

Abstract
This unit describes Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq), a method for mapping chromatin accessibility genome-wide. This method probes DNA accessibility with hyperactive Tn5 transposase, which inserts sequencing adapters into accessible regions of chromatin. Sequencing reads can then be used to infer regions of increased accessibility, as well as to map regions of transcription factor binding and nucleosome position. The method is a fast and sensitive alternative to DNase-Seq for assaying chromatin accessibility genome-wide, or to MNase for assaying nucleosome positions in accessible regions of the genome.
Keywords: ATAC-seq, Transposase, Chromatin Accessibility
Introduction
In a human cell, approximately two meters of DNA are packed within a five-micron nucleus—a spectacular topological challenge solved by the cell through the hierarchical folding of DNA around histone proteins to form nucleosomes, and the compaction of nucleosomes into chromatin (Kornberg, 1974). This hierarchical packaging sequesters inactive genomic regions and leaves biologically active regions – be they promoters, enhancers, or other regulatory elements – accessible to transcription machinery (Gross and Garrard, 1988; Bell et al., 2011). Atop this landscape of physical compaction operates a dynamic epigenetic code that includes DNA methylation, nucleosome positioning, histone composition, and modification, as well as transcription factors, chromatin remodelers, and non-coding RNAs (Kouzarides, 2007; Rinn and Chang, 2012). Cellular phenotypes are substantially governed by epigenetic mechanisms that manipulate the composition, compaction, and nucleoprotein structure of chromatin (Chen and Dent, 2014).
Methods such as MNase-seq (Zaret, 2005), ChIP-seq (Landt et al., 2012), and DNase-seq (Song and Crawford, 2010) in particular have proven to be information-rich, genome-wide analysis methods for understanding this epigenetic structure, providing information on transcription factor binding, the positions of modified and canonical nucleosomes, and chromatin accessibility at regulatory elements such as promoters, enhancers, and insulators (Thurman et al., 2012; Valouev et al., 2011; Consortium, 2012). However, current methods for assaying chromatin structure and composition often require tens to hundreds of millions of cells as input material, averaging out heterogeneity in cellular populations. In many cases, rare and important cellular sub-types cannot be acquired in amounts sufficient for genome-wide chromatin analyses. The assay of transposase accessible chromatin (Buenrostro et al., 2013) (ATAC-seq) uses hyperactive Tn5 transposase (Goryshin and Reznikoff, 1998; Adey et al., 2010) to simultaneously cut and ligate adapters for high-throughput sequencing at regions of increased accessibility. Genome-wide mapping of insertion ends by high-throughput sequencing allows for multidimensional assays of the regulatory landscape of chromatin with a relatively simple protocol that can be carried out in hours for a standard sample size of 50,000 cells.
ATAC-seq data sets can also be bioinformatically separated into reads that are shorter than the canonical length generally protected by a nucleosome and reads consistent with the approximate length of DNA protected by a nucleosome. These different populations of reads provide information about the positions of nucleosomes, as well as nucleosome-free regions. In this way, paired-end ATAC-seq data provides MNase-seq-like data sets for nucleosomes at relatively accessible regions of the genome.
ATAC-seq is compatible with a number of methods for cell separation and isolation, including cell sorting as well as disruption of intact tissues to cellular suspensions. The method has also worked across many cell types and species. However, the following protocol has been optimized for human lymphoblastoid cells, and provides guidance on modifications to the method that might allow adaptation to different cell types. In general, the method is separated into three separate components: cell lysis, transposition, and amplification. Crosslinking generally reduces library creation efficiency, and therefore we recommend starting with fresh unfixed cells for maximum sensitivity of the methodology.
Materials
Phosphate Buffered Saline (PBS)
Molecular biology grade IGEPAL CA-630
Lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630)
2x TD (2x reaction buffer, Illumina Cat #FC-121-1030)
TDE1 (Nextera Tn5 Transposase, Illumina Cat #FC-121-1030)
Qiagen MinElute PCR Purification Kit
NEBNext High-Fidelity 2x PCR Master Mix (New England Labs Cat #M0541)
25 uM Custom Nextera PCR Primer 1
25 uM Custom Nextera PCR Primer 2
100x SYBR Green I (Invitrogen Cat #S-7563)
0.2-ml PCR tubes
PCR Thermal cycler
qPCR consumables, products are specific to the instrument
Cell Preparation
-
Harvest cells, protocol to be defined by the user.
Cells should be intact and in a homogenous single cell suspension. -
Spin down 50,000 cells at 500 ×g for 5 min, 4°C.
The number of cells at this step is crucial as the transposase to cell ratio sets the distribution of DNA fragments generated. See Critical Parameters. Wash once with 50 μl of cold 1x PBS buffer. Spin down at 500 ×g for 5 min, 4°C.
-
Gently pipette to resuspend the cell pellet in 50 μl of cold lysis buffer. Spin down immediately at 500 ×g for 10 min, 4°C.
This step provides lysis of cells with non-ionic detergent and generates of a crude nuclei preparation. Discard the supernatant, and immediately continue to transposition reaction.
Transposition Reaction and Purification
Make sure the cell pellet is set on ice.
-
To make the transposition reaction mix, combine the following:
25 μl TD (2x reaction buffer)
2.5 μl TDE1 (Nextera Tn5 Transposase)
22.5 μl Nuclease Free H2O
Resuspend nuclei in the transposition reaction mix.
-
Incubate the transposition reaction at 37°C for 30 min.
Gentle mixing may increase fragment yield. Immediately following transposition, purify using a Qiagen MinElute PCR Purification Kit.
Elute transposed DNA in 10 μl Elution Buffer (10 mM Tris buffer, pH 8).
-
Purified DNA can be stored at −20°C.
This is a convenient stopping point. Please note that these DNA fragments are not PCR amplifiable if melted at this point.
PCR Amplification
-
To amplify transposed DNA fragments, combine the following in a 0.2 ml PCR tube:
10 μl Transposed DNA
10 μl Nuclease Free H2O
2.5 μl 25 μM Custom Nextera PCR Primer 1
2.5 μl 25 μM Custom Nextera PCR Primer 2 (Contains Barcode)
-
25 μl NEBNext High-Fidelity 2x PCR Master Mix
A complete list of primers is available in Buenrostro et al. Care should be taken to ensure that samples are barcoded appropriately for subsequent pooling and sequencing.
-
Thermal cycle as follows:
1 cycle of 72°C for 5 min, 98°C for 30 sec
-
5 cycles of 98°C for 10 sec, 63°C for 30 sec, 72°C for 1 min
This first 5 minute extension at 72°C is critical to allow extension of both ends of the primer after transposition, thereby generating amplifiable fragments (see figure). This short pre-amplification step ensures that downstream quantitative PCR (qPCR) quantification will not change the complexity of the original library.
-
To reduce GC and size bias in PCR, the appropriate number of PCR cycles is determined using qPCR allowing us to stop amplification prior to saturation. To run a qPCR side reaction, combine the following in qPCR compatible consumables:
5 μl of previously PCR amplified DNA
4.41 μl Nuclease Free H2O
0.25 μl 25 μM Customized Nextera PCR Primer 1
0.25 μl 25 μM Customized Nextera PCR Primer 2
0.09 μl 100x SYBR Green I
5 μl NEBNext High-Fidelity 2x PCR Master Mix
-
Using a qPCR instrument, cycle as follows:
1 cycle of 98°C for 30 sec
20 cycles of 98°C for 10 sec, 63°C for 30 sec, 72°C for 1 min
-
To calculate the additional number of cycles needed, plot linear Rn versus cycle and determine the cycle number that corresponds to ¼ of maximum fluorescent intensity.
The purpose of this qPCR step is to generate libraries that are minimally PCR amplified. Most PCR bias comes from later PCR cycles that occur during limited reagent concentrations. This determination of the optimal number of cycles to amplify the library reduces artifacts associated with saturation PCR of complex libraries. -
Run the remaining 45 μl PCR reaction to the cycle number determined by qPCR. Cycle as follows:
1 cycle of 98°C for 30 sec
-
N cycles of 98°C for 10 sec, 63°C for 30 sec, 72°C for 1 min
Cycle for an additional N cycles, where N is determined using qPCR.
-
Purify amplified library using Qiagen MinElute PCR Purification Kit. Elute the purified library in 20 μl Elution Buffer (10 mM Tris Buffer, pH 8). Be sure to dry the column before adding elution buffer.
The concentration of DNA eluted from the column ought to be approximately 30 nM, however 5-fold variation is possible and not detrimental.
Optional Library Quality Control using Gel Electrophoresis
Prior to purification, amplified libraries can be visualized using gel electrophoresis, however the low concentration of the amplified materials requires a 5% TBE polyacrylamide gel optimized for sensitivity. We find that adding 0.6x SYBR Green I to libraries provides excellent signal-to-noise without the need for post-staining. We routinely load 15 ng of 100 bp NEB ladder with 0.6x SYBR Green I as a DNA marker. Although, in principle any instrument containing a blue-light source or imaging systems equipped with a laser that emits at 488 nm can be used to visualize DNA stained with SYBR Green I dye, we use the Typhoon TRIO Variable Mode Imager from Amersham Biosciences for improved sensitivity. Images are best obtained by digitizing at 100 microns pixel size resolution with a 520 nm band-pass emission filter to screen out reflected and scattered excitation light and background fluorescence, see Figure 2a for an example.
Figure 2.

Fragment sizes for amplified ATAC-seq libraries, determined by (A) Gel electrophoresis and (B) Bioanalyzer. Bioanalyzer image contrast has been enhanced from its original image for clarity. (C) Insert sizes determined by high-throughput sequencing. Adapters are an additional 124 bps and are not included when measuring fragment size in this panel.
Alternate, Library Quality Control with Bioanalyzer
The quality of purified libraries can also be assessed using a Bioanalyzer High Sensitivity DNA Analysis kit (Agilent), see Figure 2b for an example. We have seen that a preponderance of high molecular weight DNA (>1000 bps) can create misleading and inaccurate Bioanalyzer results.
COMMENTARY
Background Information
The ATAC-seq methodology relies on library construction using the hyperactive transposase Tn5. Tn5 is a prokaryotic transposase, which endogenously functions through the “cut and paste” mechanism, requiring sequence-specific excision of a locus containing 19 base-pair inverted repeats. The Nextera DNA Sample Preparation kit provides the Tn5 transposase reagent loaded with sequencing adapters creating an active dimeric transposome complex (Adey et al., 2010). The Nextera version of the transposase harbors specific point mutants to the Tn5 backbone which significantly increases activity (Goryshin and Reznikoff, 1998; Reznikoff, 2008). This transposase preferentially inserts sequencing adapters into unprotected regions of DNA, therefore acting as a probe for measuring chromatin accessibility genome-wide.
Critical Parameters
Cell number
ATAC-seq is often robust to relatively minor variations of cell number (roughly 25k to 75k for example). In general, using too few cells causes over-digestion of chromatin and appears to create a larger fraction of reads that map to inaccessible regions of the genome (i.e. noise); using too many cells causes under-digestion and creates high molecular weight fragments, which may be difficult to sequence. However, the total number of cells also sets the fundamental diversity of the sequencing library (i.e. the number of unique DNA fragments). We note that to obtain more complex libraries, larger reaction volumes or serial reactions with the same reaction volume, can be carried out.
Cell collection
Methods for cell collection vary and may need to be optimized for ATAC-seq. We find fixatives can reduce transposition frequency and are not recommended. We also note methods involving mechanical shearing can significantly reduce signal-noise. Methods involving high purity that produce intact cells in a homogenous single cell suspension tend to produce the best data sets.
PCR and Fragmentation distribution
The PCR methods described are designed to reduce the effect of size- and GC-bias from the library construction process. These methods are particularly useful for ATAC-seq because of the highly diverse fragment sizes. We find that samples containing an excess of large fragments (>1 kb) are relatively hard to quantify and result in reduced clustering efficiencies when sequencing. If a library is enriched for long fragments, library fragmentation can be optimized using more or fewer cells per reaction. Alternatively, size selection prior to sequencing might be carried out to eliminate these potentially confounding long fragments. Although it is common to size-select a narrow interval for sequencing, we recommend excising a large fragment size window of 100–1000 bps to maintain high library complexity, and enable the richness of the inferences that can be extracted from the full fragment size distribution. The described PCR method has the additional benefit of serving as an estimate for library complexity. We find that if >6 additional cycles are needed (>11 total cycles) library complexity becomes a concern. Library complexity can be improved by optimizing the input cell number or by making libraries of technical replicates.
Library Quantitation
We use qPCR based methods to quantify our ATAC-seq libraries. We have found that other methods, such as Qubit, can potentially give misleading and inaccurate results due to variation within the fragment size distribution. We recommend quantifying libraries using the KAPA Library Quant Kit for Illumina Sequencing Platforms (KAPABiosystems). Alternately, integrated Bioanalyzer traces can be used to approximate library concentration.
Sequencing
ATAC-seq and the Nextera workflow are designed for sequencing using Illumina high-throughput sequencing instruments. When sequencing ATAC-seq libraries, Nextera-based sequencing primers and reagents must be used. Depending on the sequencing instrument, this may require modification of the standard sequencing workflow. For nucleosome mapping, paired-end sequencing is preferred. Paired-end 50-cycle reads generally provide accurate alignments with reasonable costs. For inferring differences in open chromatin within human samples we generally use >50M mapped reads and for transcription factor foot-printing we use >200M mapped reads (Neph et al., 2012). Data yield can sometimes be impacted by the large fraction of mitochondrial reads. For troubleshooting cluster densities and sequencing quality refer to Illumina’s technical support staff.
Troubleshooting
Assuming that the cells of interest are healthy and intact prior to beginning the procedure, the biggest source of failure comes from variations in cell number. In general, the addition of too many cells leads to “under transposition” (with a majority of large fragments), while the addition to too few cells leads to “over transposition” and a preponderance of short fragments on the gel, and the possible elimination of any banding pattern. When applying this protocol to non-human cells, we see large differences in the number of required cells depending on the species; however, variation in outcome can also come from different cell types as well. If results using the standard protocol are not ideal, an efficient method for optimization is to scale the reaction down 10x and to optimize the lysis methods as well as the cell number, starting from 5,000 cells per reaction. The nucleosomal banding pattern shown in Figure 2 is correlated with high quality libraries.
Anticipated results
After sequencing human libraries, we often find that approximately ½ of reads are of sub-nucleosomal length (less than approximately 150 bp) and approximately half of the reads longer than this length (Figure 2c). In general we find excellent agreement with DNase-seq methods and enrichment for regions of accessible chromatin, with ~20% of reads concentrated in ~2% of the genome. We estimate that a single 50 ul reaction volume can generate approximately 50–100M unique amplifiable fragments.
Time Considerations
ATAC-seq was designed with speed in mind; we have optimized the protocol to require a total time of 3 hours. The above protocol for nuclei extraction requires 30 minutes. Transposition and purification requires 45 minutes. PCR and purification requires 1 hour and 45 minutes. Methods for QC and library quantitation can vary. A natural stopping point exists after transposition and cleanup, at which point DNA fragments can be frozen indefinitely prior to the PCR amplification.
Figure 1.
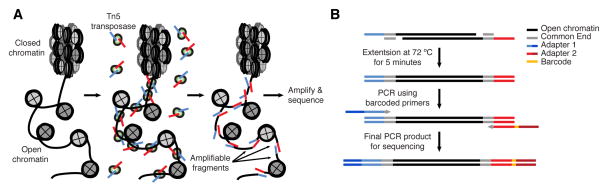
(A) Library preparation schematic. (B) Transposition results in fragmented DNA. Prior to amplification, adapters have to be completed with a 72°C extension step. During the subsequent PCR additional sequence is incorporated into the adapters, which include common sequencing ends and a sequencing barcode.
Acknowledgments
J.D.B. acknowledges support from a National Science Foundation graduate research fellowship and NIH training grant T32HG000044. H.Y.C. acknowledges support as an Early Career Scientist of the Howard Hughes Medical Institute. This work is supported by NIH (H.Y.C., W.J.G., J.D.B.), including RC4NS073015, U01DK089532, and U19AI057229, Scleroderma Research Foundation (H.Y.C.), and California Institute for Regenerative Medicine (H.Y.C.). W.J.G acknowledges support as a Rita Allen Foundation Young Scholar.
Footnotes
Competing financial interests
Stanford University has filed a provisional patent application on the methods described, and J.D.B., H.Y.C. and W.J.G. are named as inventors.
Literature Cited
- Adey AA, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biology. 2010;11:R119. doi: 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O, Tiwari VK, Thomä NH, Schübeler D. Determinants and dynamics of genome accessibility. Nature Reviews Genetics. 2011;12:554–564. doi: 10.1038/nrg3017. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Dent SYR. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nature Reviews Genetics. 2014;15:93–106. doi: 10.1038/nrg3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium TEP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryshin IY, Reznikoff WS. Tn5 in vitro transposition. The Journal of biological chemistry. 1998;273:7367–7374. doi: 10.1074/jbc.273.13.7367. [DOI] [PubMed] [Google Scholar]
- Gross DS, Garrard WT. Nuclease Hypersensitive Sites in Chromatin. Annual Review of Biochemistry. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin Modifications and Their Function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikoff WS. Transposon Tn5. Annual review of genetics. 2008;42:269–286. doi: 10.1146/annurev.genet.42.110807.091656. [DOI] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annual Review of Biochemistry. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor protocols. 2010;2010 doi: 10.1101/pdb.prot5384. pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516–520. doi: 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret K. Micrococcal nuclease analysis of chromatin structure. In: Ausubel Frederick M, et al., editors. Current protocols in molecular biology. Unit 21.1. Chapter 21. 2005. [DOI] [PubMed] [Google Scholar]