

Abstract
Cutis laxa is a connective tissue disorder with distinctive lax, redundant, and inelastic skin. It is a genetically heterogenous disorder with autosomal dominant and recessive patterns of inheritance. We report a patient with cutis laxa supported by clinical, microscopic, and ultrastructural findings. Molecular analysis of fibulin-4 and -5, of the α2 subunit of the V-type H+ ATPase, and of the component of the oligomeric Golgi complex 7 (COG7) genes excluded the type I and type II autosomal recessive forms of cutis laxa, and congenital disorders of glycosylation associated with cutis laxa. Remarkably, our patient also presented severe and lethal pulmonary hypertension as a newborn. This case with cutis laxa, severe pulmonary hypertension, and no detectable mutations in fibulin-4 and -5 genes may represent a previously unrecognized syndrome.
Keywords: cutis laxa, primary pulmonary hypertension, FBLN4, FBLN5, COG7, ATP6V0A2
Introduction
Cutis laxa (CL) is a rare connective tissue disorder characterized by lax, redundant, and inelastic skin. CL is a genetically heterogenous condition including the autosomal dominant (ADCL; OMIM 123700) and autosomal recessive (ARCL types I and II; OMIM 219100 and 219200, respectively) forms. The autosomal dominant form is caused by mutations in the elastin (ELN) gene and presents with additional manifestations such as gastrointestinal diverticula, hernias, uterine prolapse, pulmonary artery stenosis, aortic and arterial dilatation and tortuosity, Raynaud's phenomenon, bronchiectasis, and emphysema (Szabo et al. 2006; Urban et al. 2005). Homozygous mutations in ELN have been recently reported in autosomal recessive CL (Megarbane et al. 2009). The autosomal recessive forms are clinically more severe and are often accompanied by pulmonary emphysema and cardiovascular complications, which can lead to death in childhood. ARCL type I is a specific, life-threatening disorder with organ involvement, lung atelectasis and emphysema, diverticulae of the gastrointestinal and genitourinary system, and vascular anomalies (arterial aneurysms, fibromuscular artery dysplasia and stenoses, including peripheral pulmonary stenoses). Although mutations have been detected in the fibulin-5 (FBLN5) and the fibulin-4 (FBLN4) genes in a few children with the severe forms of cutis laxa syndrome (Dasouki et al. 2007; Hucthagowder et al. 2006; Loeys et al. 2002), the underlying genetic etiology in the majority of cases is still unknown. ARCL type II is associated with joint laxity, developmental delay, pre- and post-natal growth delay, large fontanels with delayed closure, and congenital hip dislocation. Congenital disorder of glycosylation type IIe owing to a defect in conserved oligomeric Golgi complex 7 (COG7) is responsible for a form of cutis laxa associated with pulmonary hypertension, growth retardation, severe microcephaly, hypotonia, adducted thumbs, feeding problems due to gastrointestinal pseudo-obstruction, failure to thrive, cardiac anomalies, and episodes of extreme hyperthermia (Morava et al. 2007; Wu et al. 2004). Moreover, recessive mutations in the LTPB4 have recently been associated with a cutis laxa syndrome with impaired pulmonary, gastrointestinal, genitourinary and muskoloskeletal development (Urban et al. 2009). In addition, impaired glycosylation caused by mutations of the α2 subunit of the V-type H+ ATPase and mutations in the pyrroline-5-carboxylate reductase 1 gene have been described in several families with ARCL type II (Guernsey et al. 2009; Hucthagowder et al. 2009; Kornak et al. 2008).
Case report
We report a male born by a spontaneous vaginal delivery after 37 weeks of gestation complicated by oligohydramnios detected a week before delivery and intrauterine growth retardation (IUGR). The parents both from South America had a previous miscarriage and were not consanguineous. The family history was otherwise unremarkable. Apgar scores were 8 at one minute and 9 at five minutes. At birth, weight was 1755 grams (below the 3rd centile), length 46 cm (25th centile), and head circumference 30.5 cm (10th centile). Shortly after birth, the patient developed respiratory distress which worsened progressively during the first hours of life requiring oxygen treatment. An echocardiogram revealed pulmonary hypertension. From the second day of life he was maintained on oxygen and inhaled nitric oxide by nasal cannula. On the sixth day of life the patient exhibited a significant worsening of the respiratory distress and of the metabolic acidosis. He was intubated, placed on 100% oxygen and inhaled nitric oxide at 20 PPM. However, despite the aggressive treatment, the patient had escalating pulmonary pressures as measured by echocardiogram. From birth he was also noted to have skin laxity. The patient’s physical examination was also remarkable for the presence of blue sclerae, redundant skin on the nasal bridge, low-set right ear, and hooked nose. The skin was transparent with prominent venous pattern and redundant with reduced elastic recoil (Figure 1A and B). He also exhibited umbilical hernia and bilateral cryptorchidism. Upong percutaneous phlebotomy the patient's skin and deep tissues were noted to be fibrous and the veins were abnormally fragile blood. Over time, the patient became more difficult to oxygenate despite maximal ventilator settings and attempts to start the patient on high frequency oscillation were unsuccessful. The chest x-ray revealed a right lower lobe infiltrate consistent with pneumonia for which intravenous antibiotics were given. The patient also required pressor support to maintain adequate blood pressures. A 2D-echocardiogram showed increased pulmonary pressures which measured above the systemic pressure values consistent with severe pulmonary hypertension. The clinical condition of the patient continued to worsen over the next few days with progressive rising in blood lactate. Following discussion with the medical team and parents, intervention was withdrawn on day of life 11. Permission for autopsy was obtained. Clinical laboratory investigations obtained prior to death revealed minimally elevated serum copper (101 µg/dl) and ceruloplasmin (28.7 mg/dl). Chromosome analysis (46,XY) and chromosomal microarray analysis (genome coverage of the microarray can be found at http://www.bcm.edu/geneticlabs/cma/tables.html) were normal.
Figure 1.
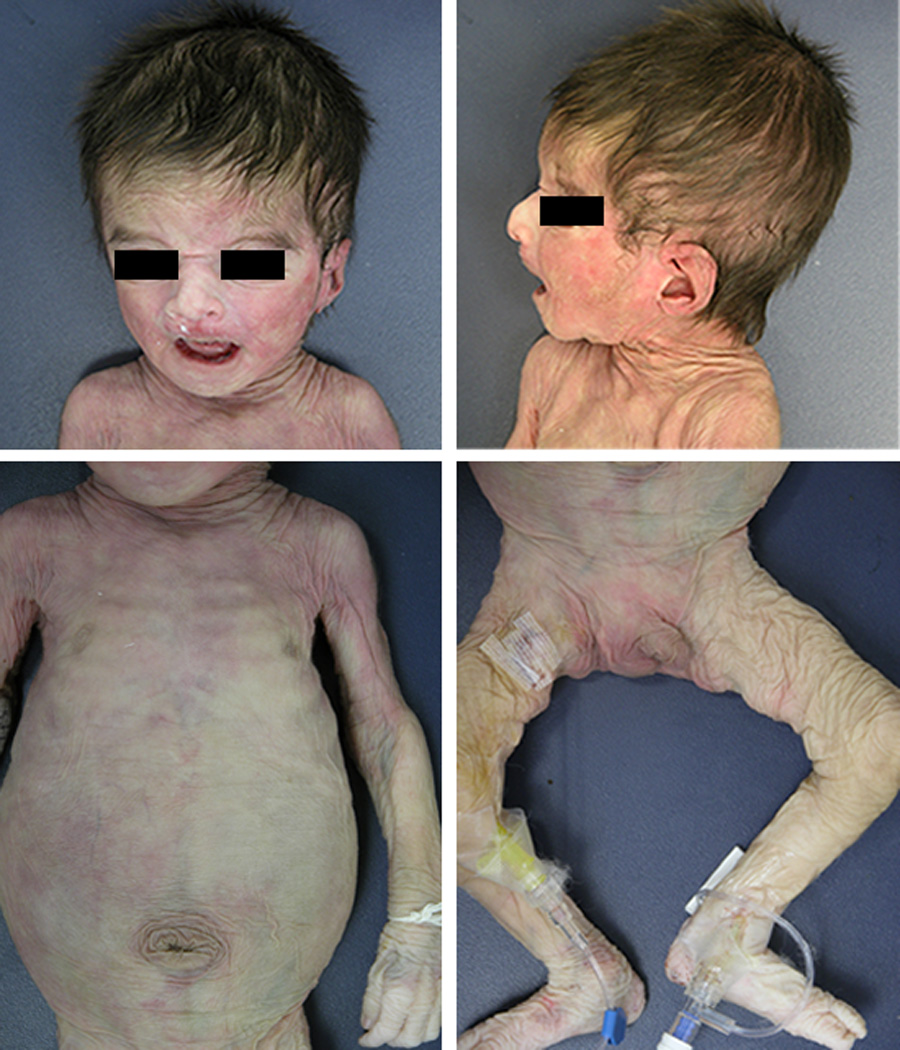
Post-mortem photograph. Note the redundancy of the skin, low-set right ear, hooked nose, and the umbilical hernia.
Methods
FBLN4, FBLN5, ATP6V0A2, COG7 molecular analyses
Mutational analysis of exons, intron/exon boundaries was performed on a genomic DNA sample of the patient by PCR and direct sequencing. Primers used for PCR amplification of FBLN4, FBLN5, ATP6V0A2, COG7 genes are available upon request.
Results
Pathological studies
Gross examination at autopsy revealed right ventricular hypertrophy, bilateral rib cartilage fusion between 10th–11th and 11th–12th ribs, and a normal appearance of the brain. The lung examination confirmed the presence of acute bronchopneumonia in the right middle lobe suspected on the chest X-ray. The lungs were congested with moderate to severe pulmonary arterial hypertensive changes and patchy parenchymal necrosis (Figure 2A and B). In addition, there was diffuse alveolar enlargement suggesting impaired lung growth and development. The severity of the pulmonary arteriopathy did not appear to be due to the alveolar enlargement alone. No microscopic abnormalities of the artery walls were detected. Histological examination of the skin showed an intact epidermis and a reduction in elastic fibers in papillary dermis and deep dermis (Figure 3). The hair appendages appeared normal. On ultrastructural examination elastic fiber fragmentation and increased granular substance were evident (Figure 4).
Figure 2.
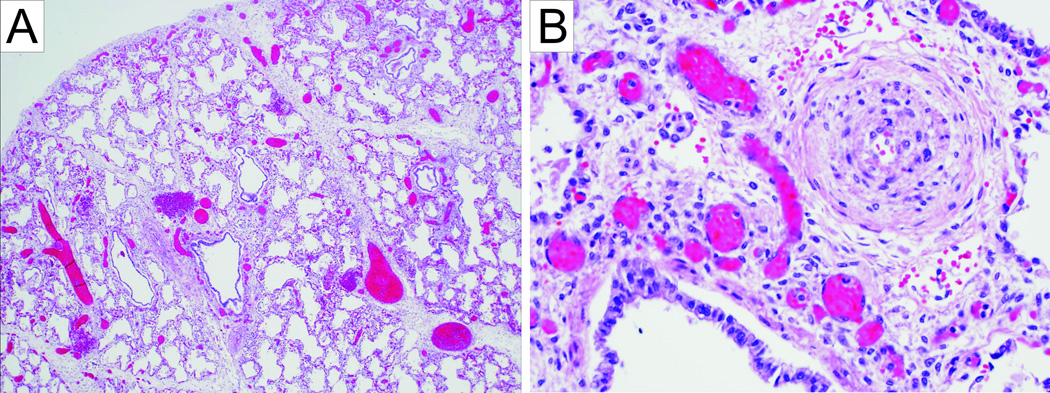
A. Enlargement of airspaces of the lung (10×). B. Lung tissue with severe proliferation of pulmonary-artery smooth muscle (50×).
Figure 3.

Hematoxylin-eosin staining of the skin (100×): intact epidermis and marked reduction in elastic fibers in papillary dermis and deep dermis in the patient (A) as compared to a normal control (B). Elastic stain of the skin of the patient shows decreased elastin fibers (C) as compared to a normal control (D) (200×).
Figure 4.
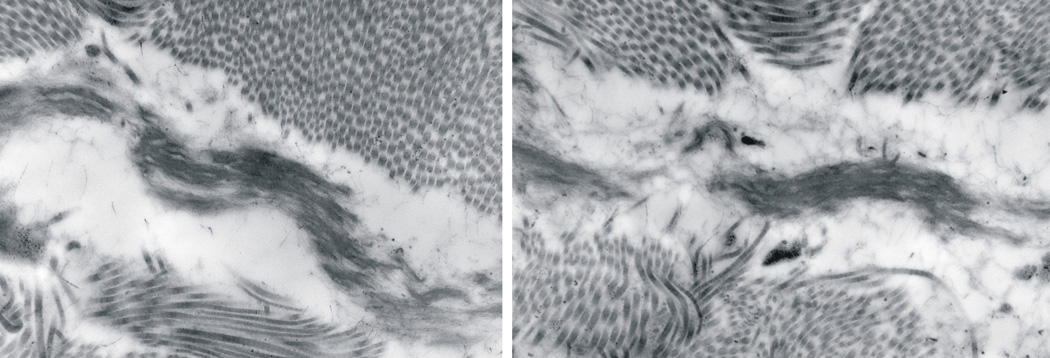
Electron microscopy of the skin showing elastic fibers fragmentation and increased granular substance.
FBLN4, FBLN5, ATP6V0A2, COG7 molecular analyses
No mutations were found in the coding sequences of the FBLN4, FBLN5, ATP6V0A2, and COG7 genes (data not shown).
Discussion
We report a patient with a clinical diagnosis of CL supported by microscopic and ultrastructural findings. By laboratory studies, we ruled out the autosomal recessive forms of CL type I due to FBLN4 and FBLN5 mutations, ARCL type II due to mutations of the α2 subunit of the V-type H+ ATPase. Blood sample for transferrin isoelectric focusing (IEF) was not obtained in our patient; however, results of transferrin IEF has been reported to be normal up to the age of 4 months even if affected individuals who later have diagnostic values (Morava et al. 2009). Therefore, we performed direct sequencing of the COG7 gene to rule out the form of CL due to congenital disorder of glycosylation. Sequence analysis of this gene was negative for mutation.
A neonatal lethal case of CL with arterial wall thickening, aneurysmal dilation of the ascending aorta and dissection of the pulmonary artery wall due to compound heterozygous mutations in FBLN4 has been previously reported (Dasouki et al. 2007). Importantly, mutations of the coding regions of the FBLN4 have been excluded in the patient we report. Stenoses of the pulmonary artery or its branches have been previously reported in children with CL (Hayden et al. 1968; Pandey et al. 2008; Tsuji et al. 1990; Weir et al. 1977) but to our knowledge, neonatally lethal pulmonary hypertension has not been described in CL.
Neonatal pulmonary hypertension is a rare, fatal condition defined by a sustained increase in pulmonary vascular resistance after birth resulting is right-to-left shunting of blood through fetal circulatory pathways. Severe cases can lead to intractable hypoxemia that fails to respond to conventional respiratory support. This condition can be associated with a variety of respiratory conditions such as meconium aspiration, pneumonia, and respiratory distress syndrome. However, the etiology of neonatal pulmonary hypertension is often undetermined (Rubin, 1997). Bone morphogenetic protein receptor type II (BMPR-II), a member of the transforming growth factor- β (TGF-β) receptor family has been involved in the familial form of primary pulmonary hypertension (PPHN) which is inherited in an autosomal dominant fashion with low penetrance (Deng et al. 2000; Lane et al. 2000; Machado et al. 2001; Thomson et al. 2000). Microscopically, PPHN is characterized by proliferation of pulmonary artery smooth muscle and endothelial cells, by intimal hyperplasia, and by in situ thrombus formation. All these features were also observed in our case (Figure 2B). We speculate that BPMR-II or other members of the TGF-β receptor family could be involved in our case because there are several experimental evidences linking genes involved in CL and those involved in PPHN. First, FBLN5 gene expression is regulated via TGF-β/Smad signaling (Kuang et al. 2006). Second latent TGF-β-binding protein 2 (LTBP-2) promote elastic fiber assembly by fibulin-5 (Hirai et al. 2007). Third, several cardiac and vascular abnormalities have been observed in fibulin-4 deficient mice (Hanada et al. 2007).
In summary, we report a patient with CL with associated neonatally lethal pulmonary hypertension which could represent a new form of CL. The description of additional cases with similar clinical presentation would help confirm and define the features of this newly recognized association.
Acknowledgments
We thank Don Singer for critical review of the manuscript. We are grateful to Prof Gert Mathijs for his support.
References
- Dasouki M, Markova D, Garola R, Sasaki T, Charbonneau NL, Sakai LY, Chu ML. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am J Med Genet A. 2007;143A:2635–2641. doi: 10.1002/ajmg.a.31980. [DOI] [PubMed] [Google Scholar]
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guernsey DL, Jiang H, Evans SC, Ferguson M, Matsuoka M, Nightingale M, Rideout AL, Provost S, Bedard K, Orr A, Dube MP, Ludman M, Samuels ME. Mutation in pyrroline-5-carboxylate reductase 1 gene in families with cutis laxa type 2. Am J Hum Genet. 2009;85:120–129. doi: 10.1016/j.ajhg.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Vermeij M, Garinis GA, de Waard MC, Kunen MG, Myers L, Maas A, Duncker DJ, Meijers C, Dietz HC, Kanaar R, Essers J. Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ Res. 2007;100:738–746. doi: 10.1161/01.RES.0000260181.19449.95. [DOI] [PubMed] [Google Scholar]
- Hayden JG, Talner NS, Klaus SN. Cutis laxa associated with pulmonary artery stenosis. J Pediatr. 1968;72:506–509. doi: 10.1016/s0022-3476(68)80341-5. [DOI] [PubMed] [Google Scholar]
- Hirai M, Horiguchi M, Ohbayashi T, Kita T, Chien KR, Nakamura T. Latent TGF-beta-binding protein 2 binds to DANCE/fibulin-5 and regulates elastic fiber assembly. Embo J. 2007;26:3283–3295. doi: 10.1038/sj.emboj.7601768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucthagowder V, Morava E, Kornak U, Lefeber DJ, Fischer B, Dimopoulou A, Aldinger A, Choi J, Davis EC, Abuelo DN, Adamowicz M, Al-Aama J, Basel-Vanagaite L, Fernandez B, Greally MT, Gillessen-Kaesbach G, Kayserili H, Lemyre E, Tekin M, Turkmen S, Tuysuz B, Yuksel-Konuk B, Mundlos S, Van Maldergem L, Wevers RA, Urban Z. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum Mol Genet. 2009;18:2149–2165. doi: 10.1093/hmg/ddp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucthagowder V, Sausgruber N, Kim KH, Angle B, Marmorstein LY, Urban Z. Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am J Hum Genet. 2006;78:1075–1080. doi: 10.1086/504304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornak U, Reynders E, Dimopoulou A, van Reeuwijk J, Fischer B, Rajab A, Budde B, Nurnberg P, Foulquier F, Lefeber D, Urban Z, Gruenewald S, Annaert W, Brunner HG, van Bokhoven H, Wevers R, Morava E, Matthijs G, Van Maldergem L, Mundlos S. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008;40:32–34. doi: 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- Kuang PP, Joyce-Brady M, Zhang XH, Jean JC, Goldstein RH. Fibulin-5 gene expression in human lung fibroblasts is regulated by TGF-beta and phosphatidylinositol 3-kinase activity. Am J Physiol Cell Physiol. 2006;291:C1412–C1421. doi: 10.1152/ajpcell.00087.2006. [DOI] [PubMed] [Google Scholar]
- Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- Loeys B, Van Maldergem L, Mortier G, Coucke P, Gerniers S, Naeyaert JM, De Paepe A. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet. 2002;11:2113–2118. doi: 10.1093/hmg/11.18.2113. [DOI] [PubMed] [Google Scholar]
- Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA, 3rd, Newman J, Williams D, Galie N, Manes A, McNeil K, Yacoub M, Mikhail G, Rogers P, Corris P, Humbert M, Donnai D, Martensson G, Tranebjaerg L, Loyd JE, Trembath RC, Nichols WC. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megarbane H, Florence J, Oliver Sass J, Schwonbeck S, Foglio M, de Cid R, Cure S, Saker S, Megarbane A, Fischer J. An Autosomal-Recessive Form of Cutis Laxa Is Due to Homozygous Elastin Mutations, and the Phenotype May Be Modified by a Heterozygous Fibulin 5 Polymorphism. J Invest Dermatol. 2009;129:1650–1655. doi: 10.1038/jid.2008.450. [DOI] [PubMed] [Google Scholar]
- Morava E, Guillard M, Lefeber DJ, Wevers RA. Autosomal recessive cutis laxa syndrome revisited. Eur J Hum Genet. 2009;17:1099–1110. doi: 10.1038/ejhg.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morava E, Zeevaert R, Korsch E, Huijben K, Wopereis S, Matthijs G, Keymolen K, Lefeber DJ, De Meirleir L, Wevers RA. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur J Hum Genet. 2007;15:638–645. doi: 10.1038/sj.ejhg.5201813. [DOI] [PubMed] [Google Scholar]
- Pandey R, Garg R, Manikandan R, Punj J, Darlong V, Singh SA. Perianesthetic management of generalized congenital cutis laxa syndrome associated with pulmonary stenosis undergoing inguinal hernia repair. Paediatr Anaesth. 2008;18:907–909. doi: 10.1111/j.1460-9592.2008.02564.x. [DOI] [PubMed] [Google Scholar]
- Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–117. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- Szabo Z, Crepeau MW, Mitchell AL, Stephan MJ, Puntel RA, Yin Loke K, Kirk RC, Urban Z. Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene. J Med Genet. 2006;43:255–258. doi: 10.1136/jmg.2005.034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, Ward K, Yacoub M, Mikhail G, Rogers P, Newman J, Wheeler L, Higenbottam T, Gibbs JS, Egan J, Crozier A, Peacock A, Allcock R, Corris P, Loyd JE, Trembath RC, Nichols WC. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet. 2000;37:741–745. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji A, Yanai J, Miura T, Shirai Y, Osano M, Hosoda Y, Sato M, Asaishi T, Oda Y, Hajikano H. Vascular abnormalities in congenital cutis laxa--report of two cases. Acta Paediatr Jpn. 1990;32:155–161. doi: 10.1111/j.1442-200x.1990.tb00802.x. [DOI] [PubMed] [Google Scholar]
- Urban Z, Gao J, Pope FM, Davis EC. Autosomal dominant cutis laxa with severe lung disease: synthesis and matrix deposition of mutant tropoelastin. J Invest Dermatol. 2005;124:1193–1199. doi: 10.1111/j.0022-202X.2005.23758.x. [DOI] [PubMed] [Google Scholar]
- Urban Z, Hucthagowder V, Schurmann N, Todorovic V, Zilberberg L, Choi J, Sens C, Brown CW, Clark RD, Holland KE, Marble M, Sakai LY, Dabovic B, Rifkin DB, Davis EC. Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am J Hum Genet. 2009;85:593–605. doi: 10.1016/j.ajhg.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir EK, Joffe HS, Blaufuss AH, Beighton P. Cardiovascular abnormalities in cutis laxa. Eur J Cardiol. 1977;5:255–261. [PubMed] [Google Scholar]
- Wu X, Steet RA, Bohorov O, Bakker J, Newell J, Krieger M, Spaapen L, Kornfeld S, Freeze H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med. 2004;10:518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]