechanism-based approach to pain management in osteoarthritis. Nat Rev Rheumatol. 2013;9:654–664. doi: 10.1038/nrrheum.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller RE, et al. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A. 2012;109:20602–20607. doi: 10.1073/pnas.1209294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Punzi L, Frigato M, Frallonardo P, Ramonda R. Inflammatory osteoarthritis of the hand. Best Pract Res Clin Rheumatol. 2010;24:301–312. doi: 10.1016/j.berh.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Zhen G, et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;2013(19):704–712. doi: 10.1038/nm.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guermazi A, et al. Synovitis in knee osteoarthritis assessed by contrast-enhanced magnetic resonance imaging (MRI) is associated with radiographic tibiofemoral osteoarthritis and MRI-detected widespread cartilage damage: The MOST Study. J Rheumatol. 2014;41:501–508. doi: 10.3899/jrheum.130541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Lange-Brokaar BJ, et al. Degree of synovitis on MRI by comprehensive whole knee semi-quantitative scoring method correlates with histologic and macroscopic features of synovial tissue inflammation in knee osteoarthritis. Osteoarthritis Cartilage. 2013;21 doi: 10.1016/j.joca.2013.12.013. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 16.Knoop J, et al. Biomechanical factors and physical examination findings in osteoarthritis of the knee: associations with tissue abnormalities assessed by conventional radiography and high-resolution 3.0 Tesla magnetic resonance imaging. Arthritis Res Ther. 2012;14:R212. doi: 10.1186/ar4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guermazi A, et al. Prevalence of abnormalities in knees detected by MRI in adults without knee osteoarthritis: population based observational study (Framingham Osteoarthritis Study) BMJ. 2012;345:e5339. doi: 10.1136/bmj.e5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambert C, et al. Gene expression pattern of synovial cells from inflammatory and normal areas of osteoarthritis synovial membrane. Arthritis Rheum. 2013;64 doi: 10.1002/art.38315. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, et al. Hyaluronan injection in murine osteoarthritis prevents TGFbeta 1-induced synovial neovascularization and fibrosis and maintains articular cartilage integrity by a CD44-dependent mechanism. Arthritis Res Ther. 2012;14:R151. doi: 10.1186/ar3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flannery CR, et al. Prevention of cartilage degeneration in a rat model of osteoarthritis by intraarticular treatment with recombinant lubricin. Arthritis Rheum. 2009;60:840–847. doi: 10.1002/art.24304. [DOI] [PubMed] [Google Scholar]
- 21.Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51:249–257. doi: 10.1016/j.bone.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chevalier X, Eymard F, Richette P. Biologic agents in osteoarthritis: hopes and disappointments. Nat Rev Rheumatol. 2013 Jul;9(7):400–10. doi: 10.1038/nrrheum.2013.44. 2013. [DOI] [PubMed] [Google Scholar]
- 23.Husa M, Liu-Bryan R, Terkeltaub R. Shifting HIFs in osteoarthritis. Nat Med. 2010 Jun;16(6):641–4. doi: 10.1038/nm0610-641. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bougault C, et al. Stress-induced cartilage degradation does not depend on the NLRP3 inflammasome in human osteoarthritis and mouse models. Arthritis Rheum. 2012;64:3972–3981. doi: 10.1002/art.34678. [DOI] [PubMed] [Google Scholar]
- 25.Hernandez-Cuellar E, et al. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol. 2012;189:5113–5117. doi: 10.4049/jimmunol.1202479. [DOI] [PubMed] [Google Scholar]
- 26.Mao K, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013;23:201–212. doi: 10.1038/cr.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu-Bryan R, Terkeltaub R. The growing array of innate inflammatory ignition switches in osteoarthritis. Arthritis Rheum. 2012;64:2055–2058. doi: 10.1002/art.34492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schelbergen RF, et al. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on Toll-like receptor 4. Arthritis Rheum. 2012;64:1477–1487. doi: 10.1002/art.33495. [DOI] [PubMed] [Google Scholar]
- 29.Zreiqat H, et al. S100A8 and S100A9 in experimental osteoarthritis. Arthritis Res Ther. 2010;12:R16. doi: 10.1186/ar2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cecil DL, et al. The pattern recognition receptor CD36 is a chondrocyte hypertrophy marker associated with suppression of catabolic responses and promotion of repair responses to inflammatory stimuli. J Immunol. 2009;182:5024–5031. doi: 10.4049/jimmunol.0803603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin C, et al. NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc Natl Acad Sci U S A. 2011;108:14867–14872. doi: 10.1073/pnas.1111101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q, et al. Identification of a central role for complement in osteoarthritis. Nat Med. 2011;17:1674–1679. doi: 10.1038/nm.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lepus CM, et al. Brief report: carboxypeptidase B serves as a protective mediator in osteoarthritis. Arthritis Rheumatol. 2014;66:101–106. doi: 10.1002/art.38213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcu KB, Otero M, Olivotto E, Borzi RM, Goldring MB. NF-kappaB signaling: multiple angles to target OA. Curr Drug Targets. 2010;11:599–613. doi: 10.2174/138945010791011938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin AC, et al. Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat Med. 2009;15:1421–1425. doi: 10.1038/nm.2055. [DOI] [PubMed] [Google Scholar]
- 36.Echtermeyer F, et al. Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat Med. 2009;15:1072–1076. doi: 10.1038/nm.1998. [DOI] [PubMed] [Google Scholar]
- 38.Pap T, Bertrand J. Syndecans in cartilage breakdown and synovial inflammation. Nat Rev Rheumatol. 2013;9:43–55. doi: 10.1038/nrrheum.2012.178. [DOI] [PubMed] [Google Scholar]
- 39.Saito T, et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med. 2010;16:678–686. doi: 10.1038/nm.2146. [DOI] [PubMed] [Google Scholar]
- 40.Yang S, et al. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010;16:687–693. doi: 10.1038/nm.2153. [DOI] [PubMed] [Google Scholar]
- 41.Kim JH, et al. Regulation of the Catabolic Cascade in Osteoarthritis by the Zinc-ZIP8-MTF1 Axis. Cell. 2014;156:730–743. doi: 10.1016/j.cell.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 42.Merz D, Liu R, Johnson K, Terkeltaub R. IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J Immunol. 2003 Oct 15;171(8):4406–15. doi: 10.4049/jimmunol.171.8.4406. [DOI] [PubMed] [Google Scholar]
- 43.Liu-Bryan R, Terkeltaub R. Chondrocyte innate immune myeloid differentiation factor 88-dependent signaling drives procatabolic effects of the endogenous Toll-like receptor 2/Toll-like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein 1 in mice. Arthritis Rheum. 2010;62:2004–12. doi: 10.1002/art.27475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yano F, Saito T, Ogata N, Yamazawa T, Iino M, Chung UI, Kawaguchi H. β-catenin regulates parathyroid hormone/parathyroid hormone-related protein receptor signals and chondrocyte hypertrophy through binding to the intracellular C-terminal region of the receptor. Arthritis Rheum. 2013;65:429–35. doi: 10.1002/art.37779. [DOI] [PubMed] [Google Scholar]
- 45.Pesesse L, Sanchez C, Delcour JP, Bellahcène A, Baudouin C, Msika P, Henrotin Y. Consequences of chondrocyte hypertrophy on osteoarthritic cartilage: potential effect on angiogenesis. Osteoarthritis Cartilage. 2013;21:1913–23. doi: 10.1016/j.joca.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 46.Konisti S, Kiriakidis S, Paleolog EM. Hypoxia–a key regulator of angiogenesis and inflammation in rheumatoid arthritis. Nat Rev Rheumatol. 2012;8:153–62. doi: 10.1038/nrrheum.2011.205. [DOI] [PubMed] [Google Scholar]
- 47.Murphy CL. HIF-2alpha–a mediator of osteoarthritis? Cell Res. 2010;20(9):977–9. doi: 10.1038/cr.2010.99. [DOI] [PubMed] [Google Scholar]
- 48.Clerigues V, Murphy CL, Guillen MI, Alcaraz MJ. Haem oxygenase-1 induction reverses the actions of interleukin-1β on hypoxia-inducible transcription factors and human chondrocyte metabolism in hypoxia. Clin Sci (Lond) 2013;125(2):99–108. doi: 10.1042/CS20120491. [DOI] [PubMed] [Google Scholar]
- 49.Thoms BL, Dudek KA, Lafont JE, Murphy CL. Hypoxia promotes the production and inhibits the destruction of human articular cartilage. Arthritis Rheum. 2013;65(5):1302–12. doi: 10.1002/art.37867. [DOI] [PubMed] [Google Scholar]
- 50.Johnson K, et al. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000;43:1560–1570. doi: 10.1002/1529-0131(200007)43:7<1560::AID-ANR21>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 51.Johnson K, et al. Mediation of spontaneous knee osteoarthritis by progressive chondrocyte ATP depletion in Hartley guinea pigs. Arthritis Rheum. 2004;50:1216–1225. doi: 10.1002/art.20149. [DOI] [PubMed] [Google Scholar]
- 52.Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7:161–169. doi: 10.1038/nrrheum.2010.213. [DOI] [PubMed] [Google Scholar]
- 53.Rego-Perez I, et al. Mitochondrial genetics and osteoarthritis. Front Biosci (Schol Ed) 2013;5:360–368. doi: 10.2741/s377. [DOI] [PubMed] [Google Scholar]
- 54.Scott JL, et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann Rheum Dis. 2010;69:1502–1510. doi: 10.1136/ard.2009.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tschopp J. Mitochondria: Sovereign of inflammation? Eur J Immunol. 2011;41:1196–1202. doi: 10.1002/eji.201141436. [DOI] [PubMed] [Google Scholar]
- 56.Misawa T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14:454–460. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 57.Gavriilidis C, Miwa S, von Zglinicki T, Taylor RW, Young DA. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum. 2013;65:378–387. doi: 10.1002/art.37782. [DOI] [PubMed] [Google Scholar]
- 58.Vaamonde-Garcia C, et al. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. 2012;64:2927–2936. doi: 10.1002/art.34508. [DOI] [PubMed] [Google Scholar]
- 59.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 60.Rath E, Haller D. Mitochondria at the interface between danger signaling and metabolism: role of unfolded protein responses in chronic inflammation. Inflamm Bowel Dis. 2012;18:1364–1377. doi: 10.1002/ibd.21944. [DOI] [PubMed] [Google Scholar]
- 61.Schiavi A, Ventura N. The interplay between mitochondria and autophagy and its role in the aging process. Exp Gerontol. 2014;49 doi: 10.1016/j.exger.2014.02.015. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 62.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 63.Witczak CA, Sharoff CG, Goodyear LJ. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci. 2008;65:3737–3755. doi: 10.1007/s00018-008-8244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Terkeltaub R, Yang B, Lotz M, Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to inflammatory cytokines IL-1β and TNFα. Arthritis Rheum. 2011;63:1928–1937. doi: 10.1002/art.30333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petursson F, Husa M, June R, Lotz M, Terkeltaub R, Liu-Bryan R. Linked decreases in Liver Kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res Ther. 2013;15:R77. doi: 10.1186/ar4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11:230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 67.Dvir-Ginzberg M, Steinmeyer J. Towards elucidating the role of SirT1 in osteoarthritis. Front Biosci. 2013;18:343–55. doi: 10.2741/4105. [DOI] [PubMed] [Google Scholar]
- 68.Gabay O, et al. Sirtuin 1 enzymatic activity is required for cartilage homeostasis in vivo in a mouse model. Arthritis Rheum. 2013;65:159–166. doi: 10.1002/art.37750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dvir-Ginzberg M, Gagarina V, Lee EJ, Hall DJ. Regulation of cartilage-specific gene expression in human chondrocytes by SirT1 and nicotinamide phosphoribosyltransferase. J Biol Chem. 2008;283:36300–36310. doi: 10.1074/jbc.M803196200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hong EH, et al. Ionizing radiation induces cellular senescence of articular chondrocytes via negative regulation of SIRT1 by p38 kinase. J Biol Chem. 2010;285:1283–1295. doi: 10.1074/jbc.M109.058628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takayama K, et al. SIRT1 regulation of apoptosis of human chondrocytes. Arthritis Rheum. 2009;60:2731–2740. doi: 10.1002/art.24864. [DOI] [PubMed] [Google Scholar]
- 72.Gagarina V, et al. SirT1 enhances survival of human osteoarthritic chondrocytes by repressing protein tyrosine phosphatase 1B and activating the insulin-like growth factor receptor pathway. Arthritis Rheum. 2010;62:1383–1392. doi: 10.1002/art.27369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gabay O, et al. Increased apoptotic chondrocytes in articular cartilage from adult heterozygous SirT1 mice. Ann Rheum Dis. 2012;71:613–616. doi: 10.1136/ard.2011.200504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matsuzaki T, et al. Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2012-202620. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 75.Moon MH, et al. SIRT1, a class III histone deacetylase, regulates TNF-α-induced inflammation in human chondrocytes. Osteoarthritis Cartilage. 2013;21:470–480. doi: 10.1016/j.joca.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 76.Matsushita T, et al. The overexpression of SIRT1 inhibited osteoarthritic gene expression changes induced by interleukin-1β in human chondrocytes. J Orthop Res. 2013;31:531–537. doi: 10.1002/jor.22268. [DOI] [PubMed] [Google Scholar]
- 77.Lei M, et al. Resveratrol inhibits interleukin 1β-mediated inducible nitric oxide synthase expression in articular chondrocytes by activating SIRT1 and thereby suppressing nuclear factor-κB activity. Eur J Pharmacol. 2012;674:73–79. doi: 10.1016/j.ejphar.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 78.Wang J, Gao JS, Chen JW, Li F, Tian J. Effect of resveratrol on cartilage protection and apoptosis inhibition in experimental osteoarthritis of rabbit. Rheumatol Int. 2012;32:1541–1548. doi: 10.1007/s00296-010-1720-y. [DOI] [PubMed] [Google Scholar]
- 79.Dave M, et al. The antioxidant resveratrol protects against chondrocyte apoptosis via effects on mitochondrial polarization and ATP production. Arthritis Rheum. 2008;58:2786–2797. doi: 10.1002/art.23799. [DOI] [PubMed] [Google Scholar]
- 80.Shakibaei M, Csaki C, Nebrich S, Mobasheri A. Resveratrol suppresses interleukin-1beta-induced inflammatory signaling and apoptosis in human articular chondrocytes: potential for use as a novel nutraceutical for the treatment of osteoarthritis. Biochem Pharmacol. 2008;76:1426–1439. doi: 10.1016/j.bcp.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 81.Csaki C, Keshishzadeh N, Fischer K, Shakibaei M. Regulation of inflammation signalling by resveratrol in human chondrocytes in vitro. Biochem Pharmacol. 2008;75:677–687. doi: 10.1016/j.bcp.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 82.Shakibaei M, John T, Seifarth C, Mobasheri A. Resveratrol inhibits IL-1 beta-induced stimulation of caspase-3 and cleavage of PARP in human articular chondrocytes in vitro. Ann NY Acad Sci. 2007;1095:554–563. doi: 10.1196/annals.1397.060. [DOI] [PubMed] [Google Scholar]
- 83.Liu TF, Brown CM, El Gazzar M, McPhail L, Millet P, Rao A, Vachharajani VT, Yoza BK, McCall CE. Fueling the flame: bioenergy couples metabolism and inflammation. J Leukoc Biol. 2012;92:499–507. doi: 10.1189/jlb.0212078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lotz M, Carames B. Autophagy: a new therapeutic target in cartilage injury and osteoarthritis. J Am Acad Orthop Surg. 2012;20:261–262. doi: 10.5435/JAAOS-20-04-261. [DOI] [PubMed] [Google Scholar]
- 86.Husa M, Petursson F, Lotz M, Terkeltaub R, Liu-Bryan R. C/EBP homologous protein drives pro-catabolic responses in chondrocytes. Arthritis Res Ther. 2013;15:R218. doi: 10.1186/ar4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Colbert RA, Tran TM, Layh-Schmitt G. HLA-B27 misfolding and ankylosing spondylitis. Mol Immunol. 2014;57:44–51. doi: 10.1016/j.molimm.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Carneiro LA, Travassos LH. The Interplay between NLRs and Autophagy in Immunity and Inflammation. Front Immunol. 2013;4:361. doi: 10.3389/fimmu.2013.00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arroyo DS, Gaviglio EA, Peralta Ramos JM, Bussi C, Rodriguez-Galan MC, Iribarren P. Autophagy in inflammation, infection, neurodegeneration and cancer. Int Immunopharmacol. 2014;18:55–65. doi: 10.1016/j.intimp.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Claudio N, Dalet A, Gatti E, Pierre P. Mapping the crossroads of immune activation and cellular stress response pathways. EMBO J. 2013;32:1214–1224. doi: 10.1038/emboj.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garg AD, et al. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med. 2012;18:589–598. doi: 10.1016/j.molmed.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 93.Liang G, et al. Endoplasmic reticulum stress-unfolding protein response-apoptosis cascade causes chondrodysplasia in a col2a1 p.Gly1170Ser mutated mouse model. PLoS One. 2014;9:e86894. doi: 10.1371/journal.pone.0086894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gualeni B, et al. A novel transgenic mouse model of growth plate dysplasia reveals that decreased chondrocyte proliferation due to chronic ER stress is a key factor in reduced bone growth. Dis Model Mech. 2013;6:1414–1425. doi: 10.1242/dmm.013342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oliver BL, Cronin CG, Zhang-Benoit Y, Goldring MB, Tanzer ML. Divergent stress responses to IL-1beta, nitric oxide, and tunicamycin by chondrocytes. J Cell Physiol. 2005;204:45–50. doi: 10.1002/jcp.20261. [DOI] [PubMed] [Google Scholar]
- 96.Takada K, et al. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int J Exp Pathol. 2011;92:232–242. doi: 10.1111/j.1365-2613.2010.00758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guo FJ, et al. ATF6 upregulates XBP1S and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage. Cell Signal. 2014;26:332–342. doi: 10.1016/j.cellsig.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 98.Liu Y, et al. XBP1S associates with RUNX2 and regulates chondrocyte hypertrophy. J Biol Chem. 2012;287:34500–34513. doi: 10.1074/jbc.M112.385922. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 99.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fukai A, Kamekura S, Chikazu D, Nakagawa T, Hirata M, Saito T, Hosaka Y, Ikeda T, Nakamura K, Chung UI, Kawaguchi H. Lack of a chondroprotective effect of cyclooxygenase 2 inhibition in a surgically induced model of osteoarthritis in mice. Arthritis Rheum. 2012;64:198–203. doi: 10.1002/art.33324. [DOI] [PubMed] [Google Scholar]
- 101.Vincent HK, Percival SS, Conrad BP, Seay AN, Montero C, Vincent KR. Hyaluronic Acid (HA) Viscosupplementation on Synovial Fluid Inflammation in Knee Osteoarthritis: A Pilot Study. Open Orthop J. 2013;7:378–84. doi: 10.2174/1874325001307010378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Henrotin Y, Lambert C, Richette P. Importance of synovitis in osteoarthritis: evidence for the use of glycosaminoglycans against synovial inflammation. Semin Arthritis Rheum. 2014;43:579–87. doi: 10.1016/j.semarthrit.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 103.Carames B, Kiosses WB, Akasaki Y, Brinson DC, Eap W, Koziol J, Lotz MK. Glucosamine activates autophagy in vitro and in vivo. Arthritis Rheum. 2013;65:1843–52. doi: 10.1002/art.37977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Leong DJ, Choudhury M, Hirsh DM, Hardin JA, Cobelli NJ, Sun HB. Nutraceuticals: potential for chondroprotection and molecular targeting of osteoarthritis. Int J Mol Sci. 2013 Nov 21;14(11):23063–85. doi: 10.3390/ijms141123063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Messier SP, et al. Effects of intensive diet and exercise on knee joint loads, inflammation, and clinical outcomes among overweight and obese adults with knee osteoarthritis: the IDEA randomized clinical trial. JAMA. 2013;310:1263–1273. doi: 10.1001/jama.2013.277669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Srinivas V, Bohensky J, Shapiro IM. Autophagy: a new phase in the maturation of growth plate chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells Tissues Organs. 2009;189:88–92. doi: 10.1159/000151428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Abou-Raya A, Abou-Raya S, Khadrawe T. Methotrexate in the treatment of symptomatic knee osteoarthritis: randomised placebo-controlled trial. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2013-204856. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 108.de Cabo R, Carmona-Gutierrez D, Bernier M, Hall MN, Madeo F. The search for antiaging interventions: from elixirs to fasting regimens. Cell. 2014 Jun 19;157(7):1515–26. doi: 10.1016/j.cell.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Uehara Y, Hirose J, Yamabe S, Okamoto N, Okada T, Oyadomari S, Mizuta H. Endoplasmic reticulum stress-induced apoptosis contributes to articular cartilage degeneration via C/EBP homologous protein. Osteoarthritis Cartilage. 2014 Jul;22(7):1007–17. doi: 10.1016/j.joca.2014.04.025. Epub 2014 May. [DOI] [PubMed] [Google Scholar]