

Abstract
The bivalent ligand approach has been utilized not only to study the underlying mechanism of G protein-coupled receptors dimerization and/or oligomerization, but also aimed to enhance ligand affinity and/or selectivity for potential treatment of a variety of diseases by targeting this process. Substance abuse and addiction have made both the prevention and the treatment of human immunodeficiency virus (HIV) infection more difficult to tackle. It has been extensively studied that morphine, a mu opioid receptor (MOR) agonist, can accelerate HIV infection through up-regulating the expression of the chemokine receptor CCR5, a well-known co-receptor for HIV invasion to the host cells. Meanwhile, two research groups have described the putative MOR/CCR5 heterodimers in their independent studies. The purpose of this paper is to report the design and synthesis of a bivalent ligand to explore the biological and pharmacological process of the putative MOR/CCR5 dimerization phenomenon. The developed bivalent ligand thus contains two distinct pharmacophores linked through a spacer; ideally one of which will interact with the MOR and the other with the CCR5. Naltrexone and Maraviroc were selected as the pharmacophores to generate such a bivalent probe. The overall reaction route to prepare this bivalent ligand was convergent and efficient, and involved sixteen steps with moderate to good yields. The preliminary biological characterization showed that the bivalent compound 1 retained the pharmacological characteristics of both pharmacophores towards the MOR and the CCR5 respectively with relatively lower binding affinity, which tentatively validated our original molecular design.
Keywords: Mu opioid receptor, Chemokine receptor CCR5, HIV, Drug abuse, Naltrexone, Maraviroc, Bivalent ligand
Introduction
The negative co-operativity among β-adrenergic receptors in frog erythrocyte membranes1 and occurrence of opioid receptors in clusters on the neuroblastoma cell surface2 were attributed to be the earliest reports of receptor dimerization/oligomerization.3–4 The exact term “dimerization” was actually coined by Gregory and co-workers in 1982,5 yet it took more than another decade for more convincing evidence being recognized. Among the evidence, the most significant ones are the co-expression studies with mutant muscarinic/adrenergic receptors conducted by Maggio et al.6 and a co-immunoprecipitation approach utilized by Bouvier group for β2-adrenergic receptor7. The “dimerization/oligomerization” concept for G protein-coupled receptors (GPCRs) was widely accepted by the end of 1990s based on the research of GABAB receptor from several groups8, and was further supported by the X-ray crystal structures of some others later on9. GPCRs dimerization/oligomerization poses a differentiated pharmacology from the monomers.10 In this regard, a number of bivalent ligands have been synthesized to explore the underlying biology and pharmacology mechanisms of GPCRs dimerization/oligomerization, as well as to develop prospective agents with enhanced affinity and/or selectivity to treat different disorders and diseases by targeting this “novel” mechanism.4a, 11
Since acquired immunodeficiency syndrome (AIDS) was identified three decades ago12, the global prevalence of AIDS has become stable at 0.8%, with over 33 million people infected with human immunodeficiency virus (HIV) in 200713. There are almost 16 million people who are injecting drug users (IDUs) worldwide and nearly 10% of HIV infection was attributed to injecting drug use through contaminated needles.14 Statistics showed IDUs account for approximately 13% of the total HIV infection in thirty-four US states during 2004 to 2007.15 Not only driving HIV transmission among IDUs, the abused substances, such as opioids, cocaine, and alcohol also accelerate the progression of AIDS and complicate the treatment of this disease.16 Moreover, HIV infection seems to increase drug addiction vulnerability as well.17 Needle-exchange programs (NEPs) have shown appreciable outcome on reducing HIV prevalence among IDUs for over a decade.18 The current available treatment for opioid-dependent HIV patients also adopts opioid substitution therapy (OST), i.e. methadone, buprenorphine and buprenorphine/naloxone, into HIV management.19 Although opioid maintenance therapy have shown to improve patients adherence and promising outcome for HIV treatment, the adverse drug-drug interaction between methadone, buprenorphine and antiretroviral agents compromise the overall effects.20 New agents and remedies are still highly demanded for the treatment of these patients.
Opiates and alcohol abuse/addiction liability is mainly associated with the mu opioid receptor (MOR)21, which is also involved in different immunomodulatory activities induced by opioids22. The chemokine receptor CCR5 was identified as a major co-receptor for HIV in 199623, and is largely expressed on activated memory CD45RO+ T cells, monocyte/macrophages, dendritic cells, granulocyte precursors, and natural killer cells24. Both receptors belong to the seven-transmembrane GPCR superfamily. Several research groups have shown that MOR agonists, such as morphine, methadone, and DAMGO, can increase CCR5 expression, thus enhance and facilitate HIV infection and replication both in vitro and in vivo.25 In light of this observation, as well as the fact that the opioid receptors and the CCR5 are all present on immune cells, Suzuki and co-workers studied the interactions between these GPCRs through a co-immunoprecipitation approach.26 Their research demonstrated for the first time that CCR5 and opioid receptors form oligomers and the oligomerization modulates the function of such a complex.26 Two years later, Chen et al. reported the heterodimerization and cross-desensitization between the MOR and the CCR5 in co-expressed Chinese hamster ovary (CHO) cells.27 The authors proposed that the MOR/CCR5 heterodimers may contribute to the observed cross-desensitization. Despite these fundamental studies, a chemical probe that is capable of interacting with both receptors simultaneously has never been developed to facilitate the study of the biological and pharmacological process of MOR/CCR5 dimerization. Herein, we report the design and synthesis of a bivalent ligand 1 (Figure 1) as a lead compound in order to test our hypothesis and to reveal the underlying mechanism of MOR/CCR5 heterodimers, eventually.
Figure 1.

Chemical structures of naltrexone, Maraviroc, designed bivalent (1) and monovalent ligands (2, 3).
Bivalent Ligand Rational Design
Receptor antagonists serve as important pharmacological probes to uncover the probable involvement of a receptor mechanism.28 Therefore, it seemed ideal to build a bivalent ligand containing a MOR-antagonist moiety as well as a CCR5-antagonist one, linked through an appropriate spacer. Naltrexone29 (Figure 1) was selected as the moiety to interact with the MOR based on the following considerations: first, naltrexone has been successfully used to investigate the dimerization of opioid receptors previously30; second, it represents an ideal treatment for alcohol and opiate addiction and has been successfully used to treat alcoholism clinically31. Maraviroc32 (Figure 1) is the only CCR5 antagonist that has been approved for HIV treatment by the FDA so far33 and thus became our first choice as the CCR5 pharmacophore. Meanwhile, both of these two ligands showed high affinity and reasonable selectivity toward the MOR and the CCR5 respectively.
It has been proved that the loci for tethering two pharmacophores through a spacer affect the binding affinities of the resulted bivalent ligands.34 In addition, the overall chemical modification of these two pharmacophores for spacer attachment should also be designed from a synthetic point of view, that is, chemical reactions should be readily accomplished. Thus, based on the successful cases from Portoghese group30a,b, the C6-position of naltrexone was selected as the attaching locus after transforming its carbonyl group to the 6β-amino group (Figure 1). Whereas the discovery process of Maraviroc revealed that both of the difluorocyclohexyl moiety and the exo-1,2,4-triazole substituted tropane core are essential to its potent antiviral activity and weak hERG inhibition.35 Additionally, an interactive docking study of Maraviroc to a rhodopsin-based CCR5 homology model demonstrated the interactions between Glu283 and the tropane core, as well as Ile198 and the difluorocyclohexyl moiety within the proposed binding pocket.36 Hence, the para-position of the phenyl ring in Maraviroc was first chosen as the linking site to avoid severe impacts to the above interactions. Since EDCI/HOBt mediated coupling reaction between carboxylic acid and amine can be easily accomplished, an amino group was then chosen as the functional group on this para-phenyl ring to hook Maraviroc up with the spacer. Thus, pharmacophore 6 (Scheme 1) was designed as the precursor of the CCR5 antagonist.
Scheme 1.

Retrosynthetic analysis of the bivalent ligand 1
Several studies indicated that a spacer with 16 to 22 atoms might be beneficial for targeting GPCR dimers, ideally with 21 atoms when both pharmacophores are antagonists of their respective receptors.30a,b,37 Therefore, the 21-atom-spacer was adopted as an initial lead in the current study. The design rationale of such a spacer is to keep a favorable balance between hydrophobicity and hydrophilicity as well as to possess a reasonable rigidity, high stability and low toxicity.38 Hence, one alkyldiamine moiety and two diglycolic units were employed to build up the spacer. Monovalent ligands 2 and 3 (Figure 1) were also designed as controls to clarify the potential effects of the spacer to the binding affinity and potency of the bivalent ligand.
Chemistry and biological studies
The retrosynthetic analysis of bivalent ligand 1 revealed three major fragments, 6β-naltrexamine 4, diacid spacer 5, and the CCR5 antagonist precursor 4-NH2-Maraviroc 6 (Scheme 1). Among them, 6β-naltrexamine 4 can be conveniently prepared from naltrexone following the reported procedure39, whereas nucleophilic reaction of 1,7-diaminoheptane with diglycolic anhydride30b can readily afford the diacid spacer 5. Similarly to Maraviroc, retrosynthetic analysis of 6 identified three key fragments: a 4,4-difluorocyclohexanecarboxylic acid 8, a β-phenylalanine ester 9, and a triazole-substituted tropane 10 (Scheme 1). As the preparation of difluoro acid 8 appeared to be challenging40, a strategy that enables a later introduction of this fragment was sought. In order to avoid the tedious reduction-oxidation procedure as well as to improve the overall yields, an amide coupling strategy instead of reductive amination40,41 was postulated to generate 7 by coupling 9 with 10. Several papers have reported the highly stereoselective introduction of an amino group through Michael addition with lithium (R)-N-benzyl-N-α-methylbenzylamide in high yields.42 Hence, the same method was adopted to prepare fragment 9. Two cinnamic acids are commercially available to synthesize the substrate 11 for a later Michael addition: 4-nitrocinnacid and 4-bromocinnacid. However, the conversion of the nitro group to the amino group poses an issue for the overall synthetic route since other functional groups, such as double bond, ester, benzyl, and amide, are present in the same molecule and reducing agents such as Na2S and SnCl2 are not environment-friendly. Therefore, 4-bromocinnacid was chosen as the starting material.
The overall synthesis of the precursor 6 is illustrated in Schemes 2 and 3. The carboxylic group protection was performed by refluxing 4-bromocinnacid in isopropanol with a few drops of concentrated sulfuric acid to give a moderate yield of 12.43 The bromide 12 was then converted to aniline 13 using Lithium hexamethyldisilazide (LHMDS) catalyzed by Pd2(dba)3 and P(t-Bu)344, which upon heating with di-tert-butyl dicarbonate furnished compound 14 in a good yield45. The diastereoselective Michael addition of 14 was achieved with lithium (R)-N-benzyl-N-α-methylbenzylamide prepared in situ42e. The stereoselectivity was confirmed by comparing with the literature reported data42f. The following saponification of the conjugate adduct 15 gave acid 16, which was then coupled with 3-(3-isopropyl-5-methyl-4H-1,2,4-triazol-4-yl)-exo-8-azabicyclo[3.2.1]octane41a 10 through HOBt/EDCI method to yield 17. The reduction of 17 with either BH3·THF or LiAlH4 at ambient temperature did not give any amide reduced product 18. Heating 17 with BH3·THF resulted a mixture of complexes, with the loss of Boc and/or two benzyl groups. The steric hindrance generated by the two benzyl groups might complicate the reduction process. Hence, the catalytic hydrogenolysis of 17 with 10% Pd/C was conducted to produce intermediate 19 instead. Although reaction of 19 with BH3·THF did afford compound 7, the majority of the product formed a complex with tetrahydrofuran (1:1), which requires acid to release the free amine46. However, Boc group may be sensitive to such acidic conditions. Replacement of BH3·THF with LiAlH4, which only needs water to decompose the intermediate formed after the reaction,47 provided 7 in a reasonable yield (Scheme 2). Reaction of 7 with 4,4-difluorocyclohexanecarboxylic acid48 8 was mediated by HOBt/EDCI and the coupling product 20 was subsequently converted to the CCR5 antagonist precursor 6 with TFA/DCM (1:10) at ambient temperature49 (Scheme 3).
Scheme 2.

Synthesis of intermediate 7a
a Regents and conditions: (a) i-PrOH, H2SO4 (conc.), reflux, 80%; (b) i) LHMDS, Pd2(dba)3, P(t-Bu)3, Toluene, rt; ii) 1N HCl, rt, 90%; (c) Boc2O, THF, reflux, 85%; (d) THF, −78 °C; (e) LiOH, MeOH/H2O (2/1), reflux, 85%, two steps; (f) EDCI, HOBt, TEA, 10, 4Å MS, DCM, 0 °C to rt, 73%; (g) 10% Pd/C, 60 psi, MeOH, 84%; (h) i) LiAlH4, THF, 0 °C to rt; ii) H2O, NaOH, 71%.
Scheme 3.
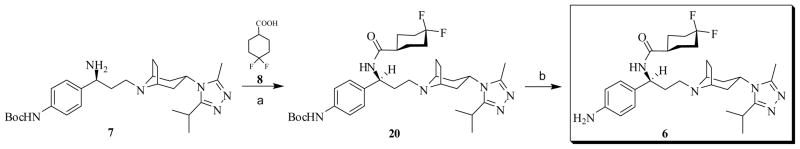
Synthesis of 4-NH2-Maraviroc (6)a
(a) EDCI, HOBt, TEA, 8, 4Å MS, DCM, 0 °C to rt, 85%; (b) CF3COOH, DCM, 0 °C to rt, 95%.
Then the bivalent ligand 1 was prepared following a linear synthetic route as shown in Scheme 4. Reaction of 1,7-diaminoheptane with 0.9 equivalent of benzyl chloroformate under ice-water bath generated mono-Cbz protected intermediate 2150, which was further condensed with diglycolic anhydride to give compound 2230b. Intermediate 23 was prepared by coupling 22 with 4 (6β-naltrexamine39) utilizing HOBt/EDCI method. Hydrogenation-deprotection of 23 with 10% Pd/C catalyst yielded amine 24. Condensation of 24 with a second molecule of diglycolic anhydride provided acid 25, which was then coupled with the CCR5 antagonist precursor 6 via HOBt/EDCI mediation to furnish bivalent ligand 1.
Scheme 4.

Synthesis of bivalent ligand 1a
a Regents and conditions: (a) CbzCl, DCM, MeOH, 5 °C, 32%; (b) THF, diglycolic anhydride, rt, 85%; (c) EDCI, HOBt, TEA, 4·2HCl, 4Å MS, DMF, 0 °C to rt, 76%; (d) 10% Pd/C, 60 psi, MeOH, 99%; (e) DMF, diglycolic anhydride, rt, 82%; (f) EDCI, HOBt, TEA, 6, 4Å MS, DMF, 0 °C to rt, 50%.
Monovalent ligand 2 was conveniently synthesized by coupling the intermediate 24 with 2630b via HOBt/EDCI peptide coupling method (Scheme 5).
Scheme 5.
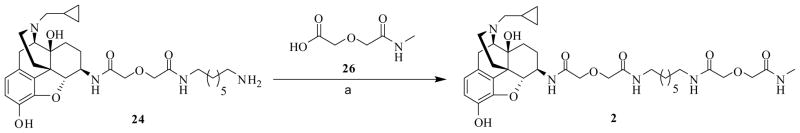
Synthesis of monovalent ligand 2a
a Regents and conditions: (a) EDCI, HOBt, TEA, 26, 4Å MS, DMF, 0 °C to rt, 65%.
From an efficient synthesis perspective, monovalent ligand 3 was prepared according to Scheme 6, considering it only involved three steps and all the reactions can be simply monitored by UV. Thus, HOBt/EDCI-mediated coupling of 22 with precursor 6 afforded intermediate 27, which underwent catalytic hydrogenolysis to yield amine 28. Monovalent ligand 3 was then obtained by coupling 28 with 26 employing HOBt/EDCI method.
Scheme 6.

Synthesis of monovalent ligand 3a
a Regents and conditions: (a) EDCI, HOBt, TEA, 6, 4Å MS, DMF, 0 °C to rt, 72%; (b) 5% Pd/C, 60 psi, MeOH, 51%; (c) EDCI, HOBt, TEA, 26, 4Å MS, DMF, 0 °C to rt, 81%.
All three ligands were then further characterized for their binding affinity and functional activity preliminarily. In a calcium mobilization assay with CCR5/MOLT-4 cells51, compound 1 showed no agonism and its antagonist property indicated by its calcium flux inhibition IC50 value as 231 ± 88 nM. Compared with the calcium flux inhibition IC50 value of Maraviroc under the same experimental condition, which was 1.57 ± 0.32 nM, apparently the introduction of the long chain spacer seemed to be influential to the binding affinity of compound 1 to the receptor CCR5, as indicted by its significant decrement on functional activity of calcium flux inhibition. This was further supported by the even lower functional property of the control compound 3, of which the calcium flux inhibition IC50 value was 833 ± 150 nM. A couple of reasons could lead to such results. First, the bulkiness of the spacer might influence the binding affinity to the receptor directly. Second, the substitution position of the spacer on the tailing aromatic ring system of Maraviroc might not be the most suitable one in preventing serious steric hindrance effect on the binding affinity. Currently the syntheses of new ligands with spacer attached at different position of this tailing ring system are underway.
Similarly, in 35S-GTP[γS] binding assays in MOR-CHO cells52, compound 1 showed very little apparent agonism (Emax = 11.7 ± 1.2%) compared to the full agonist DAMGO (100 ± 9.2%, EC50 = 13.7 ± 1.6 nM), while its binding affinity to the mu opioid receptor as indicated by Ki value was 51.8 ± 7.9 nM, which was lower than naltrexone’s binding affinity (Ki value was 0.71 ± 0.08 nM) under the same experimental condition. Correspondingly the control compound 2 also showed somewhat lower binding affinity as indicated by the Ki value of 9.18 ± 3.44 nM.
These preliminary biological activity results supported our original molecular design that the bivalent ligand did reserve the original antagonist property from both pharmacophores while its relatively lower affinity to the each corresponding receptor compared to the parent pharmacophores, which was not unusual based on previous reports from others34c,37e, certainly requires more extensive structural modification and further syntheses effort.
Conclusions
In conclusion, a bivalent ligand with 21-atom spacer was designed and synthesized as a molecular probe to study the biological and pharmacological mechanisms of the putative heterodimerization between the mu opioid receptor and the chemokine receptor CCR5. The overall 16-step synthetic route was efficient and convergent with reasonable yields. The preliminary biological data from the calcium mobilization assay and MOR-CHO binding assay showed that the bivalent ligand 1 retained the characteristics of its pharmacophores, antagonizing MOR and/or CCR5 respectively, with relatively lower binding affinity. Further characterization of these ligands and synthesis of ligands with different length of spacer and linkage at different position are undergoing right now. Based on the current pilot study, it is believed that such a bivalent ligand with a favorable length of spacer and an optimized linkage site may serve as a pharmacological probe to study the function of the putative MOR/CCR5 dimerization and help to understand the mechanism of such protein-protein interactions in various neuronal-immuno diseases, for example, HIV-infected opiate/alcohol abuse and addiction.
Experimental
Synthesis
General methods
All reagents were purchased from Sigma-Aldrich or as otherwise stated. TLC analyses were carried out on Analtech Uniplate F254 plates. Chromatographic purification was accomplished on silica gel columns (230~400 mesh, Merck). Melting points were obtained with a Fisher scientific micro melting point apparatus without further correction. All IR spectra were recorded on a Nicolet iS10 FT-IR Instrument. Proton (400 MHz) and Carbon-13 (100 MHz) nuclear magnetic resonance (NMR) spectra were acquired at ambient temperature with tetramethylsilane as the internal standard on a Bruker Ultrashield 400 Plus spectrometer. MS analysis was performed on an Applied Bio Systems 3200 Q trap with a turbo V source for TurbolonSpray. HPLC analysis of the final compounds was achieved on Varian ProStar 210 system on Microsorb-MV 100-5 C18 column (250 mm × 4.6 mm) at 254 (1 & 3) or 210 (2) nm eluting with acetonitrile (0.1% TFA)/water (50/50) at 1 mL/min over 10 min. Elemental analysis of the final compounds was conducted in Atlantic Microlab, Inc.
General procedure for amide coupling
On an ice-water bath, a solution of acid in either DCM or DMF (3 mL), was added EDCI (1.5 eq), HOBt (1.5 eq), molecular sieves, and TEA (4.0 eq) with N2 protection. After 15 min, a solution of amine (1.0 eq) in DMF or DCM (1 mL) was added dropwise. The resulted mixture was allowed to warm up to ambient temperature gradually. After completion of the reaction as monitored by TLC, the reaction mixture was filtered through celite. When DMF was used as the reaction solvent, the filtrate was concentrated in vacuum to remove DMF and the residue was then purified with column chromatography to afford the coupling product, whereas when DCM was the solvent, the filtrate was washed with brine, dried over Na2SO4, concentrated and the crude product was purified by either crystallization or column chromatography.
Bivalent ligand 1
The title compound was prepared according to the general amide coupling procedure by reacting acid 25 with amine 6 in DMF for 7 days. The crude product was purified by column chromatography using CH2Cl2/MeOH (10/1) as eluent to give 87 mg white solid, in 50% yield. 1H NMR (400 MHz, CD3OD): δ 7.60 (d, J = 8.56 Hz, 2H), 7.33 (d, J = 8.56 Hz, 2H), 6.67 (d, J = 8.08 Hz, 1H), 6.62 (d, J = 8.20 Hz, 1H), 5.03 (t, J = 7.38 Hz, 1H), 4.58 (d, J = 7.64 Hz, 1H), 4.39 (m, 1H), 4.20 (s, 2H), 4.12 (s, 2H), 4.05 (s, 2H), 4.04 (s, 2H), 3.74 (m, 1H), 3.43 (m, 2H), 3.28–3.23 (m, 5H), 2.90–2.74 (m, 3H), 2.50 (s, 3H), 2.48–2.41 (m, 2H), 2.41–2.32 (m, 2H), 2.30–2.20 (m, 2H), 2.19–2.05 (m, 4H), 2.04–1.69 (m, 15H), 1.68–1.46 (m, 8H), 1.37 (m, 6H), 1.34 (d, J = 6.84 Hz, 6H), 0.94 (m, 1H), 0.64–0.58 (m, 2H), 0.29 (m, 2H); 13C NMR (100 MHz, CD3OD): δ 176.63, 171.70, 171.55, 171.50, 170.10, 161.47, 161.43, 143.77, 143.44, 140.34, 138.06, 137.93, 128.22, 121.89, 120.63, 120.43, 119.03, 92.56, 71.99, 71.79, 71.62, 71.58 (× 2), 63.99, 60.76, 60.24, 57.25, 52.46, 52.40, 49.51, 49.30, 44.31, 43.69, 40.06, 38.17, 36.74, 36.08, 35.89, 33.88 (J 13C-19F 23 Hz), 31.20, 30.34, 30.31, 29.95, 28.03, 27.84, 27.82, 27.29, 27.19, 27.07, 27.00, 26.76, 25.17, 23.91, 22.06, 15.72, 12.45, 3.93. IR ν (Diamond, cm−1): 3275, 1652, 1532, 1128, 1107. mp 158.5–160 °C. Anal. Calcd for C64H92F2N10O11: C 63.24, H 7.63, N 11.52; Found: C 63.15, H 7.57, N 11.28. MS (ESI) m/z found 1198.4 (M + H)+, 1220.5 (M + Na)+.
Monovalent ligand 2
The title compound was prepared according to the general amide coupling procedure by reacting acid 26 with amine 24 in DMF for 8 h. The crude product was purified with chromatography using CH2Cl2/MeOH (20/1) as eluent to give 32 mg white solid, in 65% yield. 1H NMR (400 MHz, DMSO-d6): δ 9.03 (brs, 1H), 8.22 (d, J = 8.44 Hz, 1H), 8.04–7.96 (m, 3H), 6.59 (d, J = 8.08 Hz, 1H), 6.53 (d, J = 8.12 Hz, 1H), 4.90 (brs, 1H), 4.60 (d, J = 7.80 Hz, 1H), 3.95 (s, 2H), 3.94 (s, 2H), 3.91 (m, 4H), 3.57–3.48 (m, 1 H), 3.19–3.09 (m, 4H), 3.03–2.99 (m, 2H), 2.66 (d, J = 4.68 Hz, 3H), 2.63–2.58 (m, 2H), 2.40–2.29 (m, 2H), 2.32–2.09 (m, 1H), 1.99 (dt, J1 = 3.43 Hz, J2 = 11.92 Hz, 1H), 1.84–1.75 (m, 1H), 1.48–1.42 (m, 6H), 1.33–1.24 (m, 8H), 0.86 (m, 1H), 0.48 (m, 2H), 0.13 (m, 2H); 1H NMR (400 MHz, CD3OD): δ 6.67 (d, J = 8.12 Hz, 1H), 6.60 (d, J = 8.12 Hz, 1H), 4.56 (d, J = 7.56 Hz, 1H), 4.10 (s, 2H), 4.09 (s, 2H), 4.08 (s, 2H), 4.06 (s, 2H), 3.82–3.78 (m, 1H), 3.30 (q, J = 7.34 Hz, 4H), 3.18–3.13 (m, 2H), 2.82 (s, 3H), 2.73–2.64 (m, 2H), 2.48–2.40 (m, 2H), 2.32–2.25 (m, 1H), 2.18 (dt, J1 = 3.04 Hz, J2 = 11.81 Hz, 1H), 1.94 (m, 1H), 1.60–1.58 (m, 6H), 1.48–1.32 (m, 8H), 0.95 (m, 1H), 0.56 (m, 2H), 0.19 (m, 2H); 13C NMR (100 MHz, CD3OD): δ 172.10, 171.51, 171.43, 171.38, 143.71, 141.88, 132.49, 125.45, 120.06, 118.55, 92.89, 71.68, 71.57, 71.53, 71.45, 71.41, 63.72, 60.27, 52.45, 48.88, 45.38, 45.24, 40.02, 31.98, 31.22, 30.35, 29.99, 27.84, 25.87, 25.46, 23.52, 10.29, 4.45, 4.21. IR ν (Diamond, cm−1): 3291, 1652, 1544, 1124. mp 74–76 °C. Anal. Calad for C36H55N5O10: C 60.23, H 7.72, N 9.76; Found: C 60.22, H 7.74, N 9.57. MS (ESI) m/z found 701.0 (M + H)+, 722.9 (M + Na)+.
Monovalent ligand 3
The title compound was prepared according to the general amide coupling procedure by reacting acid 26 with amine 28 in DMF overnight. The crude product was purified with chromatography using CH2Cl2/MeOH (8/1) as eluent to give 106 mg white solid, in 81% yield. 1H NMR (400 MHz, CD3OD): δ 7.60 (d, J = 8.52 Hz, 2H), 7.33 (d, J = 8.56 Hz, 2H), 5.03 (t, J = 7.32 Hz, 1H), 4.44–4.35 (m, 1H), 4.20 (s, 2H), 4.12 (s, 2H), 4.02 (m, 4H), 3.42 (m, 2H), 3.30–3.20 (m, 5H), 2.78 (s, 3H), 2.50 (s, 3H), 2.47–2.40 (m, 2H), 2.39–2.30 (m, 1H), 2.30–2.17 (m, 2H), 2.15–2.03 (m, 4H), 2.02–1.95 (m, 2H), 1.90–1.65 (m, 10H), 1.60–1.45 (m, 4H), 1.40–1.30 (m, 12H); 13C NMR (100 MHz, CD3OD): δ 176.61, 172.13, 171.68, 171.46, 170.08, 161.42, 152.61, 140.33, 138.08, 128.22, 121.89, 71.97, 71.77, 71.48, 71.44, 60.71, 60.22, 52.40, 44.55, 43.70, 40.07, 40.03, 36.75, 36.10, 33.86 (J 13C-19F 23 Hz), 30.38, 30.35, 29.99, 27.86, 27.20 (J 13C-19F 9 Hz), 27.07, 27.02, 26.98, 26.76, 25.89, 22.06, 12.44. IR ν (Diamond, cm−1): 3272, 1652, 1532, 1107. mp 79–81 °C. Anal. Calcd for C45H73F2N9O9: C 58.61, H 7.98, N 13.67; Found: C 59.29, H 7.96, N 13.47. MS (ESI) m/z found 887.2 (M + H)+, 909.3 (M + Na)+.
6′β-Naltrexamine hydrochloride salt (4·2HCl)
The title compound was prepared following the reported procedure39 in 62% yield for two steps (lit., 39 71%). 1H NMR (400 MHz, DMSO-d6): δ 9.58 (s, 1H, exchangeable), 8.91 (brs, 1H, exchangeable), 8.43 (m, 3H, exchangeable), 6.80 (d, J = 8.0 Hz, 1H), 6.67 (d, J = 8.0 Hz, 1H), 6.40 (brs, 1H), 4.68 (d, J = 7.2 Hz, 1H), 3.90 (d, J = 4.8 Hz, 1H), 3.33 (m, 2H), 3.04 (dd, J1 = 6.0 Hz, J2 = 18.8 Hz, 2H), 2.90–2.70 (m, 2H), 2.50–2.40 (m, 2H), 1.99 (q, J = 12.5 Hz, 1H), 1.82 (d, J = 14.4 Hz, 1H), 1.78–1.70 (m, 1H), 1.46 (d, J = 8.8 Hz, 1H), 1.32 (m, 1H), 1.06 (m, 1H), 0.67 (m, 1H), 0.59 (m, 1H), 0.51 (m, 1H), 0.41 (m, 1H).
5,15-Dioxo-3,17-dioxa-diazanonadecane-1,19-dioic acid (5)
To the solution of 1,7-diaminoheptane (1.3 g, 10 mmol) in THF (4 mL) at 0 °C was added diglycolic anhydride (2.44 g, 21 mmol) in one portion. The resultant mixture was stirred at the same temperature for 15 min and allowed to warm to ambient temperature and stirred overnight. After removed THF under reduced pressure, the residue was crystallized by EtOAc/hexane to give 3.470 g white solid as first crop, in 96% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.78 (brs, 2H), 7.81 (t, J = 5.70 Hz, 2H), 4.10 (s, 4H), 3.94 (s, 4H), 3.08 (q, J = 6.76 Hz, 4H), 1.41 (m, 4H), 1.25 (m, 6H). 13C NMR (100 MHz, CD3OD): δ 171.47, 168.60, 70.12, 67.85, 38.07, 28.98, 28.36, 26.24. IR ν (Diamond, cm−1): 3306, 1699, 1646, 1548, 1247, 1151, 1136, 711. mp 64–67 °C. MS (ESI) m/z found 363.5 (M + H)+.
4,4-Difluoro-cyclohexanecarboxylic acid {1-(4-amino-phenyl)-3-[3-(3-isopropyl-5-methyl-[1,2,4]triazol-4-yl)-8-aza-bicyclo[3,2,1]oct-8-yl]-propyl}-amide (6)
On ice-water bath, to the solution of 20 (165 mg, 0.262 mmol) in DCM (5 mL) was added TFA (0.5 mL) dropwise. The resultant mixture was allowed to warm to ambient temperature within 15 min and stirred at the same temperature for 1.5 h. The mixture was cooled to 0°C, and saturated Na2CO3 was added. The aqueous layer was adjusted to pH = 12, and taken up with DCM (20 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, evaporated and dried in vacuum to afford 131 mg white solid, which is pure enough for the next step, in 95% yield. 1H NMR (400 MHz, CDCl3): δ 7.05 (d, J = 8.28 Hz, 2H), 6.62 (d, J = 8.24 Hz, 2H), 6.38 (brs, 1H), 4.98 (q, J = 7.17 Hz, 1H), 4.27 (m, 1H), 3.36 (m, 2H), 2.78 (seq, J = 6.84 Hz, 1H), 2.48 (s, 3H), 2.39 (m, 2H), 2.26–2.02 (m, 8H), 1.94–1.73 (m, 6H), 1.70–1.50 (m, 5H), 1.36 (d, J = 6.72 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 173.28, 159.23, 150.75, 145.98, 131.65, 127.67, 122.74 (J 13C-19F 239 Hz), 115.34, 58.98, 58.42, 51.59, 48.16, 47.38, 42.95, 35.59, 35.50, 34.84, 32.91 (J 13C-19F 24 Hz), 26.84, 26.78, 26.06 (J 13C-19F 9.7 Hz), 25.93, 21.75, 13.21. mp 109–110 °C. IR ν (Diamond, cm−1): 3334, 3230, 1636, 1517, 1105, 1031, 962, 831. MS (ESI) m/z found 529.6 (M + H)+.
(4-{1-Amino-3-[3-(3-isopropyl-5-methyl-[1,2,4]triazol-4-yl)-8-aza-bicyclo[3,2,1]oct-8-yl]-propyl}-phenyl)-carbamic acid tert-butyl ester (7)
On ice-water bath, a solution of 19 (764 mg, 1.538 mmol) in dry THF (10 mL) was added dropwise to a suspension of LiAlH4 (292 mg, 7.692 mmol) in dry THF (10 mL). The resultant mixture was stirred at the same temperature for 15 min and then 3 h at ambient temperature. The mixture was cooled in an ice bath again, and the complex was decomposed by dropwise addition of 2.4 mL H2O, 2.4 mL 4 N NaOH, and 4.8 mL H2O cautiously. The resulting white suspension was continued to stir for 1 h at ambient temperature, then filtered. The filtrate cake was washed with THF (20 mL × 3), diethyl ether (20 mL × 3). The combined filtrates were concentrated under reduced pressure and the residue was purified by silica gel using DCM/MeOH (6/1) to give 523 mg white solid, in 71% yield. 1H NMR (400 MHz, CD3OD): δ 7.39 (d, J = 8.52 Hz, 2H), 7.28 (d, J = 8.60 Hz, 2H), 4.38 (m, 1H), 3.95 (t, J = 6.90 Hz, 1H), 3.42 (m, 2H), 3.25 (seq, J = 5.83 Hz, 1H), 2.45 (s, 3H), 2.43 (m, 1H), 2.42–2.32 (m, 1H), 2.24–2.15 (m, 2H), 2.05–1.96 (m, 3H), 1.94–1.81 (m, 1H), 1.73 (m, 4H), 1.51 (s, 9H), 1.33 (d, J = 6.84 Hz, 6H). 13C NMR (100 MHz, CD3OD): δ 161.38, 155.32, 152.56, 140.28, 139.81, 128.02, 120.01, 80.86, 60.17, 60.07, 55.62, 49.97, 38.02, 36.37, 36.30, 28.76, 27.13, 26.98, 26.76, 22.05, 12.39. mp 103–105 °C. IR ν (Diamond, cm−1): 3252, 1713, 1522, 1240, 1159, 838. MS (ESI) m/z found 483.7 (M + H)+.
4,4-Difluoro-cyclohexanecarboxlic acid (8)
The title compound was prepared as described by Mackenzie et al.48, except that the ester was purified by silica gel using EtOAc/hexane (80/1) as eluent. The total yield is 52%. 1H NMR (400 MHz, CDCl3): δ 2.48 (m, 1H), 2.16–2.09 (m, 2H), 2.06–2.02 (m, 2H), 1.94–1.83 (m, 4H). 19F NMR (400 MHz, CDCl3): δ −94.45–95.09 (d, 1F), −99.19–99.80 (d, 1F). mp 98.5–99.5 °C (lit., 48 105.9 °C).
3-(3-Isopropyl-5-methyl-[1,2,4]triazol-4-yl)-exo-8-aza-bicyclo[3.2.1]octane (10)
The title compound and its precursors were synthesized following the same procedure by Haycock-Lewandowski et al.41a. The crude product (free base) was crystallized from hexane to give 1.2 g 10, in 97% yield. 1H NMR (400 MHz, CDCl3): δ 4.33 (seq, J = 6.01 Hz, 1H), 3.74 (m, 2H), 3.02 (seq, J = 6.86 Hz, 1H), 2.53 (s, 3H), 2.18 (dt, J1 = 12.6 Hz, J2 = 2.66 Hz, 2H), 1.94 (m, 2H), 1.76 (m, 4H), 1.39 (d, J = 6.88 Hz, 6H). mp 190–191 °C.
3-(4-Bromo-phenyl)-acrylic acid isopropyl ester (12)
To the solution of 4-bromocinnacid (1.135 g, 5 mmol) in isopropanol (50 mL) was added several drops of concentrated sulfuric acid. The mixture was heated to reflux for 48 hours. After cooled down, the residue was worked up with ethyl acetate. The ethyl acetate layer was washed with sat. NaHCO3 aqueous solution, dried over Na2SO4. After filtration and concentration, the resulting crude product was purified by silica column using hexane and ethyl acetate (from 100:1 to 75:1 then 50:1) as eluent to give 1.08 g white solid, in 80% yield. 1H NMR (400 MHz, CDCl3): δ 7.59 (d, J = 16.0 Hz, 1H), 7.51 (d, J = 8.48 Hz, 2H), 7.38 (d, J = 8.44 Hz, 2H), 6.40 (d, J = 16.0 Hz, 1H), 5.14 (seq, J = 6.26 Hz, 1H), 1.31 (d, J = 6.24 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 166.38, 143.04, 133.63, 132.26, 129.53, 124.51, 119.70, 68.14, 22.07. mp 64–66 °C. IR ν (Diamond, cm−1): 1703, 1637, 1304, 1172, 1104, 981, 817. MS (ESI) m/z found 268.9 (M + H)+, 271.1 (M + 2 + H)+.
3-(4-Amino-phenyl)-acrylic acid isopropyl ester (13)
The mixture of 12 (1.53 g, 5.68 mmol), Pd2(dba)3 (260 mg, 5% mmol), P(t-Bu)3 (0.23 mL, 1 M in toluene, 4% mmol) in dry toluene (30 mL) was stirred under N2 protection for 15 min. Then a solution of LHMDS in toluene (6.2 mL, 1 M in toluene, 6.2 mmol) was added dropwise. After stirred at ambient temperature overnight, the resultant dark color suspension was added 1 N hydrochloric acid (8 mL) slowly. The resulting mixture was stirred at ambient temperature for 2 hours. Then the suspension was filtered through celite and the filtrate was diluted with dichloromethane (70 mL). The organic layer was washed with saturated NaHCO3 aqueous solution, brine, and dried over Na2SO4. After filtered and concentrated, the crude product was purified by silica gel column using hexane and ethyl acetate (2:1) as eluent to give 1.03 g light yellow solid, in 90% yield. 1H NMR (400 MHz, CDCl3): δ 7.58 (d, J = 15.9 Hz, 1H), 7.35 (d, J = 8.48 Hz, 2H), 6.65 (d, J = 8.56 Hz, 2H), 6.22 (d, J = 15.9 Hz, 1H), 5.12 (seq, J = 6.24 Hz, 1H), 3.91 (brs, 2H), 1.30 (d, J = 6.24 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 167.20, 148.56, 144.55, 129.80, 124.88, 114.84, 114.35, 67.32, 22.00. mp 78–80 °C. IR ν (Diamond, cm−1): 3417, 3335, 1673, 1592, 1513, 1264, 1168, 1104, 980, 823. MS (ESI) m/z found 206.2 (M + H)+.
3-(4-tert-Butoxycarbonylamino-phenyl)-acrylic acid isopropyl ester (14)
The solution of 13 (1.351 g, 6.58 mmol) and Boc2O (1.58 g, 7.24 mmol) in dry tetrahydrofuran (30 mL) was heated to reflux overnight. After cooled down and concentrated, the residue was crystallized from DCM/hexane to give 1.711 g white solid, in 85% yield. 1H NMR (400 MHz, CDCl3): δ 7.60 (d, J = 16.0 Hz, 1H), 7.46 (d, J = 8.68 Hz, 2H), 7.38 (d, J = 8.64 Hz, 2H), 6.55 (brs, 1H), 6.32 (d, J = 16.0 Hz, 1H), 5.13 (seq, J = 6.25 Hz, 1H), 1.52 (s, 9H), 1.30 (d, J = 6.24 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 166.77, 152.41, 143.81, 140.30, 129.17, 129.00, 118.33, 117.03, 80.95, 67.63, 28.29, 21.95. mp 159–159.5 °C. IR ν (Diamond, cm−1): 3307, 1725, 1688, 1586, 1521, 1150, 1106, 981, 830. MS (ESI) m/z found 306.2 (M + H)+.
3-[Benzyl-(1-phenyl-ethyl)-amino-3-(4-tert-butoxycarbonylamino-phenyl)-propionic acid isopropyl ester (15)
On ice-water bath, under N2 protection, the solution of R-(+)-N-benzyl-α-methylbenzylamine (1.88 mL, 9.02 mmol) in dry THF was added n-butyllithium (3.47 mL, 2.5 M in hexane) dropwise. The resulting purple solution was stirred for 30 minutes, then cooled down to −78 °C, the solution of 14 (1.06 g, 3.47 mmol) in dry THF was added dropwise. Then the dark red solution was stirred for 2 hours at −78 °C. Saturated ammonium chloride aqueous was added to quench the reaction. The resultant yellow solution was allowed to warm to ambient temperature within 30 min. After worked up with ethyl acetate, the organic layer was dried, concentrated. The residue was crystallized with ethyl acetate to give 20 mg 15 as colorless crystal. The filtrate was then concentrated and dried on vacuum. Purification of the crude compound by silica gel column chromatography gave mixtures of the excess R-(+)-N-benzyl-α-methylbenzylamine and the product. Hence, the following procedure was performed to transfer the excess amine to the amide to facilitate the purification process. Benzyl chloride (644 μL, 5.55 mmol) was added dropwise into a mixture of the above filtrate and triethylamine (1.543 mL, 11.1 mmol) in 30 mL CH2Cl2 at 0 °C. After stirred for 2 hours, the reaction mixture was washed with brine. The organic layer, containing the product 15 and N-benzyl-N-(1-phenyl-ethyl)-benzamide, was dried, concentrated and used for next step without further purification. 1H NMR (400 MHz, CDCl3): δ 7.40 (d, J = 7.2 Hz, 2H), 7.35–7.15 (m, 12H), 6.43 (brs, 1H), 4.79 (seq, J = 6.25 Hz, 1H), 4.38 (dd, J1 = 5.2 Hz, J2 = 9.66 Hz, 1H), 3.97 (q, J = 6.75 Hz, 1H), 3.66 (s, 2H), 2.56 (dd, J1 = 5.12 Hz, J2 = 14.68 Hz, 1H), 2.50 (dd, J1 = 14.58 Hz, J2 = 9.82 Hz, 1H), 1.51 (s, 9H), 1.25 (d, J = 6.76 Hz, 3H), 1.05 (d, J = 6.24 Hz, 3H), 1.00 (d, J = 6.20 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 171.36, 152.77, 144.15, 141.63, 137.37, 136.34, 128.75, 128.13 (× 2), 128.00, 127.84, 126.83, 126.55, 118.16, 80.47, 67.51, 59.06, 57.05, 50.82, 37.71, 28.37, 21.62, 16.46. mp 172–174 °C. IR ν (Diamond, cm−1): 1724, 1595, 1522, 1154, 1105, 1051, 697. MS (ESI) m/z found 517.5 (M + H)+.
3-[Benzyl-(1-phenyl-ethyl)-amino-3-(4-tert-butoxycarbonylamino-phenyl)-propionic acid (16)
To the above mixture of crude product 15 and N-benzyl-N-(1-phenyl-ethyl)-benzamide in methanol (30 mL) and water (15 mL) was added lithium hydroxide (831 mg, 34.7 mmol). The resulting suspension was heated to reflux for 48 hours. After cooled downed, the mixture was concentrated to remove methanol. The water layer was taken up with CH2Cl2 (30 mL × 3). The organic layer was dried and concentrated. The residue was purified by silica gel column using hexane and ethyl acetate (2:1) as eluent to give 1.406 g white solid, in 85% yield for two steps. 1H NMR (400 MHz, CDCl3): δ 7.44 (d, J = 8.52 Hz, 2H), 7.37–7.22 (m, 12H), 6.55 (brs, 1H), 4.43 (dd, J1 = 4.46 Hz, J2 = 11.32 Hz, 1H), 4.15 (q, J = 6.87 Hz, 1H), 3.98 (d, J = 13.68 Hz, 1H), 3.64 (d, J = 13.68 Hz, 1H), 2.92 (dd, J1 = 11.34 Hz, J2 = 16.94 Hz, 1H), 2.41 (dd, J1= 4.48 Hz, J2 = 16.96 Hz, 1H), 1.54 (s, 9H), 1.28 (d, J = 6.88 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 174.39, 152.82, 141.25, 138.44, 137.70, 132.56, 129.22, 128.80, 128.62, 128.60, 128.19, 127.81, 127.53, 118.52, 80.86, 57.96, 57.82, 50.61, 36.29, 28.34, 15.68. mp 93–95 °C. IR ν (Diamond, cm−1): 3307, 1699, 1594, 1081, 698. MS (ESI) m/z found 475.6 (M + H)+.
(4-{1-[Benzyl-(1-phenyl-ethyl)-amino]-3-[3-(3-isopropyl-5-methyl-[1,2,4]triazol-4-yl)-8-aza-bicyclo[3,2,1]oct-8-yl]-3-oxo-propyl}-phenyl)-carbamic acid tert-butyl ester (17)
The title compound was prepared according to the general amide coupling procedure by reacting acid 16 with amine 10 in DCM for 4 h. The crude product was crystallized using DCM/hexane to give 986 mg white solid as first crop, in 73% yield. 1H NMR (400 MHz, CDCl3): δ 7.88 (d, J = 8.32 Hz, 1H), 7.68 (d, J = 8.28 Hz, 1H), 7.46 (d, J = 7.60 Hz, 2H), 7.41–7.35 (m, 10H), 7.33–7.12 (m, 14H), 6.91 (brs, 1H), 6.66 (brs, 1H), 4.68 (m, 1H), 4.62 (m, 1H), 4.53 (dd, J1 = 5.54 Hz, J2 = 8.10 Hz, 1H), 4.40–4.28 (m, 3H), 4.03–3.96 (m, 2H), 3.82–3.62 (m, 6H), 2.76 (m, 2H), 2.54 (m, 4H), 2.21 (s, 3H), 2.11 (s, 3H), 2.06–1.96 (m, 4H), 1.96–1.55 (m, 12H), 1.51 (s, 9H), 1.50 (s, 9H), 1.30 (m, 12H), 1.28–1.24 (m, 6H); 13C NMR (100 MHz, CDCl3): δ 167.31, 166.99, 158.87, 152.80, 150.35, 144.43, 142.17 (142.05), 137.78, 136.61, 136.32, 80.46, 61.06, 59.34, 56.53 (56.40), 53.82 (53.43), 51.11 (50.80), 50.56 (50.49), 46.67, 38.87 (38.27), 37.60 (37.46), 35.67, 28.34, 26.86 (26.61), 25.79, 21.63 (21.56), 14.59, 13.80, 12.99. mp 128–130 °C. IR ν (Diamond, cm−1): 2966, 1721, 1637, 1545, 1436, 1242, 1165, 742, 705. MS (ESI) m/z found 691.5 (M + H)+.
(4-{1-Amino-3-[3-(3-isopropyl-5-methyl-[1,2,4]triazol-4-yl)-8-aza-bicyclo[3,2,1]oct-8-yl]-3-oxyl}-phenyl)-carbamic acid tert-butyl ester (19)
A solution of 17 (500 mg, 0.725 mmol) in methanol (35 mL) was treated with palladium carbon (100 mg, 10 wt %), and the resultant slurry was shaken under an atmosphere of hydrogen at 60 psi for 4 days at ambient temperature. The reaction mixture was filtered through celite. The filtrate cake was washed with methanol and the combined filtrates were concentrated and purified by silica gel using DCM/MeOH (20/1) as eluent to give 304 mg white solid 19, in 84% yield. 1H NMR (400 MHz, CDCl3): δ 7.34 (d, J = 5.88 Hz, 8H), 6.46 (brs, 2H), 4.88 (m, 2H), 4.57–4.47 (m, 4H), 4.34–4.23 (m, 2H), 2.93 (seq, J = 7.03 Hz, 2H), 2.73–2.50 (m, 4H), 2.46 (s, 3H), 2.36 (s, 3H), 2.33–1.95 (m, 8H), 1.85–1.71 (m, 8H), 1.52 (s, 18H), 1.40–1.37 (m, 12H); 13C NMR (100 MHz, CDCl3): δ 167.71(167.41), 158.99 (158.93), 152.86, 150.51 (150.38), 139.43, 137.83 (137.73), 126.95 (126.86), 118.94 (118.88), 80.56, 53.89 (53.71), 52.36 (52.07), 50.85 (50.80), 46.85 (46.76), 43.77 (43.58), 37.67, 35.95 (35.88), 28.64 (28.56), 28.36, 26.97 (26.92), 25.92, 21.73, 21.66 (21.63), 13.13 (13.08). mp 121–122.5 °C. IR ν (Diamond, cm−1): 3273, 1713, 1609, 1521, 1413, 1158, 1028, 837. MS (ESI) m/z found 497.3 (M + H)+.
(4-{1-[(4,4-Difluoro-cyclohexancarbonyl)-amino-3-[3-(3-isopropyl-5-methyl-[1,2,4] triazol-4-yl)-8-aza-bicyclo[3,2,1]oct-8-yl]-propyl}-phenyl)-carbamic acid tert-butyl ester (20)
The title compound was prepared according to the general amide coupling procedure by reacting acid 8 with amine 7 in DCM for 4 h. The crude product was purified by silica gel using DCM/MeOH (18/1) to give 269 mg white solid, in 85% yield. 1H NMR (400 MHz, CDCl3): δ 7.34 (d, J = 8.40 Hz, 2H), 7.11 (d, J = 8.48 Hz, 2H), 6.67 (brs, 1H), 6.54 (d, J = 7.56 Hz, 1H), 5.07 (q, J = 7.01 Hz, 1H), 4.29 (m, 1H), 3.37 (m, 2H), 2.98 (seq, J = 6.48 Hz, 1H), 2.49 (s, 3H), 2.40 (t, J = 6.62 Hz, 2H), 2.26–2.13 (m, 5H), 2.06–1.94 (m, 6H), 1.93–1.63 (m, 8H), 1.51 (s, 9H), 1.38 (d, J = 6.76 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 173.24, 159.12, 152.82, 150.56, 137.75, 136.34, 127.11, 122.51 (J 13C-19F 250 Hz), 118.92, 80.63, 58.87, 58.18, 51.61, 47.78, 47.28, 42.86, 35.43, 35.29, 34.66, 32.79 (J 13C-19F 25.5 Hz), 28.32, 26.80, 26.76, 25.94 (J 13C-19F 8 Hz), 25.85, 21.64, 13.11. mp 234–235 °C. IR ν (Diamond, cm−1): 3272, 1716, 1650, 1236, 1159, 1106, 963, 836. MS (ESI) m/z found 629.6 (M + H)+.
(7-Amino-heptyl)-carbamic acid benzyl ester (21)
On an ice-water bath, to the solution of 1,7-diaminoheptane (1.433 g, 11 mmol) in CH2Cl2/MeOH (125 mL/125 mL) was added the solution of CbzCl (1.71 g, 10 mmol) in CH2Cl2 (250 mL) dropwise within 12 h while keeping the temperature below 5 °C. The mixture was allowed to stir at the same temperature for another half of an hour before concentrated under reduced pressure to remove most of the MeOH. Water (150 mL) was then added, and the aqueous layer was adjusted to pH = 2 using 6 N HCl. The layers were separated. The aqueous layer was washed with DCM (50 mL × 3), then adjusted to pH = 12 with 10 N NaOH and extracted with DCM (50 mL × 3). The combined organic layers were dried over Na2SO4, concentrated and purified by flash column using DCM/MeOH (9/1) to give 856 mg white solid in 32% yield. 1H NMR (400 MHz, CDCl3): δ 7.35–7.29 (m, 5H), 5.09 (s, 2H), 4.73 (brs, 1 H), 3.18 (q, J = 6.64 Hz, 2H), 2.67 (t, J = 6.94 Hz, 2H), 1.55–1.44 (m, 2H), 1.44–1.40 (m, 2H), 1.32 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 156.40, 136.70, 128.50, 128.09, 128.06, 66.55, 42.18, 41.06, 33.71, 29.91, 29.09, 26.76, 26.68. mp 78–80 °C. IR ν (Diamond, cm−1): 3327, 1686, 1532, 1263, 1144. MS (ESI) m/z found 264.8 (M + H)+.
[(7-Benzyloxycarbonylamino-heptylcarbamoyl)-methoxy]-acetic acid (22)
To the solution of 21 (350 mg, 1.324 mmol) in THF (4 mL) was added diglycolic anhydride (161 mg, 1.39 mmol) in one portion. The resultant mixture was stirred at ambient temperature for 12 h. After removed THF under reduced pressure, the residue was crystallized by EtOAc/hexane to give 429 mg white solid as first crop, in 85% yield. 1H NMR (400 MHz, DMSO-d6): δ 12.79 (brs, 1H), 7.81 (t, J = 5.52 Hz, 1H), 7.34–7.29 (m, 5 H), 7.21 (t, J = 5.46 Hz, 1H), 5.00 (s, 2H), 4.10 (s, 2H), 3.94 (s, 2H), 3.08 (q, J = 6.56 Hz, 2H), 2.97 (q, J = 6.28 Hz, 2H), 1.42–1.37 (m, 4H), 1.24 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 171.43, 168.53, 156.06, 137.30, 128.27, 127.65, 70.18, 67.88, 65.04, 40.22, 38.08, 29.65, 29.00, 28.38, 26.28, 26.14. mp 74–74.5 °C. IR ν (Diamond, cm−1): 3374, 3331, 1726, 1688, 1608, 1548, 1249, 1236, 1135, 956, 701. MS (ESI) m/z found 381.4 (M + H)+.
6′β-(3,13-Dioxo-1-phenyl-2,15-dioxa-4,12-diazaheptadecanamido)morphinan (23)
The title compound was prepared according to the general amide coupling procedure by reacting acid 22 with amine 4·2HCl in DMF overnight. The crude product was purified with chromatography using CH2Cl2/MeOH (40/1) as eluent to give 339 mg white solid, in 76% yield. 1H NMR (400 MHz, DMSO-d6): δ 9.01 (brs, 1H), 8.21 (d, J = 8.36 Hz, 1H), 8.01 (t, J = 5.60 Hz, 1H), 7.37–7.30 (m, 5H), 7.19 (m, 1H), 6.58 (d, J = 8.04 Hz, 1H), 6.52 (d, J = 8.04 Hz, 1H), 4.99 (s, 2H), 4.88 (brs, 1H), 4.59 (d, J = 7.64 Hz, 1H), 3.94 (s, 2H), 3.93 (s, 2H), 3.53–3.45 (m, 1 H), 3.18–3.07 (m, 2 H), 3.01–2.94 (m, 4H), 2.60–2.56 (m, 2H), 2.38–2.28 (m, 2H), 2.15 (dt, J1 = 4.89 Hz, J2 = 12.29 Hz, 1H), 1.98 (m, 1H), 1.79 (m, 1H), 1.46–1.37 (m, 6H), 1.26 (m, 8H), 0.84 (m, 1H), 0.46 (m, 2H), 0.12 (m, 2H); 1H NMR (400 MHz, CDCl3): δ 7.57 (d, J = 9.24 Hz, 1H), 7.35–7.32 (m, 5H), 6.90 (t, J = 5.66 Hz, 1H), 6.72 (d, J = 8.12 Hz, 1H), 6.55(d, J = 8.16 Hz, 1H), 5.09 (s, 2H), 4.88 (m, 1H), 4.44 (d, J = 5.48 Hz, 1H), 4.02 (m, 4H), 3.24 (m, 2H), 3.18 (AB, J = 6.64 Hz, 2H), 3.10 (d, J = 5.84 Hz, 1H), 3.03 (d, J = 18.48 Hz, 1H), 2.62 (m, 2H), 2.36 (m, 2H), 2.19 (m, 2H), 1.79 (m, 1H), 1.64 (m, 1H), 1.54–1.48 (m, 7H), 1.32–1.26 (m, 7H), 0.80 (m, 1H), 0.53 (m, 2H), 0.13 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 168.63, 168.45, 156.51, 143.19, 139.38, 136.64, 130.56, 128.53 (× 2), 128.10, 124.66, 119.23, 117.77, 92.27, 77.23, 70.89, 70.13, 66.63, 62.38, 59.40, 49.41, 47.20, 43.88, 41.02, 39.04, 31.87, 29.85, 29.50, 28.92, 28.81, 26.78, 26.55, 23.17, 22.61, 9.41, 4.00, 3.81. mp >300 °C. IR ν (Diamond, cm−1): 3670, 1700, 1560, 1136. MS (ESI) m/z found 705.5 (M + H)+.
6′β-{2-[2-(7-aminoheptylamino)-2-oxoethoxy]acetamido}morphinan (24)
A solution of 23 (120 mg, 0.167 mmol) in methanol (20 mL) was hydrogenated in the presence of 10 % Pd/C (12 mg) under a H2 atmosphere (60 psi) at room temperature for 48 h. The mixture was filtered, and the filtrate was concentrated and purified by silica gel with DCM/MeOH (7/1) to give 24 as white foam (110 mg, 99 % yield). 1H NMR (400 MHz, DMSO-d6): δ 8.22 (d, J = 8.40 Hz, 1H), 8.03 (t, J = 5.74 Hz, 1H), 6.58 (d, J = 8.04 Hz, 1H), 6.52 (d, J = 8.12 Hz, 1H), 4.59 (d, J = 7.36 Hz, 1H), 3.94 (s, 2H), 3.93 (s, 2H), 3.56–3.48 (m, 1 H), 3.19–3.09 (m, 2 H), 3.10 (d, J = 5.52 Hz, 1H), 2.97 (d, J = 18.8 Hz, 1H), 2.70 (m, 2H), 2.61–2.55 (m, 2H), 2.39–2.28 (m, 2H), 2.18–2.10 (m, 1H), 1.98 (dt, J1 = 3.56 Hz, J2 = 11.92 Hz, 1H), 1.84–1.74 (m, 1H), 1.47–1.44 (m, 6H), 1.32–1.23 (m, 8H), 0.86 (m, 1H), 0.47 (m, 2H), 0.11 (m, 2H); 1H NMR (400 MHz, CD3OD): δ 6.62 (d, J = 8.08 Hz, 1H), 6.55 (d, J = 8.12 Hz, 1H), 4.51 (d, J = 7.56 Hz, 1H), 4.06 (m, 2H), 4.05 (m, 2H), 3.76 (m, 1H), 3.26 (t, J = 7.08 Hz, 2H), 3.10 (d, J = 5.96 Hz, 1H), 3.07 (d, J = 20.64 Hz, 1H), 2.66 (m, 2H), 2.58 (m, 2H), 2.40 (m, 2H), 2.27–2.11 (m, 2H), 1.90 (m, 1H), 1.60–1.51 (m, 6H), 1.49–1.32 (m, 8H), 0.85 (m, 1H), 0.54 (m, 2H), 0.16 (m, 2H); 13C NMR (100 MHz, CD3OD): δ 171.54, 171.38, 143.90, 142.52, 132.41, 125.06, 120.09, 118.88, 92.80, 71.74, 71.60, 71.55, 63.77, 60.32, 52.54, 48.93, 47.95, 45.28, 40.07, 32.05, 31.27, 30.43, 30.28, 28.38, 27.92, 25.51, 23.55, 22.12, 10.36, 4.49, 4.27. mp 83–85 °C. IR ν (Diamond, cm−1): 3278, 3075, 1652, 1548, 1128, 1035. MS (ESI) m/z found 571.6 (M + H)+.
19-(6′β-morphinanamino)-5,15–19-trixox-3,17-dioxa-6,14-diazanonadecan-1-oic acid (25)
To the solution of 24 (113 mg, 0.198 mmol) in DMF (2 mL) was added diglycolic anhydride (23 mg, 0.198 mmol) within 15 min. The resultant mixture was stirred at ambient temperature for 2 h. After removal of DMF under reduced pressure, the residue was crystallized by EtOAc/hexane to give 112 mg light yellow solid as first crop, in 82% yield. 1H NMR (400 MHz, DMSO-d6): δ 9.22 (brs, 1H), 8.28 (d, J = 8.32 Hz, 1H), 8.06 (t, J = 5.62 Hz, 1H), 7.99 (m, 1H), 6.65 (d, J = 8.12 Hz, 1H), 6.59 (d, J = 8.12 Hz, 1H), 4.68 (d, J = 7.80 Hz, 1H), 4.06 (s, 2H), 3.95 (s, 2H), 3.94 (m, 4H), 3.56–3.47 (m, 1 H), 3.24–3.06 (m, 6H), 2.89–2.83 (m, 2H), 2.65 (m, 1H), 2.33–2.28 (m, 2H), 1.87–1.78 (m, 1H), 1.59 (m, 1H), 1.49–1.40 (m, 6H), 1.27 (m, 8H), 0.98 (m, 1H), 0.58–0.52 (m, 2H), 0.30 (m, 2H); 1H NMR (400 MHz, CD3OD): δ 6.72 (d, J = 8.20 Hz, 1H), 6.70 (d, J = 8.40 Hz, 1H), 4.65 (d, J = 7.80 Hz, 1H), 4.07 (s, 2H), 4.06 (s, 2H), 3.98 (s, 2H), 3.93 (s, 2H), 3.77–3.71 (m, 2H), 3.31–3.22 (m, 4H), 3.14–2.99 (m, 3H), 2.94–2.89 (m, 1H), 2.78 (dd, J1 = 7.34 Hz, J2 = 13.30 Hz, 1H), 2.59–2.46 (m, 2H), 2.03–1.92 (m, 1H), 1.73–1.61 (m, 2H), 1.59–1.50 (m, 6H), 1.37 (m, 6H), 1.06 (m, 1H), 0.77–0.63 (m, 2H), 0.42 (m, 2H); 13C NMR (100 MHz, CD3OD): δ 177.25, 172.58, 171.61, 171.56, 143.81, 142.81, 131.26, 128.73, 120.70, 119.42, 92.23, 72.25, 71.67, 71.58, 71.47, 71.39, 64.21, 59.23, 52.37, 49.30, 47.97, 40.10, 39.96, 31.20, 30.26, 30.21, 29.95, 29.76, 27.81, 27.79, 24.91, 24.24, 7.78, 5.67, 3.68. mp 193 °C dec. IR ν (Diamond, cm−1): 3271, 3069, 1732, 1651, 1548, 1125, 1033. MS (ESI) m/z found 687.4 (M + H)+.
Methylcarbamoylmethoxy-acetic acid (26)
The title compound was prepared using the same procedure as described by Zheng et al.30b, except that white solid instead of oil was obtained. 1H NMR (400 MHz, DMSO-d6): δ 12.77 (brs, 1H), 7.77 (brs, 1H), 4.10 (s, 2H), 3.94 (s, 2H), 2.62 (d, J = 4.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 171.32, 169.08, 70.09, 67.77, 25.11. mp 33–33.5 °C.
(7-{2-[(4-{1-[(4,4-Difluoro-cyclohexanecarbonyl)-amino]-3-[3-(3-isopropyl-5-methyl-[1,2,4]triazol-4-yl)-8-aza-bicyclo[3.2.1]oct-8yl]-propyl}-phenylcarbamoyl)-methoxy]-acetylamino}-heptyl)-carbamic acid benzyl ester (27)
The title compound was prepared according to the general amide coupling procedure by reacting acid 22 with amine 6 in DMF overnight. The crude product was purified with chromatography using CH2Cl2/MeOH (13/1) as eluent to give 260 mg white foaming, in 72% yield. 1H NMR (400 MHz, CDCl3): δ 8.96 (s, 1H), 7.57 (d, J = 8.48 Hz, 2H), 7.35–7.27 (m, 5H), 7.24 (d, J = 8.48 Hz, 2H), 6.75 (m, 1H), 6.70 (m, 1H), 5.12–5.08 (m, 3H), 4.91 (m, 1H), 4.29 (m, 1H), 4.14 (s, 2H), 4.09 (s, 2H), 3.40 (m, 1H), 3.37 (m, 1H), 3.30 (q, J = 6.68 Hz, 2H), 3.15 (q, J = 6.71 Hz, 2H), 2.99 (m, 1H), 2.48 (s, 3H), 2.43 (t, J = 6.48 Hz, 2H), 2.30–2.04 (m, 6H), 2.01–1.71 (m, 8H), 1.70–1.46 (m, 9H), 1.376 (d, J = 6.80 Hz, 3H), 1.373 (d, J = 6.84 Hz, 3H), 1.35–1.31 (m, 6H); 13C NMR (100 MHz, CDCl3): δ 173.50, 168.64, 167.15, 159.17, 156.55, 150.59, 138.31, 136.66, 136.51, 128.54, 128.12, 128.00, 127.12, 120.51, 77.24, 71.66, 71.50, 66.60, 58.90, 58.30, 51.68, 47.77, 47.24, 42.83, 40.98, 39.14, 35.33, 34.59, 32.82 (J 13C-19F 25.7 Hz), 29.82, 29.36, 28.71, 26.75 (J 13C-19F 10 Hz), 26.48, 25.87, 21.65, 13.15. mp 65–67 °C. IR ν (Diamond, cm−1): 3273, 1656, 1529, 1515, 1251, 1106, 697. MS (ESI) m/z found 891.9 (M + H)+.
4,4-Difluoro-cyclohexanecarboxylic acid {1-(4-{2-[(7-amino-heptylcarbamoyl)-meth-oxy]-acetylamino}-phenyl)-3-[3-(3-isopropyl-5-methyl-[1,2,4]triazol-4-yl)-8-aza-bicyclo[3.2.1] oct-8-yl]-propyl}-amide (28)
A solution of 27 (260 mg, 0.291 mmol) in methanol (15 mL) was hydrogenated in the presence of 5% Pd/C (26 mg) under a H2 atmosphere (60 psi) at room temperature for 48 h. The mixture was filtered, and the filtrate was concentrated to give 28 as white foaming (113 mg, 51% yield). 1H NMR (400 MHz, CDCl3): δ 8.80 (s, 1H), 7.58 (d, J = 8.48 Hz, 2H), 7.25 (d, J = 8.48 Hz, 2H), 6.61 (d, J = 7.48 Hz, 1H), 6.47 (m, 1H), 5.10 (q, J = 7.00 Hz, 1H), 4.30 (m, 1H), 4.17 (s, 2H), 4.12 (s, 2H), 3.40–3.30 (m, 4H), 2.98 (seq, J = 6.90 Hz, 1H), 2.68 (t, J = 6.84 Hz, 2H), 2.48 (s, 3H), 2.43 (t, J = 6.62 Hz, 2H), 2.26–2.12 (m, 5H), 2.08–1.53 (m, 16H), 1.44 (qu, J = 6.72 Hz, 2H), 1.383 (d, J = 6.76 Hz, 3H), 1.382 (d, J = 6.80 Hz, 3H); 1.34–1.30 (m, 6H); 1H NMR (400 MHz, CD3OD): δ 7.60 (d, J = 8.52 Hz, 2H), 7.33 (d, J = 8.56 Hz, 2H), 5.03 (t, J = 7.34 Hz, 1H), 4.39 (m, 1H), 4.20 (s, 2H), 4.12 (s, 2H), 3.41 (m, 2H), 3.30–3.21 (m, 3H), 2.69 (t, J = 7.32 Hz, 2H), 2.50 (s, 3H), 2.46–2.41 (m, 2H), 2.40–2.30 (m, 1H), 2.30–2.05 (m, 2H), 2.05–2.03 (m, 4H), 2.02–1.96 (m, 2H), 1.89–1.65 (m, 10H), 1.60–1.45 (m, 4H), 1.36–1.34 (m, 12H); 13C NMR (100 MHz, CDCl3): δ 176.59, 171.68, 170.07, 161.41, 152.60, 140.39, 138.07, 128.21, 123.91 (J 13C-19F 239 Hz), 121.88, 71.95, 71.75, 60.69, 60.19, 52.41, 43.69, 42.03, 40.07, 36.77, 36.16, 33.90 (J 13C-19F 24 Hz), 33.87 (J 13C-19F 24 Hz), 32.28, 30.38, 30.11, 27.86, 27.78, 27.20(J 13C-19F 9 Hz), 27.05(J 13C-19F 9 Hz), 26.76, 22.07, 12.45. mp 100–102 °C. IR ν (Diamond, cm−1): 3256, 1651, 1538, 1515, 1261, 1103, 743. MS (ESI) m/z found 757.9 (M + H)+.
Calcium mobilization assay
The ligands were first tested with various doses (in the range of 0.1 nM to 1 μM) for possible agonist activity. The protocol was the same for the following antagonism study, except no CCL5 (RANTES) was added.
MOLT-4/CCR5 cells were plated in black 96-well plates with transparent bottom (Greinier Bio-one) at 100,000 cells per well in 50:1 HBSS:HEPES assay buffer. They were incubated for 1 hour at 37°C and 5% CO2 with control buffer or varying concentration of ligand for a total volume of 130 μL per well. Cells were then incubated with 50 μL of Fluo-4-AM loading buffer (40 μL 2 μM Fluo-4 dye, 100 μL 2.5 mM probenacid, in 5 mL assay buffer) for an additional hour. Then 20 μL 200 nM RANTES solution in assay buffer or assay buffer alone were added to the wells right before changes in Ca2+ concentration were monitored by RFU for 90 seconds using a microplate reader (FlexStation3, Molecular Devices). Peak values were obtained using SoftMaxPro software (Molecular Devices) and non-linear regression curves were generated using Prism (GraphPad) to calculate IC50 values.
The mu opioid receptor binding and functional assay
MOR/CHO cell culture and membrane homogenate preparation followed literature report52.
Opioid Receptor Binding
Saturation binding was performed by incubating membranes for 90 minutes at 30°C with 0.5–15 nM [3H]naloxone in assay buffer in a 0.5 mL volume. Non-specific binding were determined with 5 μM naltrexone. For competition assays, membranes were incubated as above with 2 nM [3H]naloxone and various concentrations of unlabeled ligand, to determine competitor IC50 for MOR. The reaction was terminated by rapid filtration through Whatman GF/B glass fiber filters, followed by 3 washes with 3 mL ice-cold Tris buffer. Bound radioactivity will be determined by liquid scintillation spectrophotometry at 45% efficiency for [3H].
[35S]GTPγS Binding
Membranes (10 μg protein) were incubated in assay buffer at 30°C for 90 min with various drugs, 10 μM GDP (cells) and 0.1 nM [35S]GTPγS in 0.5 mL total volume for appropriate times. Basal binding was assessed in the absence of agonist, and nonspecific binding was measured with 10 μM unlabeled GTPγS. The reaction was terminated by rapid filtration as described above. Bound radioactivity was determined by liquid scintillation spectrophotometry at 95% efficiency.
Data analysis
For competition binding assay (agonists or antagonists), Hill plots linear regression analysis and the Cheng-Prusoff equation were applied to determine the IC50 and Ki values. In [35S]GTPγS binding assays, agonist concentration effect curves were fit by non-linear regression to obtain Emax and EC50 values; antagonist inhibition of agonist-stimulated [35S]GTPγS binding was analyzed by Hill analysis and AD50 values were corrected to Ki values using the Cheng-Prusoff equation. All analyses were using Prism 4.0.
Supplementary Material
Acknowledgments
We thank the funding support from National Institute on Drug Abuse DA024022.
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse, or the National Institutes of Health.
Electronic supplementary information available: The NMR spectrum for all compounds, and HPLC spectrum for the bivalent and monovalent ligands.
Notes and references
- 1.(a) Limbird LE, Lefkowitz RJ. J Biol Chem. 1976;251:5007–5014. [PubMed] [Google Scholar]; (b) Limbird LE, De Meyts P, Lefkowitz RJ. Biochem Biophys Res Commun. 1975;64:1160–1168. doi: 10.1016/0006-291x(75)90815-3. [DOI] [PubMed] [Google Scholar]
- 2.Hazum E, Chang KJ, Cuatrecasas P. Science. 1979;206:1077–1079. doi: 10.1126/science.227058. [DOI] [PubMed] [Google Scholar]
- 3.(a) Szidonya L, Cserző M, Hunyady L. J Endocrino. 2008;196:435–453. doi: 10.1677/JOE-07-0573. [DOI] [PubMed] [Google Scholar]; (b) Bouvier M. Nat Rev Neurosci. 2001;2:274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]; (c) Salahpour A, Angers S, Bouvier M. Trends Endocr Metab. 2000;11:163–168. doi: 10.1016/s1043-2760(00)00260-5. [DOI] [PubMed] [Google Scholar]
- 4.(a) Liu ZL, Zhang J, Zhang A. Curr Pharm Des. 2009;15:682–718. doi: 10.2174/138161209787315639. [DOI] [PubMed] [Google Scholar]; (b) Zhang A, Liu ZL, Kan Y. Curr Top Med Chem. 2007;7:343–345. doi: 10.2174/156802607779941279. [DOI] [PubMed] [Google Scholar]
- 5.Gregory H, Taylor CL, Hoplins CR. Nature. 1982;300:269–271. doi: 10.1038/300269a0. [DOI] [PubMed] [Google Scholar]
- 6.Maggio R, Vogel Z, Wess J. Proc Nat Acad Sci USA. 1993;90:3103–3107. doi: 10.1073/pnas.90.7.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, Bouvier M. J Biol Chem. 1996;271:16384–16392. doi: 10.1074/jbc.271.27.16384. [DOI] [PubMed] [Google Scholar]
- 8.(a) Ng GYK, Clark J, Coulombe N, Ethier N, Hebert TE, Sullivan R, Kargman S, Chateauneuf A, Tsukamoto N, McDonald T, Whiting P, Mezey É, Johnson MP, Liu QY, Kolakowski LF, Jr, Evans JF, Bonner TI, O’Neill GP. J Biol Chem. 1999;274:7607–7610. doi: 10.1074/jbc.274.12.7607. [DOI] [PubMed] [Google Scholar]; (b) Kuner R, Kohr G, Grunewald S, Kornau HC. Science. 1999;283:74–77. doi: 10.1126/science.283.5398.74. [DOI] [PubMed] [Google Scholar]; (c) Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, Tang C, Shen QR, Salon JA, Morse K, Laz T, Smith KE, Nagarathnam D, Noble SA, Branchek TA, Gerald C. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]; (d) White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]; (e) Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, Karschin A, Bettler B. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- 9.(a) Wu BL, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Salom D, Lodowski DT, Stenkamp RE, Trong IL, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Proc Nat Acad Sci USA. 2006;103:16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sacher M, Bacco AD, Lunin VV, Ye Z, Wagner J, Gill G, Cygler M. Proc Nat Acad Sci USA. 2005;102:18326–18331. doi: 10.1073/pnas.0505071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devi LA. Trends Pharmacol Sci. 2001;22:532–537. doi: 10.1016/s0165-6147(00)01799-5. [DOI] [PubMed] [Google Scholar]
- 11.For reviews, see: Berque-Bestel I, Lezoualc’h F, Jockers R. Curr Drug Discovery Technol. 2008;5:312–318. doi: 10.2174/157016308786733591.Decker M, Lehmann J. Curr Top Med Chem. 2007;7:347–353. doi: 10.2174/156802607779941297.Nowak I. Curr Top Med Chem. 2007;7:355–362. doi: 10.2174/156802607779941260.Haviv H, Wong DM, Silman I, Sussman JL. Curr Top Med Chem. 2007;7:357–387. doi: 10.2174/156802607779941215.Messers WS., Jr Curr Pharm Des. 2004;10:2015–2020. doi: 10.2174/1381612043384213.Du DM, Carlier PR. Curr Pharm Des. 2004;10:3141–3156. doi: 10.2174/1381612043383412.
- 12.Masur H, Michelis MA, Greene JB, Onorato I, Vande Stouwe RA, Holzman RS, Wormser G, Brettman L, Lange M, Murray HW, Cunningham-Rundles S. New Engl J Med. 1981;305:1431–1438. doi: 10.1056/NEJM198112103052402. [DOI] [PubMed] [Google Scholar]
- 13.Kilmarx PH. Curr Opin HIV AIDS. 2009;4:240–246. doi: 10.1097/COH.0b013e32832c06db. [DOI] [PubMed] [Google Scholar]
- 14.Mathers BM, Degenhardt L, Phillips B, Wiessing L, Hickman M, Strathdee SA, Wodak A, Panda S, Tyndall M, Toufik A, Mattick RP. Lancet. 2008;372:1733–1745. doi: 10.1016/S0140-6736(08)61311-2. [DOI] [PubMed] [Google Scholar]
- 15.Centers for Diseases Control and Prevention (CDC) MMWR Morb Mortal Wkly Rep. 2009;58:1291–1295. [PubMed] [Google Scholar]
- 16.For reviews, see: Norman LR, Basso M, Kumar A, Malow R. Curr Drug Abuse Rev. 2009;2:143–156. doi: 10.2174/1874473710902020143.Anthony IC, Arango JC, Stephens B, Simmonds P, Bell JE. Front Biosci. 2008;13:1294–1307. doi: 10.2741/2762.Hauser KF, El-Hage N, Buch S, Berger JP, Tyor WR, Nath A, Bruce-Keller AJ, Knapp PE. Neurotoxicity Res. 2005;8:63–80. doi: 10.1007/BF03033820.Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, Cass W, Turchan JT. J Acquir Immune Defic Syndr. 2002;31:S62–S69. doi: 10.1097/00126334-200210012-00006.
- 17.Chang SL, Vigorito M. Am J Infect Dis. 2006;2:98–106. [Google Scholar]
- 18.Strathdee SA, Stockman JK. Curr HIV/AIDS Rep. 2010;7:99–106. doi: 10.1007/s11904-010-0043-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Sambamoorthi U, Warner LA, Crystal S, Walkup J. Drug Alcohol Depend. 2000;60:77–89. doi: 10.1016/s0376-8716(99)00142-8. [DOI] [PubMed] [Google Scholar]; (b) Moatti JP, Carrieri MP, Spire B, Gastaut JA, Cassuto JP, Moreau J. AIDS. 2000;14:151–155. doi: 10.1097/00002030-200001280-00010. [DOI] [PubMed] [Google Scholar]; (c) Korthuis PT, Fiellin DA, Fu R, Lum PJ, Altice FL, Sohler N, Tozzi MJ, Asch SM, Botsko M, Fishl M, Flanigan TP, Boverman J, McCarty D. J Acquir Immune Defic Syndr. 2011;56:S83–S90. doi: 10.1097/QAI.0b013e31820bc9a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For reviews see: Batkis MF, Treisman GJ, Angelino AF. AIDS Patient Care STDs. 2010;24:15–22. doi: 10.1089/apc.2009.0242.Bruce RD, Altice FL, Friedland GH. Expert Rev Clin Pharmacol. 2008;1:115–127. doi: 10.1586/17512433.1.1.115.McCance-Katz EF. Clin Infect Dis. 2005;41:S89–S95. doi: 10.1086/429503.
- 21.(a) Schmidhammer H. Prog Med Chem. 1998;35:83–132. [PubMed] [Google Scholar]; (b) Zimmerman DM, Leander JD. J Med Chem. 1990;33:895–902. doi: 10.1021/jm00165a002. [DOI] [PubMed] [Google Scholar]
- 22.(a) Wetzel MA, Steele AD, Eisenstein TK, Adler MW, Henderson EE, Rogers TJ. J Immunol. 2000;165:6519–6524. doi: 10.4049/jimmunol.165.11.6519. [DOI] [PubMed] [Google Scholar]; (b) Zadina JE, Hackler L, Ge LJ, Kastin AJ. Nature. 1997;386:499–502. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]
- 23.(a) Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]; (b) Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, Larosa G, Newman W, Gerard N, Gerard C, Sodroski J. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]; (c) Doranz BJ, Rucker J, Yi YJ, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]; (d) Deng HK, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]; (e) Dragic T, Litwin V, Allaway GP, Martin SR, Huang YX, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 24.For reviews see: Gabuzda D, Wang JB. J NeuroVirol. 1999;5:643–658. doi: 10.3109/13550289909021293.Luster AD. New Eng J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706.
- 25.(a) Steele AD, Henderson EE, Rogers TJ. Virol. 2003;309:99–107. doi: 10.1016/s0042-6822(03)00015-1. [DOI] [PubMed] [Google Scholar]; (b) Mahajan SD, Schwartz SA, Shanahan TC, Chawda RP, Nair MPN. J Immunol. 2002;169:3589–3599. doi: 10.4049/jimmunol.169.7.3589. [DOI] [PubMed] [Google Scholar]; (c) Guo CJ, Li Y, Tian S, Wang X, Douglas SD, Ho WZ. J Investig Med. 2002;50:435–442. doi: 10.1136/jim-50-06-03. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li Y, Wang X, Tian S, Guo CJ, Douglas SD, Ho WZ. J Infect Dis. 2002;185:118–122. doi: 10.1086/338011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Miyagi T, Chuang LF, Doi RH, Carlos MP, Torres JV, Chuang RY. J Biol Chem. 2000;275:31305–31310. doi: 10.1074/jbc.M001269200. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki SJ, Chuang LF, Yau P, Doi RH, Chuang RY. Exp Cell Res. 2002;280:192–200. doi: 10.1006/excr.2002.5638. [DOI] [PubMed] [Google Scholar]
- 27.Chen CG, Li J, Bot G, Szabo I, Rogers TJ, Liu-Chen LY. Eur J Pharmacol. 2004;483:175–186. doi: 10.1016/j.ejphar.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 28.Portoghese PS, Nagase H, Lipkowski AW, Larson DL, Takemori AE. J Med Chem. 1988;31:836–841. doi: 10.1021/jm00399a026. [DOI] [PubMed] [Google Scholar]
- 29.Blumberg H, Pachter IJ, Matossiank Z. 3332950. US Pat. 1967
- 30.(a) Zhang SJ, Yekkirala A, Tang Y, Portoghese PS. Bioorg Med Chem Lett. 2009;19:6978–6980. doi: 10.1016/j.bmcl.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zheng YG, Akgün E, Harikumar KG, Hopson J, Powers MD, Lunzer MM, Miller LJ, Portoghese PS. J Med Chem. 2009;52:247–258. doi: 10.1021/jm800174p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Peng XM, Knapp BI, Bidlack JM, Neumeyer JL. J Med Chem. 2007;50:2254–2258. doi: 10.1021/jm061327z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Portoghese PS, Larson DL, Yim CB, Sayre LM, Ronsisvalle G, Lipkowski AW, Takemori AE, Rice KC, Tam SW. J Med Chem. 1985;28:1140–1141. doi: 10.1021/jm00147a002. [DOI] [PubMed] [Google Scholar]
- 31.(a) Comer SD, Sullivan MA, Hulse GK. Expert Opin Investig Drugs. 2007;16:1285–1294. doi: 10.1517/13543784.16.8.1285. [DOI] [PubMed] [Google Scholar]; (b) Adi Y, Juarez-Garcia A, Wang D, Jowett S, Frew E, Day E, Bauliss S, Roberts T, Burls A. Health Technol Assess. 2007;11(6) doi: 10.3310/hta11060. [DOI] [PubMed] [Google Scholar]; (c) Johansson BA, Berglund M, Lindgren A. Addiction. 2006;101:491–503. doi: 10.1111/j.1360-0443.2006.01369.x. [DOI] [PubMed] [Google Scholar]; (d) Kerkhof AJ. Eur Neuropsychopharmacol. 2006;16:311–323. doi: 10.1016/j.euroneuro.2005.11.001. [DOI] [PubMed] [Google Scholar]; (e) Garbutt JC. Curr Pharm Des. 2010;16:2091–2097. doi: 10.2174/138161210791516459. [DOI] [PubMed] [Google Scholar]; (f) Pettinati HM, O’Brien CP, Rabinowitz AR, Wortman SP, Oslin DW, Kampman KM, Dackis CA. J Clin Psychopharmacol. 2006;26:610–625. doi: 10.1097/01.jcp.0000245566.52401.20. [DOI] [PubMed] [Google Scholar]; (g) Littleton J, Zieglgänsberger W. Am J Addict. 2003;12:S3–S11. doi: 10.1111/j.1521-0391.2003.tb00492.x. [DOI] [PubMed] [Google Scholar]
- 32.Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, wood A, Perros M. Antimicrob Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.U.S. Food and Drug Administration (FDA), Center for Drug Evaluation and Research. Summary Minutes of the Antiviral Drugs Advisory Committee Meeting.2007. [Google Scholar]
- 34.(a) Rook Y, Schmidtke KU, Gaube F, Schepmann D, Wünsch B, Heilmann J, Lehmann J, Winckler T. J Med Chem. 2010;53:3611–3617. doi: 10.1021/jm1000024. [DOI] [PubMed] [Google Scholar]; (b) Liang CH, Romero A, Rabuka D, Sgarbi PWM, Marby KA, Duffield J, Yao S, Cheng ML, Ichikawa Y, Sears P, Hu CY, Hwang SB, Shue YK, Suchech SJ. Bioorg Med Chem Lett. 2005;15:2123–2128. doi: 10.1016/j.bmcl.2005.02.029. [DOI] [PubMed] [Google Scholar]; (c) Neumeyer JL, Zhang A, Xiong W-N, Gu X-H, Hilbert JE, Knapp BI, Stevens Negus S, Mello NK, Bidlack JM. J Med Chem. 2003;46:5162–5170. doi: 10.1021/jm030139v. [DOI] [PubMed] [Google Scholar]
- 35.(a) Lemoine RC, Petersen AC, Setti L, Baldinger T, Wanner J, Jekle A, Heilek G, deRosier A, Ji CH, Rotstein DM. Bioorg Med Chem Lett. 2010;20:1674–1676. doi: 10.1016/j.bmcl.2010.01.080. [DOI] [PubMed] [Google Scholar]; (b) Barber CG, Blakemore DC, Chiva JY, Eastwood RL. Bioorg Med Chem Lett. 2009;19:1075–1079. doi: 10.1016/j.bmcl.2009.01.009. [DOI] [PubMed] [Google Scholar]; (c) Price DA, Armour D, de Groot M, Leishman D, Napier C, Perros M, Stammen BL, Wood A. Curr Top Med Chem. 2008;8:1140–1151. doi: 10.2174/156802608785700007. [DOI] [PubMed] [Google Scholar]; (d) Price DA, Armour D, de Groot M, Leishman D, Napier C, Perros M, Stammen BL, Wood A. Bioorg Med Chem Lett. 2006;16:4633–4637. doi: 10.1016/j.bmcl.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 36.Li G, Haney KM, Kellogg GE, Zhang Y. J Chem Inf Model. 2009;49:120–132. doi: 10.1021/ci800356a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.(a) Harikumar KG, Akgün E, Portoghese PS, Miller LL. J Med Chem. 2010;53:2836–2842. doi: 10.1021/jm100135g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Daniels DJ, Kulkarni A, Xie ZH, Bhushan RG, Portoghese PS. J Med Chem. 2005;48:1713–1716. doi: 10.1021/jm034234f. [DOI] [PubMed] [Google Scholar]; (c) Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Proc Nat Acad Sci USA. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xie ZH, Bhushan RG, Daniels DJ, Portoghese PS. Mol Pharmacol. 2005;68:1079–1086. doi: 10.1124/mol.105.012070. [DOI] [PubMed] [Google Scholar]; (e) Portoghese PS, Larson DL, Sayre LM, Yim CB, Ronsisvalle G, Tam SW, Takemori AE. J Med Chem. 1986;29:1855–1861. doi: 10.1021/jm00160a010. [DOI] [PubMed] [Google Scholar]
- 38.Li F, Liu JC, Jas GS, Zhang JW, Qin GT, Xing J, Cotes C, Zhao H, Wang XK, Diaz LA, Shi ZZ, Lee DY, Li KCP, Li Z. Bioconjugate Chem. 2010;21:270–278. doi: 10.1021/bc900313d. [DOI] [PubMed] [Google Scholar]
- 39.Sayre LM, Portoghese PS. J Org Chem. 1980;45:3366–3368. [Google Scholar]
- 40.Price DA, Gayton S, Selby MD, Ahman J, Haycock-Lewandowski S, Stammen BL, Warren A. Tetrahedron Lett. 2005;46:5005–5007. [Google Scholar]
- 41.(a) Haycock-Lewandowski SJ, Wilder A, Åhman J. Org Process Res Dev. 2008;12:1094–1103. [Google Scholar]; (b) Åhman J, Birch M, Haycock-Lewandowski SJ, Long J, Wilder A. Org Process Res Dev. 2008;12:1104–1113. [Google Scholar]
- 42.(a) Davies SG, Smith AD, Price PD. Tetrahedron: Asymmetry. 2005;16:2833–2891. [Google Scholar]; (b) Bull SD, Davies SG, Roberts PM, Savory ED, Smith AD. Tetrahedron. 2002;58:4629–4642. [Google Scholar]; (c) Bull SD, Davies SG, Smith AD. Tetrahedron: Asymmetry. 2001;12:2941–2945. [Google Scholar]; (d) Imamura H, Shimizu A, Sato H, Sugimoto Y, Sakuraba S, Nakajima S, Abe S, Miura K, Nishimura I, Yamada K, Morishima H. Tetrahedron. 2000;56:7705–7713. [Google Scholar]; (e) Davies SG, Walters LAS. J Chem Soc, Perkin Trans 1. 1994;48:1129–1139. [Google Scholar]; (f) Davies SG, Ichihara O. Tetrahedron: Asymmetry. 1991;2:183–186. [Google Scholar]
- 43.Gonzalez-Bello C, Abell C, Leeper FJ. J Chem Soc, Perkin Trans 1:Org Bio-Org Chem. 1997;7:1017–1024. [Google Scholar]
- 44.Lee S, Jørgensen M, Hartwig JF. Org lett. 2001;3:2729–2732. doi: 10.1021/ol016333y. [DOI] [PubMed] [Google Scholar]
- 45.Stover JS, Shi J, Jin W, Vogt PK, Boger DL. J Am Chem Soc. 2009;131:3342–3348. doi: 10.1021/ja809083d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.(a) Lee MH, Kang KH, Lee YM, Kim D, Park YS. Bull Korean Chem Soc. 2008;29:1075–1078. [Google Scholar]; (b) Gallagher G, Jr, Lavanchy PG, Wilson JW, Hieble JP, DeMarins RM. J Med Chem. 1985;28:1533–1536. doi: 10.1021/jm00148a028. [DOI] [PubMed] [Google Scholar]
- 47.Nichols DE, Cassady JM, Persons PE, Yeung MC, Clemens JA. J Med Chem. 1989;32:2128–2134. doi: 10.1021/jm00129a017. [DOI] [PubMed] [Google Scholar]
- 48.Mackenzie AR, Marchington AP, Middleton DS, Meadows SD. WO97/27185. PCT Int Appl. 1997
- 49.Pinto DJP, Orwat MJ, Koch S, Rossi KA, Alexander RS, Smallwood A, Wong PC, Rendina AR, Luettgen JM, Knabb RM, He K, Xin B, Wexler RR, Lam PYS. J Med Chem. 2007;50:5339–5356. doi: 10.1021/jm070245n. [DOI] [PubMed] [Google Scholar]
- 50.Atwell GJ, Denny WA. Synthesis. 1984;12:1032–1033. [Google Scholar]
- 51.Haney KM, Zhang F, Arnatt CK, Yuan YY, Li G, Ware JL, Gewirtz DA, Zhang Y. Bioorg Med Chem Lett. 2011;21:5159–5163. doi: 10.1016/j.bmcl.2011.07.058. [DOI] [PubMed] [Google Scholar]
- 52.Li G, Aschenbach LC, Chen JY, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, Zhang Y. J Med Chem. 2009;52:1416–1427. doi: 10.1021/jm801272c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.