

Abstract
Neutrophils play a central role in the innate immune response and a critical role in bacterial killing. Most studies of neutrophil function have been conducted under conditions of ambient oxygen, but inflamed sites where neutrophils operate may be extremely hypoxic. Previous studies indicate that neutrophils sense and respond to hypoxia via the ubiquitous prolyl hydroxylase/hypoxia-inducible factor pathway and that this can signal for enhanced survival. In the current study, human neutrophils were shown to upregulate hypoxia-inducible factor (HIF)-1α–dependent gene expression under hypoxic incubation conditions (3 kPa), with a consequent substantial delay in the onset of apoptosis. Despite this, polarization and chemotactic responsiveness to IL-8 and fMLP were entirely unaffected by hypoxia. Similarly, hypoxia did not diminish the ability of neutrophils to phagocytose serum-opsonized heat-killed streptococci. Of the secretory functions examined, IL-8 generation was preserved and elastase release was enhanced by hypoxia. Hypoxia did, however, cause a major reduction in respiratory burst activity induced both by the soluble agonist fMLP and by ingestion of opsonized zymosan, without affecting expression of the NADPH oxidase subunits. Critically, this reduction in respiratory burst activity under hypoxia was associated with a significant defect in the killing of Staphylococcus aureus. In contrast, killing of Escherichia coli, which is predominantly oxidase independent, was fully preserved under hypoxia. In conclusion, these studies suggest that although the NADPH oxidase-dependent bacterial killing mechanism may be compromised by hypoxia, neutrophils overall appear extremely well adapted to operate successfully under severely hypoxic conditions.
Introduction
Cells of the innate immune system, such as neutrophils and macrophages, are required to infiltrate and operate efficiently within highly adverse environments, yet still kill invading pathogens and prevent the systemic spread of infection. Neutrophils accumulate rapidly at such infective foci, which are characterized by low oxygen tensions (1,2). This has led investigators to examine oxygen sensing and hypoxic regulation in these inflammatory cells. Such studies are informed by the observation that neutrophils depend principally on glycolysis rather than oxidative metabolism for ATP generation (and consequently have rich glycogen stores) but also need to consume oxygen to maintain the NADPH oxidase-driven respiratory burst, which generates reactive oxygen species (ROS) required for killing certain bacteria (3–5). Thus lack of molecular oxygen at sites of infection may influence profoundly the interaction between the invading pathogen and the host phagocytic cell tasked with eliminating or confining infection.
Oxygen sensing is now recognized as a universal property of all mammalian cells and is essential for physiological adaptation. It is signaled via a well-characterized pathway involving a family of oxygen-dependent hydroxylase enzymes, which regulate the cellular levels and transcriptional activity of hypoxia-inducible factor (HIF). This latter molecule is a heterodimeric protein composed of a constitutively expressed β subunit and a hydroxylase-targeted HIF-1, -2 or -3α subunit. HIF plays a fundamental role in directing the cell’s response to hypoxia, in part through modulation of multiple HIF effector genes containing hypoxia response elements. Neutrophils express a number of the HIF-regulating hydroxylases, including the prolyl hydroxylase domain-containing enzymes PHD1–3 and factor-inhibiting HIF (6,7). This pathway is clearly activated at physiologically relevant levels of hypoxia and results in profound inhibition of neutrophil apoptosis under hypoxia or anoxia (6,8). Studies using myeloid targeted HIF-1α knockout mice also reveal a central role for HIF-1α in maintaining neutrophil ATP levels, and as a consequence, preserving myeloid cell recruitment and bacterial killing (9–11). Recent studies have revealed the capacity for additional nonhypoxic regulation of HIF-1α, for example, by IKKβ (12), and for agents such as LPS to upregulate HIF-1α levels in inflammatory cells (13). The interdependence of innate immunity and the response to hypoxia is further underscored by the finding that pathogen ingestion likewise leads to HIF-1α accumulation in the phagocyte (10), conferring enhanced bactericidal activity. However, studies involving the manipulation of cellular HIF-1α levels do not fully recapitulate the potential for lack of molecular oxygen to influence phagocyte function directly, particularly with regard to the ability to mount an oxidative burst.
Despite such data and parallel clinical and animal studies showing compromised bacterial killing and wound healing under hypoxia (14,15), very few studies have examined the effects of acute hypoxia on neutrophil function in vitro. In this paper, we show that hypoxia impairs the capacity of human peripheral blood neutrophils to generate ROS and kill Staphylococcus aureus but that hypoxic challenge does not compromise their motility, migration, receptor regulation, or degranulation responses. Indeed, fMLP-stimulated elastase release was enhanced under hypoxia, which may promote tissue infiltration and potentiate histotoxic tissue injury. These data confirm the remarkable adaptation of neutrophils to hypoxic environments but importantly also reveal a selective impairment of oxidase-dependent bacterial killing under hypoxia.
Materials and Methods
Isolation and culture of peripheral blood neutrophils from healthy human volunteers
Neutrophils were purified by dextran sedimentation and discontinuous plasma-Percoll gradients as detailed previously (16). Purified cells were resuspended at 1–45 × 106 cells/ml in HBSS (Sigma-Aldrich, Ayrshire, U.K.), PBS (Sigma-Aldrich), or IMDM (Life Technologies, Invitrogen, Paisley, U.K.). Neutrophils were routinely >98% pure, and care was taken to avoid inadvertent priming of the neutrophils during purification. Hypoxia was established by resuspending cells in media that had been pre-equilibrated using an InvivO2 400 hypoxic work station with separate feeds of compressed air, N2, CO2, and 10% H/90% N2 (Ruskinn, Bridgend, U.K.); the CO2/N2 proportions were adjusted depending on the media buffering system. Normoxic incubations used IMDM equilibrated in a humidified 5% CO2/air incubator (Forma Series II 3111, Forma Scientific, Marietta, OH) or PBS and HBSS equilibrated under ambient atmospheric conditions at 37°C. The PO2, PCO2, and pH of the media were measured at the beginning and end of each experiment (ABL5 Blood Gas Analyzer; Radiometer, Copenhagen, Denmark) to confirm the delivery of a consistent hypoxic (or normoxic) environment and to ensure that appropriate pH levels were maintained. These studies were approved by the Cambridge Research Ethics Committee (06/Q0108/281).
Assessment of neutrophil apoptosis
To assess the effects of hypoxia on apoptosis, purified neutrophils were resuspended at 5 × 106 cells/ml in IMDM supplemented with 10% autologous serum and 50 U/ml streptomycin and penicillin and cultured in Falcon Flexiwell 96-well plate inserts as described (17). Neutrophils were harvested at 20 h, cytocentrifuged, fixed in methanol, stained with May-Grünwald-Giemsa (Merck, Nottingham, U.K.), and morphology was examined by oil-immersion light microscopy. Apoptotic neutrophils were defined as those with darkly stained pyknotic nuclei. Apoptosis was also assessed by flow cytometry using 1) FITC-labeled recombinant human annexin V (Annexin VFITC)/propidium iodide staining (18) and 2) the fluorescent cationic dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzamidazolocarbocyanin iodide (JC-1) (19).
TaqMan real-time PCR and Western blotting
Total RNA was isolated using TRI reagent (Sigma-Aldrich) followed by RNA purification with DNase digest, using the RNeasy microcolumn kit (Qiagen, Crawley, U.K.). cDNA was generated from 1 μg total RNA, using a High Capacity cDNA Kit (Applied Biosystems, Foster City, CA), and quantitative PCR (qPCR) (iCycler; Bio-Rad, Milpitas, CA) was performed using SYBR Green Master Mix (Sigma-Aldrich) with the appropriate primers purchased from Qiagen. Relative gene expression was determined by correcting the cycle threshold for the target gene against four housekeeping genes (β2-microglobulin, β-actin, YWHAZ, UBC) using genNORM (http://medgen.ugent.be/~jvdesomp/genorm); each of these had been determined to be stable in human neutrophils under the test conditions employed (data not shown). The ΔCT for the target gene of interest in control, normoxic, and hypoxic-stimulated neutrophils was corrected against the value obtained in freshly isolated neutrophils to give ΔΔCT values. Relative gene expression (fold change) is expressed as 2−ΔΔCT.
Neutrophil shape change and chemotaxis
For determination of shape change, neutrophils (5 × 106/ml in HBSS) were incubated under normoxic or hypoxic conditions in BD Falcon flexible 96-well plates (BD Biosciences, Durham, NC) and treated with IL-8 (1 ng/ml–1 μg/ml; R&D Systems, Minneapolis, MN), fMLP (0.1–10 nM; Sigma-Aldrich), or buffer for 5 min while being maintained in the same oxygen environment. The cell suspension (150 μl) was aspirated and added to 250 μl 8.5% v/v CellFix (BD Biosciences, Erembodegem, Belgium), 16.5% distilled H2O, and 75% v/v PBS (optimized CellFix), and analyzed by flow cytometry (FACSCalibur; BD Biosciences, Paisley, U.K.). Data were analyzed to generate both mean forward scatter values and percentage shape change for the gated neutrophils. In addition, aliquots of the fixed cells were examined by microscopy using a hemocytometer and cells scored as shape changed if they contained >1 cell surface bleb or irregularity (20). A minimum of 100 cells were counted in triplicate for each experimental point.
Neutrophil chemotaxis was measured using Neuro Probe ChemoTx microplates (Neuro Probe, Receptor Technologies, Adderbury, U.K.), as described (21). In brief, microplate wells were filled in triplicate with 30 μl media alone (IMDM plus 10% autologous serum) or varying concentrations of IL-8. The framed nitrocellulose membrane (5-μm pores, 4000/cm2) was applied and the solutions allowed to equilibrate at the appropriate oxygen tension for 1 h. Neutrophils (5 × 106/ml in IMDM containing 10% autologous serum) were cultured concurrently in normoxia or hypoxia, and 50 μl cell suspension was then added to the upper aspect of the membrane. After a further incubation for 1.5 h, the membrane was removed and the lower wells aspirated and washed twice with 30 μl 0.05% trypsin/0.53 mM EDTA in PBS; the latter step was incorporated to ensure full cell recovery. The number of cells migrating to the lower wells was quantified using a hemocytometer.
Determination of neutrophil CD11b, CD16, CD62L, IL-8R, and IL-8RB expression
Neutrophils (5 × 106/ml in HBSS) were incubated under normoxic or hypoxic conditions for 1 or 4 h prior to stimulation with fMLP (100 nM), GM-CSF (10 ng/ml), TNF-α (10 ng/ml), or buffer for 30 min. The reactions (100 μl) were stopped by the addition of 250 μl ice cold optimized BD CellFix and placed on ice. Aliquots were incubated with anti-human CD11b FITC conjugates (10 μg/ml, Clone Bear1; Beckman Coulter, High Wycombe, U.K.) and anti-human CD16 R-phycoerythrin conjugates (10 μg/ml, Clone DJ130c; DAKO, Ely, U.K.), or anti-human IL-8RA or IL-8RB FITC conjugated Abs (R&D Systems Clone 42705, 50 μg/ml, and Clone 48311, 50 μg/ml, respectively), in the dark for 30 min on ice. Samples were then diluted with 2 ml ice cold PBS and analyzed immediately using a BD FACSort flow cytometer (BD Biosciences, Oxford, U.K.) and FSC Press software (FSC Press, Cambridge, U.K.) (18). Isotype-matched controls and single-stained samples were used to set baselines and compensation.
Phagocytosis assays
Streptococcus pneumoniae type 14 organisms were cultured to log phase in Todd-Hewitt broth containing 0.5% yeast extract (Oxoid, Basingstoke, U.K.), heat inactivated at 60°C for 1 h, and labeled with FITC, as detailed previously (22). Neutrophils were allowed to equilibrate at the appropriate oxygen tension and temperature (37°C or 4°C) for 1 or 3 h prior to the addition of the bacteria (52 streptococci per neutrophil, pre-opsonized in 50% [v/v] pooled heat-inactivated human serum [Sigma-Aldrich] for 30 min at 37°C). The neutrophils were then washed three times in ice cold PBS, fixed in optimized CellFix, and analyzed by flow cytometry. The percentage of FITC+ neutrophils and the geometric mean fluorescence of the FITC+ cells were used as measures of phagocytosis, with uptake at 4°C used to control for bacterial adhesion. For electron microscopy, neutrophils were incubated with streptococci exactly as above and processed as previously described (6).
Determination of neutrophil elastase and IL-8 secretion
To quantify spontaneous and stimulated elastase release, neutrophils (1 × 106/ml in 90 μl) were incubated in pre-equilibrated HBSS in normoxia or hypoxia for 1 or 4 h. Cytochalasin B (5 μg/ml; Sigma-Aldrich) or buffer was added to the cells for 5 min, followed by fMLP (100 nM) or buffer for 10 min. After centrifugation (10,000 g; 10 s), the supernatants were collected and stored at −80°C, and elastase was measured using a commercial human elastase ELISA kit (Hycult Biotechnology, Uden, The Netherlands) according to the manufacturer’s instructions.
To assess the effects of hypoxia on IL-8 secretion, neutrophils (5 × 106 cells/ml) were incubated under normoxia or hypoxia in IMDM for 30 min prior to the addition of buffer, GM-CSF (10 ng/ml), and/or TNF-α (10 ng/ml) for a further 2, 4, 6, or 20 h. IL-8 was assayed in cell-free supernatants, using an in-house ELISA as detailed (23). To examine the effects of hypoxia on the release of a wider range of inflammatory cytokines and chemokines, neutrophils (11.6 × 106/ml) were incubated under hypoxia or normoxia for 30 min, prior to stimulation with LPS (LPS, 100 ng/ml) plus LPS binding protein 100 ng/ml for a further 3.5 h. The reaction was terminated by the addition of normoxic or hypoxic HBSS (800 μl) and harvest of the supernatants. The supernatants were analyzed using commercial Ab arrays (RayBio Human Chemokine Ab Array 1 and The RayBio Human Inflammation Ab Array 3; Raybiotech, Norcross, GA) according to the manufacturer’s instructions.
Assessment of NADPH oxidase activity
Extracellular superoxide anion generation was determined by resuspending purified neutrophils in hypoxic or normoxic HBSS at 11.1 × 106/ml and incubating for 1 or 3 h in the appropriate oxygen atmosphere prior to the addition of 100 nM fMLP or 200 nM PMA for 30 min. GM-CSF (10 ng/ml) was added 1 h prior to fMLP to ensure optimal priming of the response. The superoxide dismutase-inhibitable reduction of cytochrome c (Sigma-Aldrich) was quantified as detailed previously (24).
For assessment of hypoxia/reoxgygenation, neutrophils were incubated as above under normoxia or hypoxia in the presence or absence of GM-CSF 10 ng/ml. After 1 h, either 1) the fMLP-stimulated superoxide dismutase-inhibitable reduction of cytochrome C was quantified, or 2) unprimed cells were further incubated under normoxic or hypoxic conditions or were transferred from hypoxia to normoxia (reoxygenated cells) prior to treatment with GM-CSF 10 ng/ml for 2 h and assessment of the fMLP-stimulated oxidative burst by reduction of cytochrome c, as above.
For measuring intracellular ROS generation, neutrophils were resuspended at 1 × 107/ml in normoxic or hypoxic HBSS and incubated for a further 1 or 3 h under these conditions prior to the addition of 3 μM diphenyleneiodonium (DPI) (Sigma-Aldrich) or buffer for 5 min, followed by 0.2 μM 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Molecular Probes, Invitrogen, Paisley, U.K.) for 10 min. Aliquots of these cell suspensions (225 μl) were added to 25-μl aliquots PBS containing 22.5 × 106 particles of opsonized (50% [v/v] pooled heat-inactivated human serum, 30 min, 37°C) zymosan (10 zymosan particles/neutrophil). Incubations were terminated after 60 min by the addition of 250-μl volumes ice cold optimized CellFix. Cell suspensions were diluted in ice cold PBS and analyzed by flow cytometry (FACSCalibur; BD Biosciences).
To determine the effects of hypoxia on pyocyanin-mediated ROS generation, neutrophils were incubated (5 × 106/ml in RPMI 1640) under normoxic or hypoxic conditions in the presence or absence of 50 μM pyocyanin [prepared by the photolysis of phenazine methosulfate; Sigma, Poole, U.K.; as described (25)] for 30 min prior to the assessment of the oxidative burst exactly as before (25).
Western blotting for NADPH oxidase components
Neutrophils were incubated at 1 × 107/ml in normoxic or hypoxic HBSS for 4 h, and 2 × 107 cells per condition lysed in 1 ml ice cold buffer (0.1% Triton X-100, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 3 mM β-glycerophosphate, 30 mM NaF, 0.2% cholate, with leupeptin, aprotinin, and antipain, each at 2.5 μg/ml). After further incubation on ice for 30 min, the cells were sonicated and spun (5 min; 15,000 g), and 50 μg of each sample was analyzed by SDS/PAGE, as described (26).
Neutrophil killing of S. aureus and Escherichia coli
S. aureus (subspecies aureus rosenbach; Wood 46, ATTC-LGC Standard, Teddington, U.K.) organisms were subcultured overnight, grown to logarithmic phase, and opsonized by incubation at 37°C in 50% pooled human serum (Sigma-Aldrich). The bacteria were washed twice and resuspended in 0.9% NaCl at a density of 80 × 106 S. aureus per milliliter prior to addition to the cells. Neutrophils were incubated in HBSS at 30 × 106/ml under hypoxia or normoxia for 1 h before addition of 3 μM DPI or buffer. After 5 min, neutrophils were incubated with S. aureus for 1 h at a preoptimized ratio of 1 bacterium per 6 neutrophils. The reaction was terminated by adding 50 μl cell suspension to 2.5 ml distilled H2O (pH 11) for 5 min to achieve neutrophil lysis. Samples were diluted serially in Luria-Bertani broth (Sigma-Aldrich), and 100-μl aliquots were spread on 10-cm agar plates and left for overnight incubation at 37°C. To determine the effects of reoxygenation, neutrophils were incubated under normoxia or hypoxia, as above, for 1 h prior to transferring the hypoxic cells to normoxic conditions for 30 min and assessment of killing, as above. Data were expressed as S. aureus CFUs per plate.
To assess the effects of hypoxia on bacterial-induced neutrophil necrosis, pre-equilibrated (1 h) normoxic or hypoxic neutrophils (5 × 106/ ml in 50 μl RPMI 1640 with 10% heat-inactivated serum) were incubated with S. aureus (multiplicity of infection, 0.5) for 3 h at 37°C, and the absolute number of intact neutrophils was quantified using CountBright absolute counting beads (Molecular Probes, Invitrogen) according to the manufacturer’s instructions.
Escherichia coli (E2348169; gift from Dr. Keith Davidson, Babraham Institute, Cambridge, U.K.) organisms were subcultured overnight, grown to logarithmic phase, and opsonized by incubation at 37°C in 50% (v/v) pooled heat-inactivated human serum (Sigma-Aldrich). The bacteria were washed twice and resuspended in 0.9% NaCl at a density of 180 × 106 E. coli/ml before addition to the cells. Neutrophils (45 × 106/ml in HBSS) were incubated under hypoxia or normoxia for 1 h prior to the addition of 3 μM DPI or buffer for 5 min and then treated with E. coli for 1 h at a previously optimized ratio of 1 bacterium per neutrophil. Neutrophils were then lysed and E. coli CFU quantified, as detailed above. Data are expressed as E. coli CFU per plate.
Statistical analysis
Data are reported as mean ± SEM from (n) independent experiments and analyzed using two-way ANOVA with a Bonferroni post-test using Prism 5.0 software (GraphPad, San Diego, CA). For comparison of two sample means, paired Student t tests were used. A p value < 0.05 was considered significant.
Results
Hypoxia delays apoptosis and upregulates HIF-1α–dependent gene expression in human neutrophils
The PO2, PCO2, and pH of the media were measured at the beginning and end of each experiment (ABL5 Blood Gas Analyzer, Radiometer) and confirmed the delivery of a consistent normoxic or hypoxic environment with equivalent pH in the various media employed (Fig. 1A, Table I). In agreement with our previous observations (6,8), the degree of physiological hypoxia used in this study resulted in a significant delay in constitutive neutrophil apoptosis assessed after 20 h, using cell morphology, or Annexin VFITC/propidium iodide or JC-1 staining quantified by flow cytometry (Fig. 1B).
FIGURE 1.

Validation of hypoxic incubation. A, PO2 and pH of HBSS maintained under normoxia or hypoxia were analyzed using an ABL 500 blood gas analyzer [mean ± SEM of (n) = 20 independent experiments]. *p < 0.05 for hypoxia versus normoxia. B, The degree of hypoxia attained is sufficient to delay apoptosis. Neutrophils were incubated for 20 h in IMDM containing 10% autologous serum under normoxic or hypoxic conditions, and apoptosis was assessed by Annexin VFITC staining, JC-1 fluorescence, or cell morphology, as indicated. Data represent mean ± SEM of (n) = 3 independent experiments, each performed in triplicate. *p < 0.05 for hypoxia versus normoxia. C, Hypoxic incubation leads to induction of HIF-dependent transcription. BNIP3 mRNA was quantified by qPCR in neutrophils incubated under either normoxia or hypoxia for 1–4 h. Gene expression relative to the level at 30 min of hypoxic incubation (fold change) is expressed as 2−ΔΔCT (see Materials and Methods for details) and represent mean ± SD of (n) = 3 independent experiments, each performed in triplicate. *p < 0.05 for hypoxia versus normoxia.
Table 1.
Analysis of media pH, pO2, and pCO2
| Normoxia | Hypoxia | ||
|---|---|---|---|
| PBS | |||
| pH | 7.2 ± 0.1 | 7.16 ± 0.2 | |
| pCO2 | 0.37 ± 0.1 | 0.32 ± 0.1 | |
| pO2 | 25.98 ± 4.5 | 3.07 ± 0.6 | |
| RPMI | |||
| pH | 7.45 ± 0.1 | 7.37 ± 0.0 | |
| pCO2 | 3.25 ± 0.5 | 3.93 ± 0.3 | |
| pO2 | 21.56 ± 1.98 | 2.65 ± 0.5 | |
| HBSS | |||
| pH | 7.42 ± 0.1 | 7.41 ± 0.0 | |
| pCO2 | 0.87 ± 0.0 | 0.92 ± 0.0 | |
| pO2 | 23.70 ± 0.3 | 3.04 ± 0.0 | |
| IMDM | |||
| pH | 7.44 ± 0.0 | 7.39 ± 0.0 | |
| pCO2 | 3.87 ± 0.1 | 4.62 ± 0.1 | |
| pO2 | 20.27 ± 1.9 | 3.48 ± 0.1 |
Media type indicated was incubated under normoxia or hypoxia for 1–4 h in a 10-cm petri dish. A sample of each individual medium was taken using a 1-ml syringe with an airtight cap, and assessed immediately using an ABL 500 blood gas analyzer.
Data represent the mean 6 SEM, where n≥20.
To confirm that these incubation conditions were sufficient to activate the PHD/factor-inhibiting HIF hydroxylase/HIF-1α pathway, we examined the abundance of BNIP3 and IκB mRNA, two genes that are recognized to be regulated by hypoxia and HIF-1 in neutrophils (6), together with BclA1 and Bcl-XL, which are upregulated in response to hypoxic activation of the NF-κB pathway (27). Fig. 1C shows the substantial and time-dependent increase in BNIP3 mRNA abundance observed in purified neutrophils under hypoxia. Under identical conditions, hypoxia also caused a significant increase in IκB, BclA1, and Bcl-XL mRNA levels without affecting the chosen housekeeping genes (data not shown).
Neutrophil shape change and chemotaxis under hypoxia
Exposure of circulating neutrophils to systemic or locally generated chemotactic agents represents one of the earliest responses of these cells to an inflammatory event. To determine whether neutrophil polarization induced by nongradient chemokine challenge, or directional migration to a chemotactic gradient, was influenced by hypoxia, we examined neutrophil shape change and chemotaxis in response to IL-8. Both of these assays have been validated extensively in neutrophils by our and other groups (20,21,28). As shown in Fig. 2A–C, preincubation of neutrophils under hypoxia (3 kPa) for 1 h modified neither the magnitude nor the potency of these responses, which were both assayed under continued normoxic or hypoxic exposure. Fig. 2D demonstrates equal expression of IL-8RA and IL-8RB under normoxic and hypoxic conditions, as well as equal receptor shedding in response to GM-CSF and TNF-α stimulation. Responses to the bacterial tripeptide fMLP and to LPS were likewise unaffected by hypoxia (data not shown). These data indicate that the capacity of neutrophils to sense, polarize, and migrate toward a chemotactic stimulus is not affected by hypoxia.
FIGURE 2.

Effects of hypoxia on neutrophil shape change and chemotaxis in response to IL-8. A and B, IL-8–induced neutrophil shape change is not affected by hypoxia. Neutrophils were incubated under normoxia or hypoxia for 1 h prior to the addition of the indicated concentration of IL-8 for 5 min prior to fixation. Representative histogram (A) illustrating neutrophil shape change induced by IL-8 10−7 g/ml for 5 min and assessed by flow cytometry. Concentration response (B) of neutrophil shape change induced by IL-8 (0–10−5 g/ml) under conditions of normoxia or hypoxia assessed by flow cytometry. Results represent mean ± SEM of n = 3 independent experiments performed in triplicate. C, IL-8–induced neutrophil chemotaxis is maintained under hypoxia. Neutrophils (5 × 106/ml in IMDM containing 10% autologous serum) were preincubated for 1 h in a normoxic or hypoxic environment, and migration through a nitrocellulose membrane (5-μm pores, 4000/cm2) in response to the indicated concentrations of IL-8 was assessed after 1.5 h by manual counting. Results represent mean ± SEM of n = 3 independent experiments performed in triplicate. D, Expression of IL-8 receptors is unaffected by hypoxia. IL-8RA and IL-8RB surface expression determined by flow cytometry. Neutrophils were preincubated for 1 h under normoxia or hypoxia, and stimulated (or not) with TNF-α 10 ng/ml plus GM-CSF 10 ng/ml for 30 min prior to fixing and staining with FITC-tagged monoclonal Abs, as described in Materials and Methods. Results represent mean ± SEM of n = 3 independent experiments performed in triplicate.
Preserved neutrophil phagocytosis under hypoxia
The sialoglycoprotein CD11b plays a key role in neutrophil adhesion to endothelial surfaces and in the cell’s recognition and binding of streptococci (29,30). CD11b surface expression and avidity are also upregulated following neutrophil priming or activation (24), a response that involves significant cytoskeletal rearrangement (29). In view of this, we examined initially the ability of fMLP to upregulate the cell surface expression of CD11b and found this response to be entirely preserved under hypoxic conditions at early time points and even enhanced, compared with normoxic cells after 4 h of incubation (Fig. 3A). Basal and fMLP-stimulated CD11b surface expression varied somewhat between donors, but at 4 h there was a clear increase in CD11b detection in unstimulated hypoxic cells relative to their normoxic controls (Fig. 3A). At these later time points, normoxic neutrophils progressively lost their ability to upregulate CD11b in response to fMLP, but this response was preserved when the cells were maintained in a hypoxic environment (Fig. 3A) and was also seen when LPS was used as a stimulus (data not shown). CD16 (FcγRIII) expression declined in a time-dependent manner but over 4 h was not influenced by oxygen tension (Fig. 3B).
FIGURE 3.

Effect of hypoxia on neutrophil phagocytic function. A and B, Hypoxia preserves neutrophil surface expression of CD11b, but not CD16. Neutrophils were incubated under normoxic or hypoxic conditions for 1–4 h prior to stimulation with fMLP (100 nM) or buffer. Fixed cells were stained with FITC-labeled anti-CD11b (A) or anti-CD16 (B), as described, and analyzed by flow cytometry. Data represent mean ± SEM of n = 4 independent experiments performed in triplicate. *p < 0.05. Owing to donor variability in baseline expression, results were normalized to normoxic controls. C–F, Hypoxia does not compromise the ability of neutrophils to phagocytose opsonized streptococci. Neutrophils were preincubated under normoxia or hypoxia for 1 or 3 h prior to the addition of opsonized FITC-labeled S. pneumoniae for 1 h; cells kept at 4°C were used to control for nonspecific adhesion. Representative histograms (C) illustrating phagocytosis of FITC-labeled S. pneumoniae by normoxic and hypoxic neutrophils (1 h) assessed by flow cytometry. Electron microscope image (D) of a neutrophil (osmium tetroxide stained, original magnification ×18,000) containing ingested bacteria; arrows point to S. pneumoniae within phagosomes. Phagocytosis of FITC-labeled S. pneumonia did not differ when neutrophils were maintained under normoxia or hypoxia for 1 or 3 h, assessed by % of FITC-positive neutrophils (E) or mean fluorescence intensity (F). Data represent mean ± SEM of three independent experiments performed in triplicate.
As shown in Fig. 3C–E, incubation of neutrophils with heat-inactivated FITC-labeled opsonized streptococci at a preoptimized density of 52 organisms per cell for 60 min resulted in phagocytic uptake by ~50% of cells. Prior incubation of neutrophils in hypoxia (3 kPa) for 1 or 3 h had no significant effect on either the number of cells ingesting streptococci or the number of ingested organisms per cell (Fig. 3E, 3F), again with stable hypoxia maintained for the duration of the experiment. These data indicate that neutrophil recognition and phagocytosis of microorganisms, such as S. pneumoniae, are remarkably well preserved under hypoxia.
Effects of hypoxia on neutrophil secretion
Neutrophils have the capacity to elaborate proinflammatory cytokines and to degranulate, leading to the release of histotoxic proteases; this latter process may be important in facilitating the migration of neutrophils through endothelial cells but under pathological conditions is thought to contribute to tissue injury (31). Neutrophil secretion was assessed in three ways, namely, 1) cell surface expression of adhesion molecules contained within secretory granules (e.g., CD11b, see Fig. 3A); 2) release of IL-8 and other inflammatory mediators (Fig. 4A, 4B); and 3) secretion of elastase, a serine protease derived predominantly from azurophil granules (Fig. 4C). In line with previous studies (23), optimal IL-8 release was observed at 4 h following dual stimulation with GM-CSF and TNF-α, with an ~23-fold increase in IL-8 secretion over baseline at this time (Fig. 4A). As described above and illustrated in Fig. 3A, CD11b expression was completely preserved in the face of hypoxia at early time points and even upregulated after more prolonged (4 h) hypoxic incubation. As shown in Fig. 4A, basal and agonist-stimulated IL-8 release was unaffected by hypoxia across a wide time frame, from 2 to 20 h. In view of the known cross-talk between the HIF1-α and NF-κβ pathways, we also examined the effects of hypoxia and of LPS on neutrophil release of a range of inflammatory cytokines and chemokines; as shown in Fig. 4C, LPS induced the release of IL-8, TNF-α, IL-6, IL-10, and MIP-1β, but this was not modified by hypoxia, and the detection of a range of other inflammatory mediators was not significantly affected by either hypoxia or LPS (see Supplemental Fig. 1). Hypoxia did not modulate the spontaneous release of elastase from unstimulated neutrophils. However, elastase release from cytochalasin-B–primed fMLP-stimulated cells was increased dramatically by hypoxia at 4 h (Fig. 4D), without loss of cell viability.
FIGURE 4.

Effects of hypoxia on neutrophil secretory function. A, Hypoxia does not affect neutrophil IL-8 release. Neutrophils were incubated under normoxia or hypoxia prior to the addition of buffer, GM-CSF (10 ng/ml), and/or TNF-α (10 ng/ml) for a further 2, 4, 6, or 20 h. IL-8 was assayed in cell-free supernatants by ELISA. Data represent mean ± SEM of three independent experiments performed in duplicate. B and C, LPS, but not hypoxia, upregulates neutrophil inflammatory mediator release. Neutrophils were incubated under hypoxia or normoxia for 30 min, prior to the addition of LBP/LPS for a further 3.5 h under continued normoxia or hypoxia. The supernatants were analyzed using commercial Ab arrays (RayBio). B, Representative chemokine membranes following development; dots representing IL-8 are ringed. C illustrates signal intensity quantified by Scion Image and normalized using positive controls according to the manufacturer’s instructions (mean ± SD from n = 3 experiments performed in duplicate). Data shown represent the 5 (of 78) mediators that increased in response to LPS, with the remaining data presented in Supplemental Fig. 1. D, Neutrophils were incubated for 1 or 4 h under normoxia or hypoxia prior to priming (cytochalasin B or buffer, 5 min) and stimulation (fMLP or buffer, 10 min). Supernatants were assayed using a commercial human elastase ELISA. Data represent mean ± SEM of n = 8 experiments performed in duplicate. *p < 0.05.
Hypoxia reduces extracellular superoxide anion release and particle-induced intracellular ROS generation
At the outset of these studies, we predicted that hypoxia would reduce the capacity of neutrophils to undergo a maximal respiratory burst response owing to the limited availability of molecular oxygen. As shown in Fig. 5, this was indeed what we observed in respect to both fMLP-stimulated extracellular superoxide anion generation (Fig. 5A) and opsonized zymosan (OZ)-induced intracellular total ROS generation (Fig. 5C). Of note, the fMLP response was examined in both GM-CSF–primed and unprimed cells, and both NADPH oxidase assays were run in cells incubated for 1 and 4 h in normoxia or hypoxia. Importantly, the extent of OZ phagocytosis was identical in normoxia and hypoxia (Fig. 5B), and identical results were obtained using 2′,7′-dichlorofluorescein as a reporter (data not shown). As shown in Fig. 5C, the ROS signal generated by OZ ingestion was only partially inhibited by the oxidase inhibitor DPI; possible explanations for this subtotal inhibition include artifactual oxidation of the fluorogenic probe by DPI or its metabolites (32) or the previously reported effects of DPI on the pentose phosphate pathway (33).
FIGURE 5.

Effects of hypoxia on neutrophil respiratory burst. A, Hypoxic neutrophils have a reduced ability to generate extracellular superoxide. Neutrophils were primed either directly or after a 3-h incubation period under hypoxia or normoxia with GM-CSF (10 ng/ml) or buffer for 1 h. Neutrophils were then stimulated (or not) with fMLP (100 nM) or PMA (200 nM) for 30 min. Extracellular superoxide generation was assessed by the superoxide dismutase-inhibitable reduction of cytochrome c. Data represent mean ± SEM of three independent experiments performed in triplicate. *p < 0.05. B and C, Hypoxic neutrophils ingest zymosan but have a reduced capacity to generate intracellular ROS. Representative images (B) from May-Grünwald-Giemsa–stained cytospin preparations of normoxic or hypoxic neutrophils (original magnification ×400, oil immersion) incubated with zymosan particles for 1 h, demonstrating uptake into phagosomes. Neutrophils were incubated under hypoxia or normoxia for 1 or 3 h prior to addition of CM-H2DCFDA for 10 min. IgG-opsonized zymosan was added for 1 h in the presence or absence of DPI (0.2 μM) to inhibit NADPH oxidase. Data represent mean fluorescence intensity; mean ± SEM of at least three independent experiments, each performed in triplicate (C). D and E, Hypoxia does not reduce the expression of NADPH oxidase components. Normoxic or hypoxic (4 h) neutrophils were lysed and analyzed by Western blotting for expression of p40phox or p47phox. Single representative image (D) of n = 3. Densitometry (Scion Image) values for bands normalized to β-actin (E). Data represent mean ± SEM from three independent experiments.
Because these findings would also be compatible with a major effect of hypoxia on the expression of individual NADPH oxidase components, this was examined by qPCR and Western blotting. The only effects observed by qPCR were minor reductions in p47phox and p40phox mRNA levels under hypoxia (Supplemental Fig. 2), which did not attain statistical significance. In addition, no differences were observed at a protein level (Fig. 5D, 5E). These data indicate that the profound inhibition of respiratory burst activity in the neutrophil under hypoxia does not reflect differences in oxidase component expression.
Hypoxia inhibits the ability of neutrophils to kill S. aureus but not E. coli
The effects of hypoxia on neutrophil-mediated bacterial killing were examined using two different bacterial pathogens, namely, E. coli and S. aureus. As demonstrated in Fig. 6A, neutrophils were highly efficient at killing E. coli under both normoxic and hypoxic conditions. Consistent with this was our observation, supported by previous studies (34, 35), that E. coli killing was unaffected by the oxidase inhibitor DPI. This finding suggests that neutrophil-mediated killing of E. coli is independent of NADPH oxidase.
FIGURE 6.

Effect of hypoxia on neutrophil bactericidal capacity. A, Hypoxic neutrophils maintain the ability to kill opsonized E. coli. Bacteria opsonized with heat-inactivated serum were added to pre-equilibrated (1 h) normoxic or hypoxic neutrophils in the presence or absence of 3 μM DPI for 1 h. Surviving bacteria were enumerated by quantitative culture, as described in Materials and Methods. Data represent the mean ± SEM of four independent experiments, each performed in triplicate; *p < 0.05 for incubations with versus without neutrophils. Insets represent May-Grunwald-Giemsa–stained cytospin images (original magnification ×400, oil immersion) with arrows pointing to internalized bacteria. B, Hypoxia impairs the ability of neutrophils to kill opsonized S. aureus. Serum opsonized bacteria were added to pre-equilibrated (1 h) normoxic or hypoxic neutrophils in the presence or absence of 3 μM DPI for 1 h. Surviving bacteria were enumerated by quantitative culture, as described in Materials and Methods. Data represent the mean ± SEM of six independent experiments performed in triplicate; *p < 0.05. Insets represent cytospin images with arrows pointing to internalized bacteria. C, Hypoxia does not affect the ability of S. aureus to cause neutrophil death. Bacteria were added to pre-equilibrated (1 h) normoxic or hypoxic neutrophils in RPMI 1640, 10% heat-inactivated serum for 3 h with a multiplicity of infection of 0.5 (i.e., 2 neutrophils per bacterium). Intact neutrophils were quantified using CountBright absolute counting beads. Data represent mean ± SEM of n = 3 experiments, each performed in duplicate.
In contrast, neutrophil-mediated killing of S. aureus was markedly impaired under hypoxic conditions, perhaps reflecting the oxidase-dependent (DPI-sensitive) nature of this effect (Fig. 6B). Cytospin analysis confirmed that hypoxia did not compromise the ability of neutrophils to phagocytose staphylococci (Fig. 6B, inset and data not shown). It is also apparent from the data in Fig. 6B that S. aureus bacteria themselves form fewer colonies after hypoxic than normoxic incubation, even in the absence of neutrophils. Remarkably, when S. aureus organisms were incubated with neutrophils under hypoxia, an intracellular survival benefit was observed, which, as expected from Fig. 5, was not further increased by DPI. Examination of cytospin preparations confirmed that both E. coli and S. aureus were internalized during the course of the assay, and that this process was not affected by hypoxia (Fig. 6A, 6B and data not shown).
Because prolonged exposure of neutrophils to staphylococci and other bacteria may lead to pathogen-induced cell death, we sought to determine the effects of hypoxia on this process. S. aureus organisms were shown able to induce neutrophil death to a similar extent under normoxia and hypoxia after 3 h (Fig. 6C), with no neutrophil death seen at 1 h (data not shown). These data sets concur with the above oxidase findings and indicate that hypoxia impairs the ability of neutrophils to kill bacteria by oxidase-dependent pathways. By contrast, the ability of S. aureus to kill human neutrophils appears equally effective in normoxic and hypoxic conditions.
The effect of hypoxia on neutrophil oxidative burst and staphylococcal killing reflects lack of molecular oxygen
Because hypoxic impairment of the neutrophil oxidative burst and oxidase-dependent bacterial killing was not associated with altered expression of NADPH oxidase components, we investigated whether the lack of molecular oxygen as a substrate for the oxidase was responsible. Initially, we studied the effect of hypoxia on the intracellular ROS generation induced by pyocyanin, a phenazine virulence factor produced by Pseudomonas aeruginosa. Pyocyanin oxidizes intracellular pools of NADPH, NADH, and reduced glutathione by accepting electrons, which are then available for donation to molecular oxygen to generate O2− and H2O2 in a NADPH-independent fashion (36, 37). As can be seen from Fig. 7A, and consistent with our previous data (25), aerobic incubation of neutrophils in the presence of pyocyanin led to an increase in the detection of intracellular ROS; this increase was abolished when the incubation was performed under hypoxic conditions (Fig. 7A), confirming that hypoxia can inhibit ROS generation in a manner that is independent of the NADPH oxidase. Furthermore, when hypoxic neutrophils were reoxygenated by restoring them to the ambient oxygen tension, extracellular O2− generation was fully restored to the level of cells kept continuously normoxic (Fig. 7B). Similarly, the ability to kill S. aureus was also fully restored by reoxygenation (Fig. 7C).
FIGURE 7.
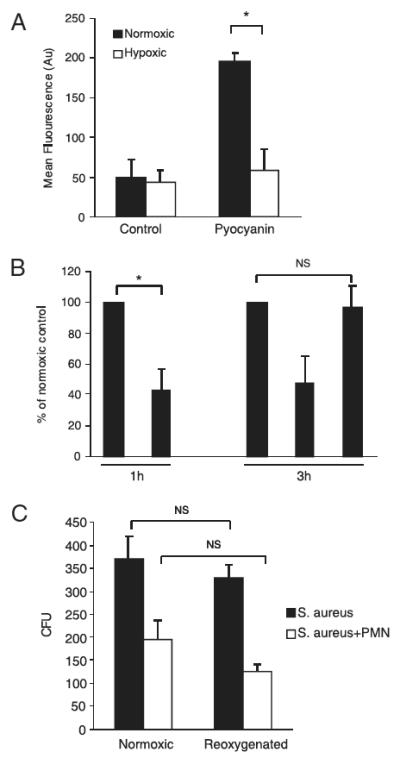
Effect of reoxygenation on neutrophil oxidative burst and oxidase-dependent killing. A, Pyocyanin-induced ROS generation is inhibited by hypoxia. Neutrophils (5 × 106/ml in RPMI 1640) were incubated under normoxia or hypoxia in the presence of pyocyanin (50 μM) or vehicle plus 3% CM-H2DCFDA for 30 min prior to assessment for ROS generation. Data represent mean fluorescence intensity; mean ± SEM of at least three independent experiments, each performed in triplicate. *p < 0.05. B, Reoxygenation restores the neutrophil oxidative burst. Neutrophils were primed (or not) with GM-CSF under normoxia or hypoxia. After 1 h, normoxic and hypoxic primed neutrophils were stimulated with fMLP (100 nM) for 30 min, and extracellular superoxide generation was assessed by the superoxide dismutase-inhibitable reduction of cytochrome c (1 h). Also at 1 h, aliquots of unprimed cells were moved from hypoxia to normoxia (or not moved), and unprimed hypoxic, normoxic, and reoxygenated cells were then primed with GM-CSF 10 ng/ml. After a further 2 h, the fMLP-stimulated generation of superoxide was determined for normoxic, hypoxic, and reoxygenated cells, as above (3 h). Data represent mean ± SEM of n = 3 experiments, each performed in triplicate. NS = p > 0.05; *p < 0.05. C, Reoxygenation restores the ability of neutrophils to kill S. aureus. Neutrophils were incubated under normoxia or hypoxia for 1 h, prior to the transfer (or not) of hypoxic cells to ambient oxygen tension. After 30 min further incubation, the ability of continuously hypoxic, continuously normoxic, or reoxygenated neutrophils to kill S. aureus was determined exactly as described. Data represent mean ± SEM of n = 3 experiments, each performed in duplicate. NS = p > 0.05.
Discussion
Many inflamed tissues offer a hostile microenvironment in which neutrophils and macrophages are required to operate. These sites are characteristically hypoxic and nutrient deprived, with high levels of reactive oxygen and nitrogen radicals. In this setting, it is now recognized that the PHD/HIF-1α pathway plays a key role in triggering adaptive cellular responses that support the function of immune cells within such environments. Hence HIF-1α accumulation, which can be triggered by pathogen-derived signals in addition to hypoxia, supports several innate immune functions in dendritic cells and macrophages and promotes the bactericidal function of phagocytes. The HIF-1α and NF-κB pathways also act cooperatively to regulate the transcription of several enzymes involved in anaerobic metabolism. Of interest, in conditions such as diabetes, in which tissue ischemia and infection are major clinical problems, the HIF-1α pathway has been found to be defective owing to impaired HIF-1α binding to the coactivator p300 (38). This latter event reflects covalent modification of p300 by the dicarbonyl metabolite methylglyoxal.
The capacity of neutrophils and other phagocytic cells to sense hypoxia via activation of the PHD/HIF-1α pathway, and for this to delay constitutive apoptosis, is firmly established. Recently, even acute global hypoxia equivalent to that of altitudes of 4300 m has been shown to be a sufficient trigger to increase HIF-1α accumulation and HIF-1α DNA in circulating leukocytes (39). However, few studies have detailed the effects of hypoxia on other aspects of neutrophil function. Our findings extend the current literature by 1) demonstrating preservation of chemotaxis, receptor regulation, and phagocytosis under severe hypoxia, and 2) demonstrating a global reduction in respiratory burst capacity under hypoxia, which impairs the capacity of neutrophils to kill bacteria such as S. aureus.
Results arising from the few previous studies that have addressed the effects of hypoxia on neutrophil function have been confusing and in some cases contradictory; this may reflect important methodological differences, including the degree of hypoxia achieved in the media bathing the cells, the use of hypoxia-mimetics or genetic manipulation of hypoxic signaling pathways rather than true hypoxia, the effects of hypoxia on PCO2/pH as well as PO2, and cellular reoxygenation during the experimental protocol. We analyzed the PO2 and pH of the media in each experiment to ensure consistent hypoxia sufficient to activate HIF-1α responses (Fig. 1) (6) and equal pH in normoxic and hypoxic incubations (Fig. 1). We were also assiduous in ensuring that all reactions were stopped (by lysis, freezing, or fixation) prior to removing cells from the hypoxic chamber. Hypoxia/reoxygenation and its clinical correlate, sleep apnea, have been shown to prime neutrophils for enhanced superoxide anion release (40), potentially contributing to the oxidative stress and endothelial dysfunction found in this common disorder (41); our data clearly demonstrate that cellular reoxygenation is essential for this augmentation of ROS production. The lack of effect of hypoxia on basal or agonist-stimulated shape change or spontaneous or fMLP-stimulated extracellular superoxide anion release demonstrates that our cells have not been inadvertently reoxygenated, and that continuous hypoxia per se does not induce neutrophil priming.
We also found that hypoxia did not compromise neutrophil chemotaxis or the phagocytosis of opsonized bacteria. Previous work has suggested that hypoxia may actually upregulate these processes in macrophages via HIF-1α stabilization (42), but deletion of HIF-1α also caused a far more profound depletion of ATP in these cells than in neutrophils (9), suggesting that energy stores in mononuclear cells are more intimately linked to the levels of this transcription factor than is the case in neutrophils. Exposure of neutrophils to hypoxia for a period of 4 h (but not shorter incubation periods) increased the expression of CD11b on the surface of unstimulated cells, and prevented the time-dependent impairment of fMLP-stimulated upregulation of this molecule seen with normoxic incubation. These results are consistent with the previously reported finding that the CD11b gene sequence incorporates a hypoxia-response element (43). Again after 4 h of hypoxia, we observed a dramatic (~4-fold) enhancement of elastase secretion from primed fMLP-stimulated cells. Elastase is an important component in the neutrophil microbicidal arsenal available to counter Gram-negative (but not Gram-positive) bacterial sepsis (44) and fungal infection (45), but may also contribute to endothelial (46) and epithelial (47) cell injury and the pathogenesis of inflammatory diseases, including lung injury (45); thus, whether enhanced elastase release in the hypoxic environment is harmful or beneficial may be highly context dependent. In the setting of hypoxia, inhibition of neutrophil elastase has been shown to attenuate intestinal injury in a rabbit model of alveolar hypoxia (48) and to reduce alveolar-capillary damage in a rabbit model of ischemia-reperfusion injury (49), suggesting that hypoxia-driven elastase release could contribute to tissue injury. Reeves et al. (50) have demonstrated that ROS are required to fully enable protease function within the phagosome; therefore, the protective bactericidal function of elastase may be compromised even in the setting of enhanced (and potentially harmful) extracellular elastase release.
Our findings of impaired bacterial killing of S. aureus under hypoxia are consistent with much earlier observations that wound healing and bacterial clearance from the skin are impaired under hypoxia (15, 51–53). It is also entirely consistent with the dramatic reduction of both extracellular and, more importantly, intracellular ROS generation displayed by neutrophils when subjected to hypoxia. The lack of effect of hypoxia on the components of the NADPH oxidase suggests that this reduction is mediated by lack of available substrate, i.e., molecular oxygen. This explanation is supported both by the fact that pyocyanin-induced ROS generation (which is independent of the NADPH oxidase) is also abolished by hypoxia (Fig. 7A) and more directly by the fact that hypoxic reduction of both ROS generation and staphylococcal killing is completely restored by re-exposure of the hypoxic neutrophil to ambient oxygen. Furthermore, the ability of hypoxic (or DPI-treated) neutrophils to kill E. coli as efficiently as normoxic neutrophils suggests that nonoxidant-dependent killing mechanisms are fully preserved.
Although hypoxia led to a temporary reduction of bacterial growth in Luria-Bertani broth (Fig. 6A, 6B and data not shown), the presence of neutrophils caused a seemingly paradoxical enhancement in staphylococcal survival in the hypoxic environment. This observation implies that the interior of a hypoxic neutrophil is a more favorable environment for bacterial survival than is hypoxic growth medium alone, a remarkable finding given the known importance of these cells in staphylococcal clearance. Even normoxic neutrophils failed to kill approximately half of the S. aureus organisms presented to them (Fig. 6B), despite the low multiplicity of infection (1 bacterium per 6 neutrophils) used; at higher pathogen/neutrophil ratios, staphylococci induced neutrophil lysis, independent of the ambient oxygen tension (Fig. 6C). These findings are in keeping with previous demonstrations that S. aureus can adapt to evade the innate immune response. Thus, in a murine model of i.p. staphylococcal infection (54), neutrophils isolated from the site of infection contained viable intracellular staphylococci, and these infected neutrophils could transfer the pathogen to infect a naive animal. Furthermore, partial, but not complete, ablation of neutrophil migration into the site of infection enhanced host defense in the setting of S. aureus infection, whereas a complete lack of neutrophils converted a nonlethal to a lethal infection. Thus, abscess formation may establish the hypoxic milieu that favors staphylococcal survival rather than bacterial elimination.
S. aureus is capable of adapting to various hostile host environments, and inhabits or infects skin, mucous membranes, blood, and deeper tissues. This flexibility is conferred by several gene regulatory systems that control the expression of S. aureus virulence genes. S. aureus displays adaptive responses following phagocytosis by neutrophils, leading to global changes in gene expression; 26.8–38.8% of S. aureus genes, including those encoding catalase and superoxide dismutase, were differentially regulated within 30 min after ingestion, depending on the precise strain analyzed (55). Although anoxia has also been shown to regulate the transcription of staphylococcal virulence factors (55), this did not occur within a time frame relevant to our assays.
Thus, it seems that although the outcome of the interaction between the neutrophil and the pathogen S. aureus depends on a number of factors, including the neutrophil/pathogen ratio, our data suggest that the ambient oxygen tension also has a profound influence on this interaction and that because most inflamed sites are highly hypoxic, this may be an important factor determining the outcome of clinical staphylococcal infections. Neutrophil functions other than the generation of the respiratory burst are not impaired by hypoxia; on the contrary, release of elastase may be significantly enhanced, with the potential for increased “bystander” tissue injury.
Supplementary Material
Acknowledgments
We thank Keith Davidson (Babraham Institute, Cambridge, U.K.) for the gift of E. coli and for assistance with bacterial killing assays, Dr. Jeremy Skepper (University of Cambridge) for assistance with electron microscopy, and Dr. Lynne Prince (Academic Unit of Respiratory Medicine, University of Sheffield) for providing the pyocyanin.
This work was supported by the Medical Research Council, the Wellcome Trust, a Raymond and Beverly Sackler Studentship, Asthma-UK, and the Cambridge National Institute for Health Research Biomedical Research Centre. N.N.M. is a Medical Research Council Capacity Building Student, C.S. is a Wellcome Trust Clinical Research Training Fellow, S.R.W. is a Wellcome Trust Intermediate Research Fellow, and A.A.R.T. is a Medical Research Council Clinical Training Fellow.
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Simmen HP, Blaser J. Analysis of pH and pO2 in abscesses, peritoneal fluid, and drainage fluid in the presence or absence of bacterial infection during and after abdominal surgery. Am. J. Surg. 1993;166:24–27. doi: 10.1016/s0002-9610(05)80576-8. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett JG, Finegold SM. Anaerobic infections of the lung and pleural space. Am. Rev. Respir. Dis. 1974;110:56–77. doi: 10.1164/arrd.1974.110.1.56. [DOI] [PubMed] [Google Scholar]
- 3.Kempner W. The nature of leukemic blood cells as determined by their metabolism. J. Clin. Invest. 1939;18:291–300. doi: 10.1172/JCI101045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levene PA, Meyer GM. The action of leucocytes on glucose. J. Biol. Chem. 1912;11:361–370. [Google Scholar]
- 5.Levene PA, Meyer GM. On the action of leucocytes on glucose, second communication. J. Biol. Chem. 1912;12:265–273. [Google Scholar]
- 6.Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER. Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-kappaB activity. J. Exp. Med. 2005;201:105–115. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walmsley SR, Cowburn AS, Clatworthy MR, Morrell NW, Roper EC, Singleton V, Maxwell P, Whyte MKB, Chilvers ER. Neutrophils from patients with heterozygous germline mutations in the von Hippel Lindau protein (pVHL) display delayed apoptosis and enhanced bacterial phagocytosis. Blood. 2006;108:3176–3178. doi: 10.1182/blood-2006-04-018796. [DOI] [PubMed] [Google Scholar]
- 8.Mecklenburgh KI, Walmsley SR, Cowburn AS, Wiesener M, Reed BJ, Upton PD, Deighton J, Greening AP, Chilvers ER. Involvement of a ferroprotein sensor in hypoxia-mediated inhibition of neutrophil apoptosis. Blood. 2002;100:3008–3016. doi: 10.1182/blood-2002-02-0454. [DOI] [PubMed] [Google Scholar]
- 9.Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1α expression regulates the bactericidal capacity of phagocytes. J. Clin. Invest. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009;9:609–617. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J. Immunol. 2007;178:7516–7519. doi: 10.4049/jimmunol.178.12.7516. [DOI] [PubMed] [Google Scholar]
- 14.Allen DB, Maguire JJ, Mahdavian M, Wicke C, Marcocci L, Scheuenstuhl H, Chang M, Le AX, Hopf HW, Hunt TK. Wound hypoxia and acidosis limit neutrophil bacterial killing mechanisms. Arch. Surg. 1997;132:991–996. doi: 10.1001/archsurg.1997.01430330057009. [DOI] [PubMed] [Google Scholar]
- 15.Jönsson K, Hunt TK, Mathes SJ. Oxygen as an isolated variable influences resistance to infection. Ann. Surg. 1988;208:783–787. doi: 10.1097/00000658-198812000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haslett C, Guthrie LA, Kopaniak MM, Johnston RB, Jr., Henson PM. Modulation of multiple neutrophil functions by preparative methods or trace concentrations of bacterial lipopolysaccharide. Am. J. Pathol. 1985;119:101–110. [PMC free article] [PubMed] [Google Scholar]
- 17.Murray J, Barbara JA, Dunkley SA, Lopez AF, Van Ostade X, Condliffe AM, Dransfield I, Haslett C, Chilvers ER. Regulation of neutrophil apoptosis by tumor necrosis factor-alpha: requirement for TNFR55 and TNFR75 for induction of apoptosis in vitro. Blood. 1997;90:2772–2783. [PubMed] [Google Scholar]
- 18.Cowburn AS, Cadwallader KA, Reed BJ, Farahi N, Chilvers ER. Role of PI3-kinase-dependent Bad phosphorylation and altered transcription in cytokine-mediated neutrophil survival. Blood. 2002;100:2607–2616. doi: 10.1182/blood-2001-11-0122. [DOI] [PubMed] [Google Scholar]
- 19.Mancini M, Sedghinasab M, Knowlton K, Tam A, Hockenbery D, Anderson BO. Flow cytometric measurement of mitochondrial mass and function: a novel method for assessing chemoresistance. Ann. Surg. Oncol. 1998;5:287–295. doi: 10.1007/BF02303787. [DOI] [PubMed] [Google Scholar]
- 20.Qu J, Condliffe AM, Lawson M, Plevin RJ, Riemersma RA, Barclay GR, McClelland DBL, Chilvers ER. Lack of effect of recombinant platelet-derived growth factor on human neutrophil function. J. Immunol. 1995;154:4133–4141. [PubMed] [Google Scholar]
- 21.Parmar JS, Mahadeva R, Reed BJ, Farahi N, Cadwallader KA, Keogan MT, Bilton D, Chilvers ER, Lomas DA. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am. J. Respir. Cell Mol. Biol. 2002;26:723–730. doi: 10.1165/ajrcmb.26.6.4739. [DOI] [PubMed] [Google Scholar]
- 22.Clatworthy MR, Smith KGC. FcgammaRIIb balances efficient pathogen clearance and the cytokine-mediated consequences of sepsis. J. Exp. Med. 2004;199:717–723. doi: 10.1084/jem.20032197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cowburn AS, Deighton J, Walmsley SR, Chilvers ER. The survival effect of TNF-alpha in human neutrophils via NF-kappa B-dependent IL-8 release. Eur. J. Immunol. 2004;34:1733–1743. doi: 10.1002/eji.200425091. [DOI] [PubMed] [Google Scholar]
- 24.Condliffe AM, Chilvers ER, Haslett C, Dransfield I. Priming differentially regulates neutrophil adhesion molecule expression/function. Immunology. 1996;89:105–111. doi: 10.1046/j.1365-2567.1996.d01-711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Usher LR, Lawson RA, Geary I, Taylor CJ, Bingle CD, Taylor GW, Whyte MK. Induction of neutrophil apoptosis by the Pseudomonas aeruginosa exotoxin pyocyanin: a potential mechanism of persistent infection. J. Immunol. 2002;168:1861–1868. doi: 10.4049/jimmunol.168.4.1861. [DOI] [PubMed] [Google Scholar]
- 26.Condliffe AM, Webb LM, Ferguson GJ, Davidson K, Turner M, Vigorito E, Manifava M, Chilvers ER, Stephens LR, Hawkins PT. RhoG regulates the neutrophil NADPH oxidase. J. Immunol. 2006;176:5314–5320. doi: 10.4049/jimmunol.176.9.5314. [DOI] [PubMed] [Google Scholar]
- 27.Chen C, Edelstein LC, Gélinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol. Cell. Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bryan SA, Jose PJ, Topping JR, Wilhelm R, Soderberg C, Kertesz D, Barnes PJ, Williams TJ, Hansel TT, Sabroe I. Responses of leukocytes to chemokines in whole blood and their antagonism by novel CC-chemokine receptor 3 antagonists. Am. J. Respir. Crit. Care Med. 2002;165:1602–1609. doi: 10.1164/rccm.200111-059OC. [DOI] [PubMed] [Google Scholar]
- 29.Anderson SI, Hotchin NA, Nash GB. Role of the cytoskeleton in rapid activation of CD11b/CD18 function and its subsequent downregulation in neutrophils. J. Cell Sci. 2000;113:2737–2745. doi: 10.1242/jcs.113.15.2737. [DOI] [PubMed] [Google Scholar]
- 30.Urano-Tashiro Y, Yajima A, Takashima E, Takahashi Y, Konishi K. Binding of the Streptococcus gordonii DL1 surface protein Hsa to the host cell membrane glycoproteins CD11b, CD43, and CD50. Infect. Immun. 2008;76:4686–4691. doi: 10.1128/IAI.00238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cepinskas G, Sandig M, Kvietys PR. PECAM-1, alpha6 integrins and neutrophil elastase cooperate in mediating neutrophil transmigration. J. Cell Sci. 2005;118:2067–2076. doi: 10.1242/jcs.02340. [DOI] [PubMed] [Google Scholar]
- 32.Balcerczyk A, Soszynski M, Rybaczek D, Przygodzki T, Karowicz-Bilinska A, Maszewski J, Bartosz G. Induction of apoptosis and modulation of production of reactive oxygen species in human endothelial cells by diphenyleneiodonium. Biochem. Pharmacol. 2005;69:1263–1273. doi: 10.1016/j.bcp.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Riganti C, Gazzano E, Polimeni M, Costamagna C, Bosia A, Ghigo D. Diphenyleneiodonium inhibits the cell redox metabolism and induces oxidative stress. J. Biol. Chem. 2004;279:47726–47731. doi: 10.1074/jbc.M406314200. [DOI] [PubMed] [Google Scholar]
- 34.Decleva E, Menegazzi R, Busetto S, Patriarca P, Dri P. Common methodology is inadequate for studies on the microbicidal activity of neutrophils. J. Leukoc. Biol. 2006;79:87–94. doi: 10.1189/jlb.0605338. [DOI] [PubMed] [Google Scholar]
- 35.Rada BK, Geiszt M, Káldi K, Timár C, Ligeti E. Dual role of phagocytic NADPH oxidase in bacterial killing. Blood. 2004;104:2947–2953. doi: 10.1182/blood-2004-03-1005. [DOI] [PubMed] [Google Scholar]
- 36.Müller PK, Krohn K, Mühlradt PF. Effects of pyocyanine, a phenazine dye from Pseudomonas aeruginosa, on oxidative burst and bacterial killing in human neutrophils. Infect. Immun. 1989;57:2591–2596. doi: 10.1128/iai.57.9.2591-2596.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Malley YQ, Reszka KJ, Spitz DR, Denning GM, Britigan BE. Pseudomonas aeruginosa pyocyanin directly oxidizes glutathione and decreases its levels in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;287:L94–L103. doi: 10.1152/ajplung.00025.2004. [DOI] [PubMed] [Google Scholar]
- 38.Thangarajah H, Yao D, Chang EI, Shi Y, Jazayeri L, Vial IN, Galiano RD, Du XL, Grogan R, Galvez MG, Januszyk M, et al. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc. Natl. Acad. Sci. U. S. A. 2009;106:13505–13510. doi: 10.1073/pnas.0906670106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tissot van Patot MC, Serkova NJ, Haschke M, Kominsky DJ, Roach RC, Christians U, Henthorn TK, Honigman B. Enhanced leukocyte HIF-1alpha and HIF-1 DNA binding in humans after rapid ascent to 4300 m. Free Radic. Biol. Med. 2009;46:1551–1557. doi: 10.1016/j.freeradbiomed.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 40.Schulz R, Mahmoudi S, Hattar K, Sibelius U, Olschewski H, Mayer K, Seeger W, Grimminger F. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am. J. Respir. Crit. Care Med. 2000;162:566–570. doi: 10.1164/ajrccm.162.2.9908091. [DOI] [PubMed] [Google Scholar]
- 41.Prabhakar NR, Kumar GK. Oxidative stress in the systemic and cellular responses to intermittent hypoxia. Biol. Chem. 2004;385:217–221. doi: 10.1515/BC.2004.015. [DOI] [PubMed] [Google Scholar]
- 42.Anand RJ, Gribar SC, Li J, Kohler JW, Branca MF, Dubowski T, Sodhi CP, Hackam DJ. Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1alpha-dependent manner. J. Leukoc. Biol. 2007;82:1257–1265. doi: 10.1189/jlb.0307195. [DOI] [PubMed] [Google Scholar]
- 43.Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc. Natl. Acad. Sci. USA. 2004;101:10440–10445. doi: 10.1073/pnas.0401339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat. Med. 1998;4:615–618. doi: 10.1038/nm0598-615. [DOI] [PubMed] [Google Scholar]
- 45.Tkalcevic J, Novelli M, Phylactides M, Iredale JP, Segal AW, Roes J. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity. 2000;12:201–210. doi: 10.1016/s1074-7613(00)80173-9. [DOI] [PubMed] [Google Scholar]
- 46.Inauen W, Granger DN, Meininger CJ, Schelling ME, Granger HJ, Kvietys PR. Anoxia-reoxygenation-induced, neutrophil-mediated endothelial cell injury: role of elastase. Am. J. Physiol. 1990;259:H925–H931. doi: 10.1152/ajpheart.1990.259.3.H925. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki T, Yamashita C, Zemans RL, Briones N, Van Linden A, Downey GP. Leukocyte elastase induces lung epithelial apoptosis via a PAR-1-, NF-kappaB-, and p53-dependent pathway. Am. J. Respir. Cell Mol. Biol. 2009;41:742–755. doi: 10.1165/rcmb.2008-0157OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakamura N, Morisaki H, Suzuki T, Yajima S, Katori N, Kotake Y, Funakoshi Y, Kawabata K, Yamada S, Ishizaka A, Takeda J. Inhibition of neutrophil elastase attenuates gut mucosal injury evoked by acute alveolar hypoxia in rabbits. Shock. 2007;28:101–105. doi: 10.1097/shk.0b013e31802fa1b2. [DOI] [PubMed] [Google Scholar]
- 49.Kishima H, Takeda S, Miyoshi S, Matsumura A, Minami M, Utsumi T, Omori K, Nakahara K, Matsuda H. Microvascular permeability of the non-heart-beating rabbit lung after warm ischemia and reperfusion: role of neutrophil elastase. Ann. Thorac. Surg. 1998;65:913–918. doi: 10.1016/s0003-4975(98)00076-9. [DOI] [PubMed] [Google Scholar]
- 50.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 51.Ahn ST, Mustoe TA. Effects of ischemia on ulcer wound healing: a new model in the rabbit ear. Ann. Plast. Surg. 1990;24:17–23. doi: 10.1097/00000637-199001000-00004. [DOI] [PubMed] [Google Scholar]
- 52.Kivisaari J, Niinikoski J. Effects of hyperbaric oxygenation and prolonged hypoxia on the healing of open wounds. Acta Chir. Scand. 1975;141:14–19. [PubMed] [Google Scholar]
- 53.Knighton DR, Halliday B, Hunt TK. Oxygen as an antibiotic. A comparison of the effects of inspired oxygen concentration and antibiotic administration on in vivo bacterial clearance. Arch. Surg. 1986;121:191–195. doi: 10.1001/archsurg.1986.01400020077009. [DOI] [PubMed] [Google Scholar]
- 54.Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J. Immunol. 2000;164:3713–3722. doi: 10.4049/jimmunol.164.7.3713. [DOI] [PubMed] [Google Scholar]
- 55.Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Saïd-Salim B, Porcella SF, Long RD, Dorward DW, Gardner DJ, Kreiswirth BN, et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J. Immunol. 2005;175:3907–3919. doi: 10.4049/jimmunol.175.6.3907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.