

Abstract
Septins are guanine nucleotide-binding proteins that form hetero-oligomeric complexes, which assemble into filaments and higher-order structures at sites of cell division and morphogenesis in eukaryotes. Dynamic changes in the organization of septin-containing structures occur concomitantly with progression through the mitotic cell cycle and during cell differentiation. Septins also undergo stage-specific post-translational modifications, which have been implicated in regulating their dynamics, in some cases via purported effects on septin turnover. In our recent study, the fate of two of the five septins expressed in mitotic cells of budding yeast (Saccharomyces cerevisiae) was tracked using two complementary fluorescence-based methods for pulse-chase analysis. During mitotic growth, previously-made molecules of both septins (Cdc10 and Cdc12) persisted through multiple successive divisions and were incorporated equivalently with newly synthesized molecules into hetero-oligomers and higher-order structures. Similarly, in cells undergoing meiosis and the developmental program of sporulation, pre-existing copies of Cdc10 were incorporated into new structures. In marked contrast, Cdc12 was irreversibly excluded from septin complexes and replaced by another septin, Spr3. Here, we discuss the broader implications of these results and related findings with regard to how septin dynamics is coordinated with the mitotic cell cycle and in the yeast life cycle, and how these observations may relate to control of the dynamics of other complex multi-subunit assemblies.
Keywords: septin, yeast, inheritance, assembly, stability, recycling, sporulation, octamer, filaments
Coordination of Septin Dynamics with the Cell Cycle in Budding Yeast
The septin family of proteins was first identified in S. cerevisiae, and much research on septins has been performed using this model eukaryote. In this organism, five septins are expressed during vegetative division (Cdc3, Cdc10, Cdc11, Cdc12 and Shs1/Sep7), which co-localize at the mother-bud neck and co-purify in stable hetero-oligomeric complexes.1–3 Consistent with septin localization to the site where cytokinesis occurs, the most prominent phenotype of cells carrying temperature-sensitive mutations in septin genes is a defect in cell division at the restrictive temperature, resulting in multi-budded or chain-like multinucleate cells that arise from cycles of budding and nuclear division without concomitant cytokinesis.4 The phenotype of such mutants likely reflects the failure to recruit to the neck various other proteins that have important structural and catalytic roles in cytokinesis, including components of the actomyosin contractile ring (e.g., Myo1, a type II myosin)5 and enzymes needed for septation (e.g., Chs3, a chitin synthase responsible for deposition of new cell wall material).6 Accordingly, one essential function of the septin structures at the neck may be to serve as a platform for the proper positioning of the necessary molecular machinery at the site of division. Indeed, much like the scaffolds workmen erect at sites of home remodeling or new construction, septins assemble into what appears as an ordered array of evenly spaced filaments along the plasma membrane at the bud neck, as first revealed in 1976 by Byers and Goetsch using thin-section electron microscopy (EM).7 Purified septin complexes, which are rod-shaped (see below), are themselves capable of end-on-end polymerization in the test tube into filaments with dimensions that approximate those seen at the neck.8,9 As visualized by fluorescence microscopy using appropriate antibodies in fixed cells or using fluorescently-tagged septins in live cells, the septin-containing structure at the neck appears as a circumferential hourglass-shaped “collar” (Fig. 1A).
Figure 1.

Recycling of budding yeast septin subunits during the mitotic cell division cycle revealed by fluorescent pulse-chase analysis. (A) The mitotic cell division cycle of budding yeast is schematized with the location of septins (green), spindle pole bodies (red), and the nucleus (blue) indicated as landmarks of cell cycle progression. Also indicated are the approximate stages when post-translational modifications are added to septins and, where known, when removed. Center, arrangement of individual septin subunits within the hetero-octameric rod. (B) The SNAP-tag™, a domain (black) of the human O6-alkyl-guanine-DNA alkyltransferase (hAGT), shown fused in-frame to the C terminus of the yeast septin Cdc12 (orange), can be covalently labeled in vivo by reaction of its active site Cys with a benzyl guanine (BG) derivatized with a fluor (red). (C) Use of the SNAP-tag for one-color-labeling for pulse-chase analysis and for two-color labeling to monitor potential age-related differences. For pulse-chase analysis, cells expressing a SNAP-tagged septin are incubated for a short time in medium containing a cell-permeable fluorescently labeled BG derivative (the “pulse”), the cells are then washed to remove any unreacted dye, and the fate of the population of labeled protein (red) is monitored under the fluorescence microscope over the course of multiple subsequent cell divisions (the “chase”). For two-color labeling to compare the behavior of older and newer molecules, the cells are allowed to propagate for only 2–3 divisions after the first labeling, which provides sufficient time for synthesis of new unlabeled molecules without reducing too drastically the level of the previously labeled molecules. The new molecules can then be labeled by incubating the cells in medium containing a second BG derivative containing a fluor of a different color (green). (D) Use of differential gene expression for two-color labeling to monitor potential age-related differences. Expression of Cdc10-GFP from the HO promoter (“PHO”) is repressed in diploid cells, whereas expression of Cdc10-mCherry from the CDC10 promoter (“PCDC10”) is constitutive. Following meiosis and sporulation, the HO promoter only becomes active after the haploid spore has undergone its first mitotic division and, hence, Cdc10-GFP is produced during the second mitotic division.
Like construction-site scaffolding in the macroscopic world, once their function has been executed, septins disappear from the sites where they are no longer needed. The septins first disassemble from the center of the collar, leaving behind a pair of septin-containing rings flanking the center of the neck,10,11 which presumably makes way for constriction of the contractile ring and concomitant septum formation. In addition, at this point, the septin rings presumably act as a security barrier to restrict the diffusion out of the neck of the integral membrane proteins involved in cytokinesis (e.g., Chs2, another chitin synthase required for formation of the primary septum).12 Furthermore, the final acts of cytokinesis, including plasma membrane abscission and chitinase-dependent degradation of the primary septum, also fail without properly assembled septin rings.
Thus, the security fence function of the septin rings also confines these acts of demolition to the proper site. Once cell separation has been properly completed, the septin rings also disappear, and concomitantly the precursor to a new septin-containing collar is assembled at the next site where bud emergence will occur.10,11 Thus, the location and supramolecular architecture of the septin-containing structures in yeast undergo dynamic changes that are coupled to the events required for progression of the cell division cycle. The distinct septin-based structures formed presumably reflect the various stage-specific functions that these septin-containing structures perform. During the early stages of collar assembly, septins (probably hetero-oligomers and not free septins) are mobile within this incipient bud site, based on the rapid recovery of fluorescence after photobleaching this region of a cell that contains fluorescently-tagged septins.13,14 Once a bud has emerged, however, septins are “frozen” into place (the photobleached area of the collar does not recover), suggesting that by this time the hetero-oligomers have become fixed by their assembly into filaments,13,14 in analogy to our metaphor about erection of a scaffold. Not unexpectedly, in the region of the collar that disassembles to permit plasma membrane ingression behind the contractile ring, the septins are again mobilized, whereas those remaining in the split ring/security fence form are still immobile.13,14
What is the precise nature of the hetero-oligomeric septin complexes of which the bud site, the collar, and the split rings are constructed? How does the cell use those building blocks to assemble different structures, and take them apart, in the proper place and in the correct order during each cell cycle, and start all over again during the next cycle? Co-expression in E. coli of Cdc3, Cdc10, Cdc11 and Cdc12 followed by purification using an affinity tag on any of the four proteins results in a stable multi-subunit complex with apparent 2:2:2:2 stoichiometry that behaves indistinguishably from the hetero-oligomeric complexes purified analogously from yeast cells.1,8 When this purification is conducted at relatively high ionic strength (>150 mM salt), the resulting purified complex appears as an octameric rod in the EM. When the salt concentration is lowered (<50 mM), these rods polymerize into filaments that are aligned in parallel pairs, like railroad tracks.8,9 We now know that the order of the septins within a rod is: Cdc11-Cdc12-Cdc3-Cdc10-Cdc10-Cdc3-Cdc12-Cdc11, and that filaments are formed by end-on-end polymerization of the rods mediated by a salt-sensitive Cdc11-Cdc11 interaction.8 The observed filament pairing is mediated by the extended carboxy termini of the Cdc3-Cdc12 heterodimers in one filament extending perpendicularly from the filament axis to interact with their counterparts in the adjacent filament. Formation of the heterotetrameric four-helix bundle that presumably results from these contacts is likely mediated by the hydrophobic heptad repeat sequences present near the ends of Cdc3 and Cdc12, which are predicted to have the propensity to form coiled-coils.8 Sequence relatedness,15 as well as genetic16 and physical1,17–19 interactions, all suggest that Shs1 can occupy the same terminal position(s) within the rod as Cdc11 and does so in vivo (Fig. 1A). In this regard, although the SHS1 locus was not identified as a cdc gene, and even a null mutation (shs1Δ) has a rather mild phenotype, it is nonetheless a strong candidate for a septin subunit with a significant role in integrating septin dynamics with other cellular processes (see below).
Intriguingly, a short time before cytokinesis, as mitosis commences, three of the five septins (Cdc3, Cdc11 and Shs1) become covalently modified by the small ubiquitin-like modifier (SUMO) protein, and then this modification is abruptly removed as the collar is split into the two rings.20 It is tempting to speculate that this modification helps drive septin collar splitting. Shs1 is also subject to phosphorylation with similar timing. At least as judged by gel mobility shift, Shs1 is more extensively phosphorylated in cells held in M phase (by treatment with the microtubule-directed spindle poison nocodazole) than when cells are held in G1 (by exposure to the peptide mating pheromone α-factor).2,21 The kinase responsible for phosphorylation of Shs1 during mitosis appears to be Gin4,2,21 and, as cytokinesis begins, these modifications are promptly removed by a specific phosphatase (Rts1-containing PP2A isoform).14 An entirely different pattern of phosphorylation applies to Cdc3, which at the time of old septin ring disassembly in early G1 reportedly becomes a substrate for a cyclin-dependent kinase (Cln-bound Cdc28).22 Cdc10, too, is phosphorylated in dividing cells,23 probably in late G1, since the kinase responsible for the majority of Cdc10 phosphorylation, Cla4, reportedly leaves the future bud site just prior to S phase.24 Obviously, periodic modifications of particular septin subunits could provide mechanisms by which rearrangements of septin-based structures could be coordinated with cell cycle progression.
Are Septins Subject to Periodic Cell Cycle-Regulated Destruction?
The observations described immediately above paint a picture of septin dynamics in which direct septin modifications regulate the cell cycle-dependent changes in septin organization. Many of the most crucial stage-specific events in the cell cycle are driven by proteins whose existence is restricted to the particular stage at which they function. This restriction is imposed by periodic synthesis and ubiquitin- and proteasome-mediated destruction, and the targets for such degradation are frequently earmarked, or the regulatory factors involved are controlled, by their phosphorylation. Cyclins are the most obvious example of critical cell cycle components that must undergo periodic synthesis and degradation. Ectopic expression in G1 of the mitotic cyclin Clb2 blocks chromosome duplication,25 illustrating how the appearance of the wrong protein at the wrong time can cripple the function of a multiprotein complex (in this case, the DNA replication machinery) that must execute a stage-specific function.
Hypothetically, modification of a given septin(s) could be causal in dictating the formation of a particular septin-based structure. Such effects could be direct (for example, Cla4-dependent phosphorylation of Cdc10 could drive polymerization of rods into filaments) or indirect (Cdk1-dependent phosphorylation of Cdc3 could allow it to recruit a factor that helps takes filaments apart). Thereafter, one of two possible fates must ensue—either the phosphorylated septin is destroyed, making way for synthesis of new unmarked molecules, or the phosphates are removed and the septin is recycled and utilized again. How can one determine experimentally whether a given septin is destroyed periodically during each cell cycle, or whether its modification is added and then erased periodically during each cell cycle? The former scenario predicts that septins will be unstable, whereas the latter predicts they will be stable.
Evidence against periodic septin destruction comes from experiments in which yeast cells were exposed to a pulse of 15N, allowed to divide asynchronously in the absence of 15N for about one full cell cycle, and then septin complexes were harvested and the fraction of 15N-labeled protein calculated.3 There appeared to be no significant turnover of Cdc3, Cdc10, Cdc11 or Cdc12 during roughly one cell doubling.3 Somewhat similarly, when the disappearance of an epitope-tagged allele of Cdc3 was monitored following a pulse of its overexpression, the tagged wild-type protein exhibited a half-life considerably longer than a cell cycle, whereas a mutant Cdc3 unable to be phosphorylated by Cdk1 persisted even longer, which was interpreted to mean that, alhough Cdc3 is a relatively long-lived protein, Cdk1-mediated phosphorylation promotes its degradation.22 In fact, under certain conditions, yeast septins can perform a dramatic vanishing act. If, after disassembling the split rings following cell separation, cells are prevented from progressing further through the cell cycle [either by genetically inactivating Cdk1 function,10 or by inhibiting exit from G1 via nutrient withdrawal (McMurray MA, Thorner J, unpublished observations)], every GFP-tagged septin becomes essentially undetectable by fluorescence microscopy, suggesting that, in G1, septins are targeted for rampant destruction. The above observations do not resolve whether cell cycle-regulated turnover of septin polypeptides actually occurs during passage through a normal cell cycle. Indeed, the fact that Cid et al.10 did not observe any defect in disassembly of the septin rings when cdc28-4ts cells were shifted to the non-permissive temperature indicates that G1 Cdk1 activity is not required for that process, in contrast to the conclusion reached by Tang & Reed22 that Cdc28-mediated phosphorylation of Cdc3 plays a role in breaking down the old septin rings prior to initiation of a new bud. Likewise, the observed G1 Cdk1-mediated phosphorylation of Shs12,21 cannot have a role in that process either. Consistent with that view, in cells expressing an Shs1 mutant in which all of its apparent Cdc28 phosphorylation sites were mutated to alanine, no accumulation of “extra” septin rings was reported.2,21 Thus, if phosphorylation of certain septins is involved in dissolution of the septin rings at the end of each cell cycle, it must be some other protein kinase that does so in early G1. One attractive candidate to fulfill this role is the Cdk5-like kinase Pho85.26 This kinase displays elevated activity under conditions, like nutrient deprivation, that prevent cells from efficiently exiting G1, 27 has a demonstrated role in affecting septin-based cell morphogenesis,28 and, as we have found in collaborative studies, is capable, when bound to certain of its ten alternative cyclin partners (Pcl1, Pcl2 and Pcl9), of directly phosphorylating several purified septins (Cdc3, Cdc10 and Cdc12; Shs1 was not tested) in vitro (Sopko R, Versele M, Moffat J, Andrews BJ, Thorner J, unpublished results). In fact, cells lacking all late G1 cyclins (including Pcl1 and Pcl2) are retarded in their ability to disassemble the old septin rings and, perhaps for that reason, also defective in assembling the new septin scaffold at the next incipient bud site.28 A similar phenotype is observed for a Cdc3 mutant in which its apparent CDK phosphorylation sites have been mutated to alanine,22 but not for cdc28 mutants10 or cells lacking Cln1 and Cln2,21 arguing that Cdc3 (like Shs1) may be a target of the Pcl1- and Pcl2-bound forms of Pho85 in vivo, consistent with our in vitro observations.
Pulse-Chase Analysis of Septin Stability
To determine definitively whether a pre-made septin persists through multiple successive cell cycles and is able to be incorporated into the full spectrum of septin-based structures in each ensuing cell generation, we devised two independent fluorescence-based pulse-chase methods that allow the fate of a given septin polypeptide to be monitored throughout its lifetime. The first technique used a covalent labeling approach that exploits the SNAP-tag™,29 a small (17-kDa) domain of a human DNA damage repair protein that removes alkyl adducts from the DNA base guanine by attaching the alkyl group permanently to a cysteine residue in the SNAP-tag active site, releasing free guanine (Fig. 1B). If the SNAP-tag is fused to a protein of interest, providing a benzyl guanine (BG) derivative with a fluorophore attached to the benzyl group29 in the growth medium results in intracellular fluorescent labeling of that chimeric protein (Fig. 1C). After washing out unbound dye, the labeled molecules can be tracked microscopically as the cells divide or differentiate. If passage through the cell division cycle involves destruction of existing molecules, the fluorescent SNAP-tagged molecules will disappear rapidly in a population of dividing cells, but if the tagged molecule is stable, its fluorescence will persist and it will occupy its normal subcellular location(s) periodically during each cell cycle.
In the second pulse-chase approach we devised, we exploited a promoter (that of the HO gene30) that is only expressed in haploid cells and used it to drive expression of a GFP-tagged septin. By mating the haploid to a form a diploid, any further expression was shut off and, hence, the pool of pre-made GFP-labeled septin molecules and their ability to be incorporated into the septin-containing collar at the bud neck could be followed through multiple subsequent cell divisions of the diploid cells. Again, if septin proteolysis occurs in a cell cycle-dependent manner, the pool of GFP-tagged proteins should be wiped out as the diploid cells divide, especially over the course of several generations; if not, the labeled molecules will persist for many cell divisions.
When each of these approaches was applied to two representative septins, Cdc10 and Cdc12, the result was the same—the fluorescently-tagged septin persisted through many cell cycles.31 Indeed, turnover seemed to occur at a negligible rate; for example, in experiments in which labeled SNAP-tagged Cdc10 or Cdc12 was tracked, fluorescence was still detectable even five generations after the cells were originally exposed to the fluorescent dye.31 Because the fluorescent molecules were inherited approximately equally by mother and daughter cells, each cell division represents roughly a two-fold reduction of the pool of labeled molecules. Hence, after five cell divisions, growth alone should have caused a thirty-two-fold reduction in the content of the fluorescent molecules in each cell, whereas if concomitant proteolytic destruction of the labeled molecules was also occurring, the level of the fluorescent signal should have disappeared even more rapidly. However, every bud neck was still faintly labeled even after five cell doublings. Thus, for at least these two septins, periodic wholesale turnover of old molecules did not seem to occur as the pool of new non-fluorescent molecules was being synthesized. These findings may provide a satisfying explanation for the earlier finding that the apparent half-life of Cdc3 was extended by alteration of a CDK phosphorylation site in Cdc3, when it is considered that this mutation makes cells divide more slowly.22 If it is assumed that the behavior of Cdc3 is like that of Cdc10 and Cdc12, in that its rate of proteolysis is negligible, and if, as seems likely, the cells overexpressing the Cdc3 mutant divided more slowly than cells overexpressing wild-type Cdc3, the mutant protein would appear to persist longer simply because its division-dependent dilution would take longer (not because the mutation stabilizes Cdc3 against degradation).
Old and New Septins Are Incorporated Equivalently into Septin Structures in Mitotic Cells
We also devised two means to examine the fate of septin molecules of different ages in the same cell. After labeling the entire population of a SNAP-tagged septin with a dye of one color and washing away the excess dye, the cells were grown for a sufficient period of time to permit synthesis of new unreacted SNAP-tagged molecules of the same septin that were then labeled with another dye of a different color (Fig. 1C). After washing out the excess second dye, the fate of the pre-made (“older”) and more recently made (“newer”) molecules could be followed simultaneously under the fluorescence microscope using the appropriate band-pass filters. To accomplish the same thing exploiting the regulated transcription approach, we used haploid cells carrying two copies of the CDC10 gene, one producing Cdc10-mCherry constitutively from the native CDC10 promoter and the other producing Cdc10-GFP under the control of the HO promoter, which has another useful feature, namely this promoter is only operational in haploid cells after they have undergone one previous division.30 Thus, we could forestall expression of newer (Cdc10-GFP) molecules and then compare their behavior to that of the older (Cdc10-mCherry) molecules once a new-born cell divided (Fig. 1D).
When examined using either approach, localization of the differentially-tagged older and newer septins was largely congruent in every septin-based structure examined in mitotically cycling cells.31 This observation is important for several reasons. First, it was theoretically possible that, due to some of the stage-dependent post-translational modifications already mentioned, older and newer molecules of a given septin might differ in their capacity to be incorporated into certain structures or parts of structures. The fact that there was little distinction provides additional support for the idea that all such “marks” are reversible and quite dynamic, rather than permanent. Second, it was also theoretically possible that some septin structures, once assembled, might serve as the seeds or templates for assembly of subsequent structures and, if so, older septins might display spatial separation from newer septins, as has been suggested by others as a way to impose some asymmetry in structures composed from an inherently non-polar building block.32 However, at least at the resolution of the fluoresence microscope, we did not observed any reproducible age-based pattern of spatial segregation of older and newer septins within the collar.31 In fact, based on the aforemention studies of septin exchange rate using fluorescence recovery after photobleaching (FRAP), which demonstrated mobility of septins within septin-based structures at multiple stages in the cell cycle, it was perhaps to be expected that older and newer proteins would exhibit an intermixed distribution.
Collectively, our findings imply that if any stage-specific modification during the cell cycle is responsible for dictating the formation of a particular septin-based structure, that modification must be reset each cell cycle, in order to restore the capability of older molecules to be utilized for all of the same assemblies as newer molecules. However, not all modifications maybe erasable. For example, in response to DNA damage, Cdc12 and Shs1 are phosphorylated on residues normally unmodified in cycling cells,33 and shs1Δ cells are hypersensitive to DNA replication stress.34 In this way, effects on the septin collar may be part of how appropriate cell cycle delays (“checkpoints”) are imposed and, because septins are so long-lived, they could serve as carriers of a kind of “molecular memory” so that the downstream effects of the response to genotoxic stresses persist for multiple generations after the initiating event.
Our fluorescent pulse-chase analysis and the FRAP approach could not resolve another fundamental issue about septin organization and dynamics, namely at what level the observed intermixing occurs. If, once assembled, a Cdc11-Cdc12-Cdc3-Cdc10-Cdc10-Cdc3-Cdc12-Cdc11 hetero-octamer is a stable complex, then the exchange observed is due to mixing of older and newer hetero-octamers. In the test tube, septin hetero-octamers resist dissociation in high salt (e.g., 1 M NaCl),1,2,8,9 suggesting that, once assembled, the subunits within a hetero-octamer remain tightly associated. However, it seemed possible that the cell contains chaperones or other factors that may permit the exchange of subunits into and out of individual hetero-octamers; if so, then the observed intermixing of older and newer septins could occur at that level.
The available evidence, especially from the ability to produce recombinant complexes in bacteria,1,19 suggests that hetero-octamer assembly is co-translational. If a hetero-octamer, once made, is stable in vivo, and given the time delay between the production of previously made and more newly synthesized septins in our experiments, then both subunits of each of the four constituent septins in a hetero-octamer should be the same (either old or new). In other words, there should be few, if any, hetero-octamers that have one old and one new molecule of a given septin. To test this hypothesis, the pulse-chase method was applied to cells expressing SNAP-tagged Cdc12. These cells were labeled first with a biotinylated BG derivative and then, following a chase period of a few divisions in which the cells could produce unlabeled SNAP-tagged Cdc12 molecules, were lysed in 1 M salt to dissociate septin filaments into free hetero-octamers. These biotin-tagged hetero-octamers were recovered by affinity capture on Streptavidin-coated beads. The question then was whether the older pre-made hetero-octamers contained any free untagged SNAP-Cdc12 molecules, whose presence could be revealed via their ability to react with a BG derivative conjugated to an infrared dye. By our estimates, a substantial fraction (perhaps as high as 50%) of the biotinylated hetero-octamers could be tagged with the infrared dye, indicating that they must contain one old and one new Cdc12.31 Thus, regardless of whether intact hetero-octamers intermix during new rounds of collar assembly, collar splitting, and split ring disassembly every cell cycle, our findings suggest that individual septin subunits can also undergo substitution within an individual hetero-octamer. Whether intra-octamer subunit exchange occurs continually throughout the cell cycle or is coincident with some specific event (e.g., assembly at the newest bud site) remains a subject for future experiments.
Individual Septin Subunits Have Different Fates During a Developmental Transition
The genomes of most eukaryotes encode multiple septins, some of which are expressed primarily at certain stages of development or in certain types of differentiated cells. For example, the closely related human septins SEPT10 and SEPT14 are most highly expressed in non-dividing cells (neurons or testes, respectively).35,36 Our observation that, in cycling cells, newly synthesized septins can exchange into pre-formed complexes provided a ready explanation for how new development-specific septin subunits could be incorporated into pre-existing septin complexes during cell differentiation. We used the yeast system to explore some related questions. When cells differentiate, and some new septin genes turn on, while others turn off, how long do septins from the previous developmental stage persist and are they utilized for ensuing developmental events?
Under conditions of nutrient scarcity, a diploid yeast cell undergoes meiosis and produces four haploid spores (Fig. 2). During sporulation, several of the same septins expressed during mitotic division surround the growing prospore membranes at the time that these membranes are engulfing each of the four haploid nuclei and their peri-nuclear cytoplasm. Additionally, two septins expressed only during sporulation, Spr3 and Spr28,37,38 co-localize with the others and have been presumed (heretofore without any biochemical evidence) to populate hetero-oligomeric complexes analogous to those found in mitotic cells. By fluorescently labeling vegetatively-growing diploid cells expressing a SNAP-tagged version of Cdc10 or Cdc12, we could then monitor what happened to these proteins when these cells were induced to sporulate. We found that Cdc10 molecules made when the cells were grown mitotically persisted through meiosis and were co-localized with the sporulation-specific septins at prospore membranes.31 Moreover, when the spores so formed were returned to growth medium, and began to germinate (initiate their first mitotic division), the same pre-made fluorescently-labeled Cdc10 molecules were incorporated into the collar at the bud neck. Thus, in keeping with its demonstrated stability, Cdc10 molecules persist unscathed and remain fully functional through two distinct developmental transitions.31
Figure 2.

Certain septin subunits are recycled, whereas others are discarded, during developmental transitions in the budding yeast life cycle. Localization of Cdc10 (green) is also representative of the localization of Cdc3 and Cdc11. Septins form a broad and very diffuse band at the base of the mating projection, or shmoo. Depiction of the organization of the septin subunits within the hetero-octamers present in sporulating cells is inferred from biochemical and genetic evidence, but has not been confirmed by methods for ultrastructural analysis. It is not known whether Cdc12 and Shs1 are free or remain associated with each other when they are excluded from septin hetero-octamers during sporulation, or whether Spr3 and Spr28 are free or remain associated with each other when they are displaced from hetero-octamers during the resumption of mitotic growth.
In stark contrast, despite their stability and persistence during sporulation, fluorescently-labelled SNAP-tagged Cdc12 molecules pre-made in the mitotically-growing diploids remained in the cytosol and were not localized at the prospore membrane, were no longer found in hetero-oligomeric complexes (as judged by loss of association with Cdc3), and were absent from the bud necks of germinating spores.31 Lack of destruction of the SNAP-tagged Cdc12 during this developmental process was confirmed by immunoblotting of lysates of a culture undergoing synchronous sporulation. Thus, Cdc12 is exiled to a portion of the cytoplasm that is not efficiently encapsulated within spores and hence little is available to be utilized when spores germinate.31 This fate befalls other cellular components not destined for inheritance by newly formed spores.39,40 As might be expected based on these observations, transcription of CDC12 is repressed during sporulation, whereas that of CDC3, CDC10, SPR3 and SPR28 are induced.37,41,42 SHS1 expression is also repressed;41,42 and, indeed, we found that Shs1, like Cdc12, is excluded from the prospore membrane.31 Consistent with these results, it seems likely that Spr3 replaces Cdc12 in the hetero-octameric rod (Fig. 2), as judged by our ability to detect the presence of Spr3 in Cdc3-containing complexes in sporulating cells.31 Likewise, Shs1 is probably replaced by Spr28 (Fig. 2), which bears strong resemblance to Shs1 at the primary sequence level.15
Our findings establish that during developmental transitions certain pre-existing septin molecules are not recycled into new structures, whereas others are. The excluded septin subunits are replaced by newly-made molecules of a distinct developmentally-specific septin. Thus, in addition to the new subunits, the resulting complexes contain pre-existing molecules of subunits that can be utilized in both mitotic and meiotic cells. We have not yet explored experimentally the fate of Spr3 and Spr28 in germinating spores. When the spore initiates a mitotic division, do active mechanisms degrade these two septins and/or exclude them from the bud neck? Alternatively, are Shs1 and Cdc12 simply incorporated preferentially into hetero-octamers, thereby displacing Spr3 and Spr28, which are then diluted away upon subsequent divisions? Two previous observations support the latter possibility. First, eliminating competition by Cdc12 and Shs1 could explain the need for the transcriptional repression of CDC12 and SHS1 that occurs in sporulating cells. Second, ectopic expression of Spr3 during mitotic division has no effect on wild-type cells, suggesting that Spr3 is no match for Cdc12 when Cd12 is present.38 In agreement with that conclusion, ectopic expression of Spr3 only interferes with septin function in cells bearing mutations in Cdc12 or in the Cdc12-binding subunit Cdc11,38 both of which presumably weaken the efficiency of Cdc12 incorporation into hetero-octamers.
If septins are highly stable and recycled during successive cell cycles, what explains the apparent temporary disappearance of fluorecently-tagged septins in cells arrested pre-START, as mentioned earlier, or the apparent disappearance of Cdc10-GFP prior to the germination (first budding) of a spore?31,43 The simplest explanation is that individual fluorescently-labeled septins (and/or individual hetero-octamers) are not readily detectable in the fluorescence microscope, whereas structures containing multiple copies of that same septin (presumably in the form of hetero-octamers and filaments) are well visualized. Thus, when septin building blocks are dispersed diffusely in the cytoplasm, they cannot be easily distinguished from background fluorescence. Once assembly starts to occur, septin-containing structures then come into view. What controls when and where septins will congregate and where (and what kind of) higher-order structures will be formed? Interaction of septins with plasma membrane phospholipids44 and/or with membrane-tethered proteins that affect their state of assembly, like the Cdc42-regulated kinase Cla4,23 are good candidates for factors that regulate septin localization and supramolecular organization. However, at present, it is not known how septins are recruited to the sites of budding and spore formation. Elucidation of these important facets of septin biology awaits additional research.
Additional Perspectives
It appears that subunit exchange into septin hetero-octamers occurs in vivo, indicating that, even after it is assembled, a hetero-octamer is likely to be a dynamic entity (Fig. 3). Consistent with this view, we found that during the developmental transition from mitosis to meiosis and sporulation that the terminal (Shs1) and penultimate (Cdc12) subunits of the hetero-octameric rod are replaced by alternative developmentally-specific subunits (Spr28 and Spr3, respectively; Fig. 2). However, Cdc3 and Cdc10 have not yet been examined in the same way. Thus, it remains possible that, once formed, the Cdc10-Cdc10 homodimer, the Cdc3-Cdc10 heterodimer, and/or the Cdc3-Cdc10-Cdc10-Cdc3 heterotetramer remain stably associated and do not undergo subunit exchange during successive mitotic divisions or during sporulation. Future studies using the SNAP-tag in combination with the benzylcytidine-reactive CLIP-tag45 to simultaneously monitor in the same cell two different subunits will help further define hetero-octamer dynamics and shed further light on septin inheritance and the reorganization of septin structures in both mitosis and meiosis.
Figure 3.
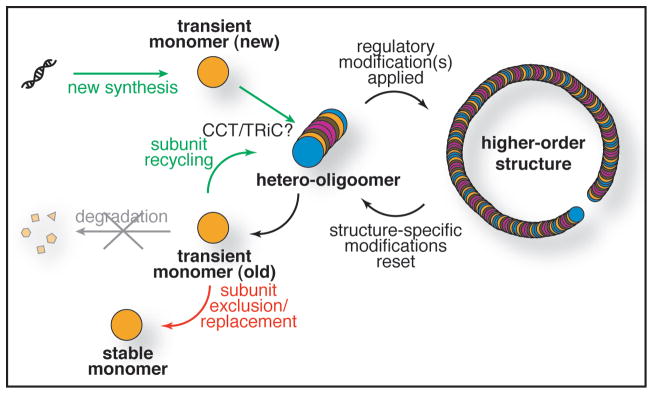
Alternative fates of a septin subunit in budding yeast. A generic septin subunit is followed diagrammatically from its initial translation and early folding steps (“new synthesis”) through subsequent incorporation with other subunits into hetero-octamers. Regulatory modifications directing assembly into higher-order structures are presumably exerted on the hetero-octamer and their removal may promote disassembly (or other active modifications may do so). In any event, upon disassembly, all such modifications must be erased or reset to allow recycling of pre-made hetero-octamers into higher-order structures in the next cell cycle. Replacement of damaged subunits or substitution of developmentally-specific subunits presumably involves partial disassembly of hetero-octamers and the incorporation of new subunits. Whether disassembly can proceed to the level of free monomers is not known. During developmental transitions, when certain subunits are excluded from hetero-octamers and replaced by other specialized subunits, this exclusion is not accompanied by degradation (and whether the excluded subunit is monomeric or in complex with other excluded subunits remains unknown). The cytosolic chaperonin complex CCT (also called TRiC) may be involved in the folding of individual septin subunits and/or in the assembly of subunits into hetero-octamers and/or in the exchange of subunits into and out of hetero-octamers. GTP binding (not shown) has an important role in stabilizing the tertiary structure of each septin and in promoting stable hetero-octamer assembly, but genetic and biochemical evidence indicates that active cycles of GTP binding and hydrolysis by septins play a minimal, if any, role in septin hetero-octamer dynamics and in the formation of higher-order structures.54
As complicated as these issues have been to sort out for budding yeast, the complexity is significantly greater for human cells. At last count, the Homo sapiens genome encodes 14 septin genes35 many of which are known to be subject to tissue-specific expression, alternative transcriptional and translational starts, and differential splicing.46 Thus, there is the potential for enormous combinatorial diversity in the nature of the septin complexes formed in different human cell types. If a given subunit is found together with different partners in different cell types, as we have observed in yeast, regulatory mechanisms ensuring both the persistence of that common subunit during differentiation and its association with the appropriate cell type-specific subunits will likely be critical for proper septin function. Indeed, misregulation of septin subunit stoichiometry and dysfunction of septin-septin interactions have pathological consequences. A single point mutation in the SEPT9 gene that causes an increase in the level of a particular isoform46 and alters its interactions with other septin subunits47 appears to be the cause of hereditary neuralgic amyotrophy (HNA). Similarly, many tumor types display misexpression of a subtly different SEPT9 isoform that is thought to perturb septin hetero-oligomer assembly in a dominant-negative fashion.48
Septin filaments are not unique as long-lived cytoskeletal polymers with cell type-specific isoforms or alternative subunits. To cite just one other example, microtubules are polymers of salt-stable heterodimers of two structurally related subunits subject, in most cellular situations, to little or no degradation.49 The α- and β-tubulins comprise the most common heterodimers, but alternative subunits, like γ-tubulins (which serve to nucleate microtubule assembly), populate certain tubulin-based structures, and some cell types express specific variants, such as the neuron-specific class III β-tubulin.50 Tubulins are decorated with an even more extensive panoply of post-translational modifications than are the septins (including acetylation, tyrosination and detyrosination, phosphorylation, glutamylation and glycylation). Many of these modifications are demonstrably regulatory; for example, periodic phosphorylation of budding yeast γ-tubulin seems to be a contributory factor in coordinating microtubule growth with cell cycle progression.51 To our knowledge, the question of whether a given tubulin subunit can populate during its lifetime the entire range of different microtubule forms found in the cytoplasm and in the mitotic spindle has not been investigated. Likewise, whether newly synthesized tubulins can exchange into preformed heterodimers in vivo has not been explored. In vitro, such tubulin subunit exchange can occur only in the presence of the CCT/TriC chaperone complex normally involved in proper tubulin protein folding;52 Interestingly, it has recently been reported that yeast septins co-purify with the CCT/TriC complex,53 providing a candidate activity potentially involved in septin subunit folding and in the exchange of subunits into hetero-octamers that we observed in vivo (Fig. 3).
Acknowledgments
This work was supported by a postdoctoral fellowship (#61-1295) from the Jane Coffin Childs Memorial Fund for Medical Research (to M.A.M.) and by NIH R01 Research Grant GM21841 (to J.T.).
References
- 1.Farkasovsky M, Herter P, Voss B, Wittinghofer A. Nucleotide binding and filament assembly of recombinant yeast septin complexes. Biol Chem. 2005;386:643–56. doi: 10.1515/BC.2005.075. [DOI] [PubMed] [Google Scholar]
- 2.Mortensen EM, McDonald H, Yates J, 3rd, Kellogg DR. Cell cycle-dependent assembly of a Gin4-septin complex. Mol Biol Cell. 2002;13:2091–105. doi: 10.1091/mbc.01-10-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vrabioiu AM, Gerber SA, Gygi SP, Field CM, Mitchison TJ. The majority of the Saccharomyces cerevisiae septin complexes do not exchange guanine nucleotides. J Biol Chem. 2004;279:3111–8. doi: 10.1074/jbc.M310941200. [DOI] [PubMed] [Google Scholar]
- 4.Hartwell LH. Genetic control of the cell division cycle in yeast IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res. 1971;69:265–76. doi: 10.1016/0014-4827(71)90223-0. [DOI] [PubMed] [Google Scholar]
- 5.Bi E, Maddox P, Lew DJ, Salmon ED, McMillan JN, Yeh E, et al. Involvement of an actomyosin contractile ring in Saccharomyces cerevisiae cytokinesis. J Cell Biol. 1998;142:1301–12. doi: 10.1083/jcb.142.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt M, Varma A, Drgon T, Bowers B, Cabib E. Septins, under Cla4p regulation, and the chitin ring are required for neck integrity in budding yeast. Mol Biol Cell. 2003;14:2128–41. doi: 10.1091/mbc.E02-08-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byers B, Goetsch L. Highly ordered ring of membrane-associated filaments in budding yeast. J Cell Biol. 1976;69:717–21. doi: 10.1083/jcb.69.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertin A, McMurray MA, Grob P, Park SS, Garcia G, 3rd, Patanwala I, et al. Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proc Natl Acad Sci USA. 2008;105:8274–9. doi: 10.1073/pnas.0803330105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frazier JA, Wong ML, Longtine MS, Pringle JR, Mann M, Mitchison TJ, et al. Polymerization of purified yeast septins: evidence that organized filament arrays may not be required for septin function. J Cell Biol. 1998;143:737–49. doi: 10.1083/jcb.143.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cid VJ, Adamikova L, Sanchez M, Molina M, Nombela C. Cell cycle control of septin ring dynamics in the budding yeast. Microbiology. 2001;147:1437–50. doi: 10.1099/00221287-147-6-1437. [DOI] [PubMed] [Google Scholar]
- 11.Lippincott J, Li R. Dual function of Cyk2, a cdc15/PSTPIP family protein, in regulating actomyosin ring dynamics and septin distribution. J Cell Biol. 1998;143:1947–60. doi: 10.1083/jcb.143.7.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobbelaere J, Barral Y. Spatial coordination of cytokinetic events by compartmentalization of the cell cortex. Science. 2004;305:393–6. doi: 10.1126/science.1099892. [DOI] [PubMed] [Google Scholar]
- 13.Caviston JP, Longtine M, Pringle JR, Bi E. The role of Cdc42p GTPase-activating proteins in assembly of the septin ring in yeast. Mol Biol Cell. 2003;14:4051–66. doi: 10.1091/mbc.E03-04-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobbelaere J, Gentry MS, Hallberg RL, Barral Y. Phosphorylation-dependent regulation of septin dynamics during the cell cycle. Dev Cell. 2003;4:345–57. doi: 10.1016/s1534-5807(03)00061-3. [DOI] [PubMed] [Google Scholar]
- 15.Pan FF, Malmberg RL, Momany M. Analysis of septins across kingdoms reveals orthology and new motifs. BMC Evol Biol. 2007;7:103–19. doi: 10.1186/1471-2148-7-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwase M, Luo J, Bi E, Toh EA. Shs1 plays separable roles in septin organization and cytokinesis in Saccharomyces cerevisiae. Genetics. 2007;177:215–29. doi: 10.1534/genetics.107.073007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mino A, Tanaka K, Kamei T, Umikawa M, Fujiwara T, Takai Y. Shs1p: A novel member of septin that interacts with Spa2p, involved in polarized growth in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1998;251:732–6. doi: 10.1006/bbrc.1998.9541. [DOI] [PubMed] [Google Scholar]
- 18.Sung H, Han KC, Kim JC, Oh KW, Yoo HS, Hong JT, et al. A set of epitope-tagging integration vectors for functional analysis in Saccharomyces cerevisiae. FEMS Yeast Res. 2005;5:943–50. doi: 10.1016/j.femsyr.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Versele M, Gullbrand B, Shulewitz MJ, Cid VJ, Bahmanyar S, Chen RE, et al. Protein-protein interactions governing septin heteropentamer assembly and septin filament organization in Saccharomyces cerevisiae. Mol Biol Cell. 2004 doi: 10.1091/mbc.E04-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson ES, Blobel G. Cell cycle-regulated attachment of the ubiquitin-related protein SUMO to the yeast septins. J Cell Biol. 1999;147:981–94. doi: 10.1083/jcb.147.5.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egelhofer TA, Villen J, McCusker D, Gygi SP, Kellogg DR. The septins function in G1 pathways that influence the pattern of cell growth in budding yeast. PLoS ONE. 2008;3:2022. doi: 10.1371/journal.pone.0002022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang CS, Reed SI. Phosphorylation of the septin Cdc3 in G1 by the Cdc28 kinase is essential for efficient septin ring disassembly. Cell Cycle. 2002;1:42–9. [PubMed] [Google Scholar]
- 23.Versele M, Thorner J. Septin collar formation in budding yeast requires GTP binding and direct phosphorylation by the PAK, Cla4. J Cell Biol. 2004;164:701–15. doi: 10.1083/jcb.200312070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holly SP, Blumer KJ. PAK-family kinases regulate cell and actin polarization throughout the cell cycle of Saccharomyces cerevisiae. J Cell Biol. 1999;147:845–56. doi: 10.1083/jcb.147.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Detweiler CS, Li JJ. Ectopic induction of Clb2 in early G1 phase is sufficient to block prereplicative complex formation in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1998;95:2384–9. doi: 10.1073/pnas.95.5.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang D, Patrick G, Moffat J, Tsai LH, Andrews B. Mammalian Cdk5 is a functional homologue of the budding yeast Pho85 cyclin-dependent protein kinase. Proc Natl Acad Sci USA. 1999;96:14445–50. doi: 10.1073/pnas.96.25.14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang D, Moffat J, Andrews B. Dissection of a complex phenotype by functional genomics reveals roles for the yeast cyclin-dependent protein kinase Pho85 in stress adaptation and cell integrity. Mol Cell Biol. 2002;22:5076–88. doi: 10.1128/MCB.22.14.5076-5088.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moffat J, Andrews B. Late-G1 cyclin-CDK activity is essential for control of cell morphogenesis in budding yeast. Nat Cell Biol. 2004;6:59–66. doi: 10.1038/ncb1078. [DOI] [PubMed] [Google Scholar]
- 29.Keppler A, Pick H, Arrivoli C, Vogel H, Johnsson K. Labeling of fusion proteins with synthetic fluorophores in live cells. Proc Natl Acad Sci USA. 2004;101:9955–9. doi: 10.1073/pnas.0401923101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nasmyth K. Regulating the HO endonuclease in yeast. Curr Opin Genet Dev. 1993;3:286–94. doi: 10.1016/0959-437x(93)90036-o. [DOI] [PubMed] [Google Scholar]
- 31.McMurray MA, Thorner J. Septin stability and recycling during dynamic structural transitions in cell division and development. Curr Biol. 2008;18:1203–8. doi: 10.1016/j.cub.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vrabioiu AM, Mitchison TJ. Symmetry of septin hourglass and ring structures. J Mol Biol. 2007 doi: 10.1016/j.jmb.2007.05.100. [DOI] [PubMed] [Google Scholar]
- 33.Smolka MB, Albuquerque CP, Chen SH, Zhou H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA. 2007;104:10364–9. doi: 10.1073/pnas.0701622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smolka MB, Chen SH, Maddox PS, Enserink JM, Albuquerque CP, Wei XX, et al. An FHA domain-mediated protein interaction network of Rad53 reveals its role in polarized cell growth. J Cell Biol. 2006;175:743–53. doi: 10.1083/jcb.200605081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peterson EA, Kalikin LM, Steels JD, Estey MP, Trimble WS, Petty EM. Characterization of a SEPT9 interacting protein, SEPT14, a novel testis-specific septin. Mamm Genome. 2007;18:796–807. doi: 10.1007/s00335-007-9065-x. [DOI] [PubMed] [Google Scholar]
- 36.Sui L, Zhang W, Liu Q, Chen T, Li N, Wan T, et al. Cloning and functional characterization of human septin 10, a novel member of septin family cloned from dendritic cells. Biochem Biophys Res Commun. 2003;304:393–8. doi: 10.1016/s0006-291x(03)00601-6. [DOI] [PubMed] [Google Scholar]
- 37.De Virgilio C, DeMarini DJ, Pringle JR. SPR28, a sixth member of the septin gene family in Saccharomyces cerevisiae that is expressed specifically in sporulating cells. Microbiology. 1996;142:2897–905. doi: 10.1099/13500872-142-10-2897. [DOI] [PubMed] [Google Scholar]
- 38.Fares H, Goetsch L, Pringle JR. Identification of a developmentally regulated septin and involvement of the septins in spore formation in Saccharomyces cerevisiae. J Cell Biol. 1996;132:399–411. doi: 10.1083/jcb.132.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurtz S, Lindquist S. Subcellular differentiation in sporulating yeast cells. Cell. 1986;45:771–9. doi: 10.1016/0092-8674(86)90791-9. [DOI] [PubMed] [Google Scholar]
- 40.Suda Y, Nakanishi H, Mathieson EM, Neiman AM. Alternative modes of organellar segregation during sporulation in Saccharomyces cerevisiae. Eukaryot Cell. 2007;6:2009–17. doi: 10.1128/EC.00238-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, et al. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 42.Friedlander G, Joseph-Strauss D, Carmi M, Zenvirth D, Simchen G, Barkai N. Modulation of the transcription regulatory program in yeast cells committed to sporulation. Genome Biol. 2006;7:20. doi: 10.1186/gb-2006-7-3-r20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joseph-Strauss D, Zenvirth D, Simchen G, Barkai N. Spore germination in Saccharomyces cerevisiae: global gene expression patterns and cell cycle landmarks. Genome Biol. 2007;8:241. doi: 10.1186/gb-2007-8-11-r241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Casamayor A, Snyder M. Molecular dissection of a yeast septin: distinct domains are required for septin interaction, localization and function. Mol Cell Biol. 2003;23:2762–77. doi: 10.1128/MCB.23.8.2762-2777.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gautier A, Juillerat A, Heinis C, Correa IR, Jr, Kindermann M, Beaufils F, et al. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–36. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 46.McDade SS, Hall PA, Russell SE. Translational control of SEPT9 isoforms is perturbed in disease. Hum Mol Genet. 2007;16:742–52. doi: 10.1093/hmg/ddm003. [DOI] [PubMed] [Google Scholar]
- 47.Sudo K, Ito H, Iwamoto I, Morishita R, Asano T, Nagata K. SEPT9 sequence alternations causing hereditary neuralgic amyotrophy are associated with altered interactions with SEPT4/SEPT11 and resistance to Rho/Rhotekin-signaling. Hum Mutat. 2007;28:1005–13. doi: 10.1002/humu.20554. [DOI] [PubMed] [Google Scholar]
- 48.Russell SE, Hall PA. Do septins have a role in cancer? Br J Cancer. 2005;93:499–503. doi: 10.1038/sj.bjc.6602753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fridovich-Keil JL, Bond JF, Solomon F. Domains of beta-tubulin essential for conserved functions in vivo. Mol Cell Biol. 1987;7:3792–8. doi: 10.1128/mcb.7.10.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dutcher SK. The tubulin fraternity: alpha to eta. Curr Opin Cell Biol. 2001;13:49–54. doi: 10.1016/s0955-0674(00)00173-3. [DOI] [PubMed] [Google Scholar]
- 51.Vogel J, Drapkin B, Oomen J, Beach D, Bloom K, Snyder M. Phosphorylation of gamma-tubulin regulates microtubule organization in budding yeast. Dev Cell. 2001;1:621–31. doi: 10.1016/s1534-5807(01)00073-9. [DOI] [PubMed] [Google Scholar]
- 52.Tian G, Bhamidipati A, Cowan NJ, Lewis SA. Tubulin folding cofactors as GTPase-activating proteins. GTP hydrolysis and the assembly of the alpha/beta-tubulin heterodimer. J Biol Chem. 1999;274:24054–8. doi: 10.1074/jbc.274.34.24054. [DOI] [PubMed] [Google Scholar]
- 53.Dekker C, Stirling PC, McCormack EA, Filmore H, Paul A, Brost RL, et al. The interaction network of the chaperonin CCT. EMBO J. 2008;27:1827–39. doi: 10.1038/emboj.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McMurray MA, Thorner J. Biochemical properties and supramolecular architecture of septin hetero-oligomers and septin filaments. In: Hall PA, Russell SEG, Pringle JR, editors. The Septins. Chicester, West Sussex, UK: John Wiley & Sons, Ltd; 2008. pp. 49–100. [Google Scholar]