

Abstract
A/J mice develop progressive hearing loss that begins before one month of age and is attributed to cochlear hair cell degeneration. Screening tests indicated this strain also develops early onset vestibular dysfunction and has otoconial deficits. The purpose of this study was to characterize the vestibular dysfunction and macular structural pathology over the lifespan of A/J mice. Vestibular function was measured using linear vestibular evoked potentials (VsEPs). Macular structural pathology was evaluated using light microscopy, SEM, TEM, confocal microscopy and Western blotting. Individually, vestibular functional deficits in mice ranged from mild to profound. On average, A/J mice had significantly reduced vestibular sensitivity (elevated VsEP response thresholds and smaller amplitudes), whereas VsEP onset latency was prolonged compared to age-matched controls (C57BL/6J). A limited age-related vestibular functional loss was also present. Structural analysis identified marked age-independent otoconial abnormalities in concert with some stereociliary bundle defects. Macular epithelia were incompletely covered by otoconial membranes with significantly reduced opacity and often contained abnormally large or giant otoconia as well as normal appearing otoconia. Elevated expression of key otoconins [i.e., otoconin 90, otolin and keratin sulfate proteoglycan] ruled out the possibility of reduced levels contributing to otoconial dysgenesis. The phenotype of A/J was partially replicated in a consomic mouse strain (C57BL/6J-Chr 17A/J/NaJ), thus indicating that Chr 17A/J contained a trait locus for a new gene variant responsible to some extent for the A/J vestibular phenotype. Quantitative trait locus analysis identified additional epistatic influences associated with chromosomes 1, 4, 9 and X. Results indicate that the A/J phenotype represents a complex trait and the A/J mouse strain presents a new model for the study of mechanisms underlying otoconial formation and maintenance.
Keywords: A/J mouse strain, gene variants, utricle, saccule, otoconia, inner ear
Introduction
The mammalian inner ear houses five organs in the vestibular labyrinth that detect head motion and contribute to balance function. Receptors within the two maculae and three ampullae encode linear and angular motion of the head, respectively. Deficits in one or more peripheral structures can cause vestibular dysfunction leading to dizziness, vertigo or imbalance. Several mouse strains with selective peripheral vestibular deficits have been described and the genes responsible for these deficits have been identified (e.g., Dickie and Deol, 1966; Sweet, 1980; Lane, 1986, 1987; Ornitz et al., 1998; Bergstrom et al., 1998; Ahituv and Avraham, 2000; Hurle et al., 2003; Cryns et al., 2004; Paffenholz et al., 2004; Beisel et al., 2005; Kiss et al., 2006; Zhao et al., 2007; Nakano et al., 2008; Goodyear et al., 2012; Lee et al., 2013).
Identifying genes critical for normal vestibular structure and function is important not only because such information leads to a better understanding of how particular genes and their protein products contribute to vestibular function, but it also can lead to the discovery of new and potentially valuable genetic markers for human and animal inner ear diseases. Such markers can be used to identify individuals at risk for developing inner ear dysfunction, improve accuracy of diagnosis and, with time, could suggest treatment strategies offering significant benefits to patients.
Efforts have been underway to identify new inbred mouse strains evidencing early onset impaired gravity receptor function (e.g., onset between birth and young adult ages, <90 days-old; Jones et al., 2006, 2008). It is clear from these efforts that gravity receptor function can vary considerably among inbred mouse strains. Mouse strains identified with vestibular deficits in initial screening studies (Jones et al., 2006) have been evaluated in more detail and the findings have been reported in cursory form (Jones et al., 2008). Preliminary vestibular testing in the AHe/J and A/J strains were reported for two ages in these studies (Jones et al., 2008). Both strains evidenced elevated vestibular thresholds and otoconial abnormalities were noted for the A/J strain.
The present report provides a detailed characterization of vestibular function, morphology and quantitative trait locus (QTL) analysis for the A/J mouse. The results show that the A/J strain develops early onset (<90-days-old) gravity receptor dysfunction, highly variable otoconial dysgenesis, a modest progressive elevation of vestibular thresholds, prolonged VsEP onset latencies and a decrease in vestibular response amplitudes with age. QTL analysis revealed a robust relationship between vestibular functional measures (P1 latency) and chromosome 9 (Chr 9) with an interaction between Chr 9 and Chr X and epistatic influences from Chr 1 and 4. Additional study of C57BL/6J mice consomic for the A/J Chr 17 (C57BL/6J-Chr 17A/J/NaJ) suggested that the A/J Chr 17 may carry a new gene variant that contributes to vestibular deficits in A/J mice.
Methods
Animals
Colonies of A/J mice were established at East Carolina University (ECU) and Boys Town National Research Hospital (BTNRH) by breeding A/J sibling pairs (stock # 000646) purchased from The Jackson Laboratory (Bar Harbor, ME). A/J offspring (n =197, both sexes) ranging from E18.5 to approximately 19 months of age were studied. Consomic C57BL/6J mice with substituted A/J Chr 17 were purchased from The Jackson Laboratory (C57BL/6J-Chr 17A/J/NaJ; stock# 004395). A separate colony of C57BL/6J-Chr 17A/J/NaJ (stock # 004395) was established at ECU (offspring n = 57) and animals studied at ages ranging from 1.4 to 12.7 months-old. Seven normal C57BL/6J mice were obtained from Jackson Laboratory and used to provide normative data for quantitative comparison of otoconial organs. Animals were caged in groups by sex in a temperature-controlled (21 +3/−4 °C) room under a 12-h light/dark cycle (lights on at 7:00 a.m.), with free access to food and water. All procedures were approved by the Institutional Animal Care and Use Committee of East Carolina University and Boys Town National Research Hospital and were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals. In addition to data collected from the experimental animals noted above, data collected previously for normal C57BL/6J animals studied over the entire lifespan of the mouse (1 to 24 months-of-age, Mock 2008) were used for comparison to A/J strains reported here. These data serve as our standard laboratory control database.
Measurement of vestibular sensory evoked potentials (VsEPs)
Mice were deeply anesthetized by intraperitoneal injection of ketamine/xylazine (18:2 mg/ml; 5–7μl/g body weight) followed by maintenance doses of 0.05 ml every 60 minutes as needed to maintain adequate anesthesia. Core body temperature was maintained at 37.0 ± 0.2°C using a homeothermic heating blanket system (FHC, Inc., Bowdoin, ME, USA). The linear VsEP is a compound action potential produced by eighth nerve neurons innervating gravity receptors and their central neural relays in the brainstem (Jones et al., 1999, 2004; Jones and Jones 2014; Nazareth and Jones 1998). Recording electrodes were placed subcutaneously at the nuchal crest (noninverting electrode), behind the right pinna (inverting electrode) and at the right hip (ground electrode). Mice were placed supine on a stationary platform and the head was secured within a spring clip coupled to a voltage-controlled mechanical shaker. The head was oriented with nose up and linear translation stimuli were presented in the naso-occipital axis parallel to the Earth-vertical axis. Vestibular stimuli consisted of 2 ms linear rectangular jerk pulses, delivered to the cranium using two stimulus polarities [normal (initial upward jerk) and inverted (initial downward jerk)] at a rate of 17 pulses/s. Stimuli were presented at the maximum level (i.e., +6 dB re: 1.0 g/ms, where 1 g = 9.8 m/s2), first without and then with auditory masking to confirm the absence of auditory components. The acoustic masker used was a broadband forward masker (50–50,000 Hz, 87.5 dB SPL, Jones and Jones, 1999) that was presented continuously, except for a 15 ms period beginning at the onset of each jerk stimulus. Stimulus level was then dropped to −18 dB and raised in 3 dB steps to complete a response threshold profile series across all stimulus levels for each animal. Response threshold was determined from this response profile. VsEP thresholds were defined as the stimulus level midway between the minimum level that produced a discernible response and the maximum level that failed to produce a response.
Signal averaging was used to extract the VsEP responses from the background electrophysiological activity. Ongoing electroencephalographic (EEG) activity was amplified (200,000X), filtered (300–3000 Hz, −6 dB points), and digitized beginning at the onset of each jerk stimulus (1024 points, 10 μs/pt) to produce one primary response trace. For each stimulus intensity and polarity, 128 primary responses were averaged to produce an averaged response waveform. Four averaged response waveforms were recorded for each stimulus intensity (i.e., two waveforms recorded for normal stimulus polarity and two for inverted polarity). Final individual response traces were produced by summing one averaged response to each stimulus polarity and dividing the result by two, thus producing two response traces for each stimulus intensity for each animal.
Tissue dissection and processing
In 100 A/J mice (ages 0.7 to 18 months) following VsEP testing and in 70 additional A/J mice at various ages (E18.5, P0, P4, P11, P22, 1.5 months, 3 months, 5 months, 7 months, and 10 months), temporal bones were harvested for morphological study. Otic capsules (bilateral) were removed from the skull of each animal; generally one ear was processed for light microscopy and the opposite ear was processed for SEM, TEM, confocal microscopy or Western blotting.
Light microscopy
Otic capsules were fixed overnight in 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB; pH 7.4) at room temperature (RT). Bones were then dehydrated through a graded series of ethanol washes (30%, 50%, 70% and 80% ethanol, each for 1 hour, then 100% ethanol overnight) at RT and cleared in methyl salicylate (MP Biomedicals; Solon, OH) overnight at RT. Otoconial membranes (OMs) overlaying the utricle and saccule were viewed under polarized light on a light microscope (Olympus BX50; 40–48X magnification) and images were acquired using a digital camera (Spot Insight; Diagnostic Instruments Inc., Sterling Heights, MI).
Scoring Otoconia
Each epithelium was assigned an integer otoconial coverage score of 0 to 4, where a score of 4 represented the normal ear with the entire OM covered with otoconia and 0 represented the absence of otoconia on the OM. The outer border of each sensory patch was visible under the microscope and it was used to define the epithelial outer boundary (figure 1). If this boundary could not be discerned, then the epithelium was scored as “not visible” and a coverage score was not assigned. Under the light microscope, an otoconial membrane with the normal complement of otoconia is opaque and appears dark or black (figure 1), whereas in the absence of otoconia, the OM appears light gray or white (transparent otoconial membrane). Coverage scores were assigned based on the estimated percentage of OM area covered by otoconia (opaque regions) within the outer epithelial boundary. In the normal macula, the OM covers the entire region within the epithelial boundary. Coverage scores were assigned to percentages as follows: 4 = normal coverage by otoconia (i.e., no otoconia missing), 3 = 75% ≥ coverage > 50%, 2 = 50% ≥ coverage > 25%, 1 = 25% ≥ coverage > 0 and 0 = 0% coverage (i.e., no otoconia). Examples of scoring are shown in figure 1. To characterize otoconial membrane opacity and area of otoconial coverage, the outer edge of visible otoconial membrane was marked and quantitative digital image analysis (ImageJ, v1.41; NIH; http://rsb.info.nih.gov/ij/) was used to calculate the area, the average gray scale level for the marked region (gray scale, 8 bit: 0 = black, 255 = white) and the opacity of the marked region (opacity level) was determined using the following formula: opacity level = 255 minus the average gray scale level (0 = transparent = white, 255 = opaque = black). Examples are illustrated in figure 1. The largest otoconia visible on the macular surface were also described in comments and given scores as follows: of normal size (score = 0) or abnormally large or giant (score of 1).
Figure 1.

Illustration of the method used for otoconia coverage scoring in the present paper. Shown are representative images of otoconial membranes (OM) overlaying the utricle and saccule in cleared temporal bones. Outer edges of OM have been outlined in black. A, B & C: AJ animals with varying grades of otoconial dysgenesis. A was given OM scores of 3 and 4 for the utricle and saccule respectively. B was scored as 2 for both the utricle and saccule. C represents an OM score of 1 for both utricle and saccule. C Inset: Utricular OM showing three giant crystals of otoconia. D & E: represent C17 animals. D had no coverage deficits for the utricle or saccule and was given an OM score of 4 for both macular organs. E had mild coverage deficits for both utricle and saccule (score 3). Inset shows coverage deficits and large abnormal otoconia in the lateral extra-striolar region of the utricle. F) C57 animal with normal utricle and saccule OMs with scores of 4. The resolution of pictures shown in this compressed collage is much less than that available during scoring of original micrographs.
Scanning electron microscopy (SEM)
Deeply anesthetized animals underwent transcardial systemic perfusion with 0.1 Msodium cacodylate buffer (pH 7.2), followed by 10–20 ml of 3% glutaraldehyde + 3 mM CaCl2 in 0.1 M sodium cacodylate buffer (pH7.2). Dissected utricles and saccules were then immersion-fixed for 24 hours in the same fixative, washed, and post-fixed in 1% osmium tetroxide for 1 hour. Samples were then dehydrated, critical point dried in CO2, mounted, sputter coated with gold palladium and viewed on a Jeol (JSM-848) scanning electron microscope at 15 kV.
Transmission electron microscopy (TEM)
Anesthetized animals were perfused (as described for SEM) and fixed in 3% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH7.2) for 1 hour at 4°C. After three washes in 0.1 M sodium cacodylate buffer, samples were post-fixed with 1% osmium tetroxide for 1 hour followed by three rinses in 0.1 M sodium cacodylate buffer. Tissues were dehydrated in graded ethanol and infiltrated in 50% propylene oxide + 50% resin for 1 hour, then placed in resin for another hour and embedded in resin based mold for polymerization overnight in a 37°C oven. Semi-thin sections were stained with Toluidine Blue. Ultra-thin sections (85 nm) were taken from each sample and analyzed on a Jeol (JEM-1011) transmission electron microscope at 60 kV.
Confocal microscopy
Phalloidin staining
Fixed (4% PFA in 0.1 M PB, pH 7.4) and decalcified vestibules were permeabilized with 0.1% Triton X-100 in PBS for 30 minutes, washed with PBS, and stained with a 50 μg/ml FITC-conjugated phalloidin solution (Sigma, St. Louis, MO, USA) for 40 minutes at RT. Unbound conjugated phalloidin was washed off with PBS and tissues were viewed using a Zeiss LSM 510 confocal microscope.
Fluorescent immunostaining
Frozen fresh tissue sections were blocked in blocking solution containing 5% goat serum and 0.2% Triton-X-100 at RT for 30 minutes. Primary antibodies (otoconin 90 (Oc90) [Zhao et al., 2007], otolin [Zhao et al., 2007], or keratin sulfate proteoglycan (KSPG) [Millipore, Billerica, MA, USA]) were added at a dilution of 1:200 to 1:400 and incubated at 4°C overnight. Primary antibodies were either purchased (KSPG, Millipore, Massachusetts, USA) or generated as described by Zhao et al., 2007 (Oc90 and Otolin). After 3 washes in 1xPBS, Alexa-488 or 568 (Molecular Probes, Carlsbad, CA, USA) conjugated secondary antibodies were added at a dilution of 1:600 and incubated at RT for 2 hours in the dark. Non-immune sera instead of primary antibodies were used in some sections as negative controls. Slides were mounted in Fluoromount-G and signals were viewed using a Zeiss LSM 510 confocal microscope.
To minimize measurement variation, all tissue sections were processed strictly under the same conditions (e.g., identical immunostaining procedures, identical confocal scanning parameters, and the same number of fluorescent exposures). Cross sections that covered both the striola and peri-macular region were analyzed to take into consideration possible intensity differences caused by the position and orientation of cells and sections. Results from these analyses were confirmed by semi-quantitative Western blotting or quantitative real-time RT-PCR.
Assaying for total protein content and protein concentrations
Protein concentrations were measured using a Micro-BCA (bicinchoninic acid) Protein Assay Kit (Pierce, Rockford, IL, USA) following the manufacturer’s protocol. Briefly, 50 μl of each diluted BSA standard and protein sample were added into microplate wells; 50 μl of freshly prepared working reagent (A:B:C @ 25:24:1) was added into each well and mixed gently. Duplicates or triplicates were done on each standard or sample. The reaction was incubated at 37°C for 2 hours with rocking. Absorbance was measured at 562 nm on a Quant MQX200 plate reader (Bio-Tek Instruments, Inc., Winooski, VT, USA). Sample protein concentrations were calculated according to standard curves.
Western blotting
Proteins from utricles, saccules, and otoconia were extracted in lysis buffer containing 40 mM Tris, 4% CHAPS, 8 M urea, 10 mg/ml DTT and 0.1 M EDTA. EDTA, buffer, and salts in otoconia were removed by passing through Centricon columns (Millipore, Billerica, MA, USA) after 20-fold dilution. Protein concentrations were determined using a Micro-BCA protein assay kit as described above. An aliquot of each sample containing an equal amount of total protein was mixed with 2X sample loading buffer (0.13 M Tris-HCl, 20% glycerol, 46 mg/ml SDS, 0.2 mg/ml bromophenol blue, 20 mg/ml DTT), boiled for 5 minutes, loaded onto a 4–15% gradient Tris-HEPES-SDS gel (Pierce, Rockford, IL, USA), electrophoresed at 150 V in Tris-HEPES-SDS running buffer (100 mM Tris, 100 mM HEPES, 3mM SDS) for 45 min and transferred to a PVDF membrane (Millipore) at 100V for 1 hour. The blot was washed once with TBST buffer (50 mM Tris, 150 mM NaCl, 0.1% Tween-20) and treated with 1% blocking buffer (Roche Applied Science, Indianapolis, IN, USA) for 2 hours at RT. The membrane was incubated with primary antibodies at 1:1000 dilutions for 3 hours, followed by 3 washes in TBST buffer and incubated with a specific peroxidase-conjugated secondary antibody (Sigma) at 1:10,000 dilutions for another 3 hours. After 3 washes, detection solution was added for 1 minute and the blot was exposed to a Kodak X-ray film and imaged using a Canoscan 8400F imaging station. Protein band intensities were analyzed using ImageJ software. The membrane was stripped several times in 25 mM glycine-HCl (pH 2) containing 1% (w/v) SDS before re-detection with several antibodies (separately) against different proteins.
Real-time quantitative RT-PCR (qPCR)
Total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA, USA). First-strand cDNA was synthesized from 1μg total RNA with random hexamer primers using the SuperScript III First-Strand Synthesis System (Invitrogen). TaqMan gene expression assays were purchased from Applied Biosystems (Foster City, CA, USA) and real-time quantitative PCR reactions (40 cycles) were performed with an ABI Prism 7900HT Sequence Detection System (Applied Biosystems), according to the manufacturer’s protocols. Mouse β-actin (Actb) gene was used as an endogenous control for normalization. The probe sequences were:
Oc90 – CCCTGGATAGGTGCTGTCTGTCCCA (ABI i.d. Mm01200956_m1),
otolin – AAGGAGAGAAAGGACTAAAGGGAGA (ABI i.d. Mm01222538_m1), and
β-actin – TTACTGAGCTGCGTTTTACACCCTT (Mm00607939_s1).
Statistics
VsEP waveforms obtained from 123 A/J mice were organized according to decreasing stimulus level and plotted to form a threshold response series for each animal. VsEP thresholds, peak latencies, and peak-to-peak amplitudes were determined from the plots. The first positive (P1) and negative (N1) peaks were scored for each waveform pair, and response peak latencies (P1 and N1) and peak-to-peak amplitude (P1-N1) were calculated. Peak latencies were measured in milliseconds (ms) from stimulus onset to the time of occurrence of each peak. Peak-to-peak amplitudes were calculated in microvolts (μV) by subtracting the amplitude of the negative peak from its corresponding positive peak (labeled P1-N1). The first two peaks (P1, N1) have been shown to reflect activity of the eighth nerve, whereas later peaks (e.g., P2, N2, etc., not reported here) reflect neural activity in central brainstem relays (Nazareth and Jones, 1998). Unless otherwise stated, descriptive metrics are expressed in the form of mean +/− SD (n), where SD is standard deviation and n is the sample size.
Univariate and multivariate analysis of variance (ANOVA and MANOVA, respectively), repeated measures MANOVA and ANOVA (rmMANOVA, rmANOVA) as well as non-parametric tests (SPSS for Windows, v.22, Chicago, IL) were used to evaluate the effects of age and to compare response parameters and otoconial distributions of A/J mice with our laboratory control mouse strain (C57BL/6J). The appropriate non-parametric tests were used for paired (related samples) versus independent means evaluations (e.g., the Wilcoxin Signed Rank Test for related means was indicated by “related-Wilcoxin RST”). Similar designations for other tests were used. Unless otherwise stated, statistical tests were two-tailed and evaluated in terms of summary values (e.g., means) for each animal, thus sample sizes for statistical tests generally reflected numbers of animals involved rather than individual ears or end organs. Descriptive data were also presented in terms of individual end organs represented (e.g., up to two utricles and two saccules may be available in a given animal). Generally, the least significant difference test (LSD) was used for post hoc tests, although other tests were also used as noted. Linear regression, ANOVA, MANOVA and non-parametric tests were used to characterize vestibular function in A/J mice over the ages from about 1 to 19 months. Changes in VsEP threshold, P1 and N1 latencies, and P1-N1 amplitudes were evaluated. Unless stated otherwise, values for P1 and N1 were summed to provide a single metric representing peripheral activation latency. Descriptive statistics were presented for each peak latency (P1, N1) separately. Running averages were also calculated and used to illustrate functional trends with aging. For this metric, values for individual animals were listed in chronological order according to age. The mean age and response parameter (e.g., response threshold) of the first 10 animals were calculated. Then incrementing by one, the new animal and previous 9 animals made up the next mean age and response parameter value. Incrementing by one each time and calculating mean age and response parameter for each group of 10 animals produced a series of means sliding (running) across the age axis. Running averages for VsEP thresholds, latencies and amplitudes were calculated using Microsoft Office Excel, 2010.
Results
Vestibular Function
VsEP responses in A/J mice were variable (A/J mice: n =123; ages 0.7–18.6 months, two animals had no response). Figure 2 illustrates the variation in response waveform morphology for a wide range of ages and identifies response peaks scored in the present study. The assortment of representative VsEP response pairs in figure 2 is ordered from the top with the best (relatively normal responses) to the bottom showing the worst (absent responses). Figure 3 illustrates thresholds, latencies and amplitudes for all A/J animals as a function of age. Numerical findings for A/J mice at approximately 3-month intervals are summarized in Table 1. The wide variation in A/J VsEP threshold is clearly evident in figure 3A. In individual animals, vestibular deficits ranged from mild to profound loss of function. Threshold ranged from greater than 6dB (i.e., absent responses for two animals) to −13.5 dB re:1 g/ms in two animals. This wide range of threshold values (~20 dB) in A/J animals is not seen in mouse strains known to have robust VsEP responses throughout the life span, such as the C57BL/6J strain. The mean threshold for A/J animals between the ages of 0.7 and 6 months-old was −7.75 +/− 2.61 (48) dB re: 1 g/ms. A/J thresholds were higher (MANOVA F1,84 = 22.797; P = 8.0 × 10−6) and distributed significantly differently than age-matched laboratory controls (C57BL/6J, −10.5 +/− 2.67 (38) dB re: 1 g/ms; Mann-Whitney U = −4.244, P < 0.001). Notice that the running average for A/J thresholds (figure 3A) was consistently above those of control C57BL/6J thresholds. VsEP amplitudes (P1-N1) over the same age range were somewhat smaller and distributed differently than those for C57BL/6J controls (Figure 3C and Table 1, MANOVA F1,84 = 6.127, P = 0.015; Mann-Whitney U = 2.291, P = 0.022). VsEP onset latencies were also longer and distributed differently than those for C57BL/6J controls (MANOVA F1,84 = 12.361, P = 0.001; Mann-Whitney U = −4.317, P < 0.001); although collectively, as may be appreciated from figure 3B and Table 1, the differences in mean latency were small. Thus, the most prominent vestibular collective deficits were elevated thresholds and reduced amplitudes in A/J mice. General temporal trends also can be gleaned from figure 3. Functional deficits appeared early (before 3 months: threshold: MANOVA F1,36 = 15.477, P = 3.65 × 10−4; amplitudes: MANOVA F1,36 = 4.109, P = 0.05 and latencies: MANOVA F1,36 =38.8, P = 7.34 × 10−7; compared to controls), collectively were of relatively modest magnitude and remained stable or increased somewhat at older ages. Linear regression analysis confirmed a modest aging effect on A/J VsEP responses. Thresholds increased (figure 3A, Regression F1,117 = 13.221, P = 4.13 × 10–4, R2 = 0.102; slope = 0.220 dB/month; intercept = −8.989 dB) and P1-N1 amplitudes decreased with age (figure 3C, Regression F1,119 = 33.110, P = 6.8013 × 10-8; R2 = 0.218; slope = −0.020 μV/month; intercept = 0.653 μV). The onset latency of the peripheral response also increased slightly with age (Regression F1,119 = 4.178, P = 0.043) however, the rate of increase accounted for little of the latency variation indicating age was of little practical import for latency (Figure 3B, R2 = 0.034; slope = 7.733 μs/month). Threshold increased an average of 0.22 dB per month and amplitude decreased by 0.02 microvolts per month.
Figure 2.

VsEP waveforms from A/J mice. An assortment of VsEP response pairs illustrating representative examples of relatively normal (top), poor (middle) and absent responses (bottom). The range of responses shown represent normal to profound deficits in gravity receptor function ordered from top to bottom. Severity of the functional deficit was unrelated to age. Age of each animal is shown to the left of each response pair. Mo = months.
Figure 3.

A/J VsEP response metrics as a function of age. A/J VsEP thresholds (A), latencies (B) and amplitudes (C) are contrasted with response values for control C57 mice represented as a running average (black solid line [C57 ave]). Individual values for A/J mice are shown as open circles (AJ data). The running average for A/J findings are represented as a red solid line (AJ ave). Two A/J animals had no responses at the highest stimulus level (open stars in A). Regression lines for A/J data are shown as the dotted black line coursing with the A/J running averages.
Table 1.
VsEP functional measures for the A/J strain. Data summary for ages up to 19 months-old. Threshold in dBre:1g/ms. Amplitude: P1N1 in microvolts. Latency in microseconds. Mean +/− SD (n).
| Age (months) | Threshold | P1N1 | P1 latency | N1 latency |
|---|---|---|---|---|
| 0–3 | −7.69 +/− 1.33 (16) | 0.623 +/− 0.249 (16) | 1387 +/− 63 (16) | 1679 +/− 59 (16) |
| 3–6 | −7.78 +/− 3.08 (32) | 0.589 +/− 0.176 (34) | 1418 +/− 103 (34) | 1748 +/− 110 (34) |
| 6–9 | −8.36 +/− 2.43 (28) | 0.522 +/− 0.154 (28) | 1434 +/− 102 (28) | 1762 +/− 98 (28) |
| 9–12 | −7.03 +/− 3.2 (19) | 0.355 +/− 0.152 (19) | 1422 +/− 105 (19) | 1702 +/− 109 (19) |
| 12–15 | −7.04 +/− 4.03 (13) | 0.439 +/− 0.16 (13) | 1456 +/− 78 (13) | 1736 +/− 81 (13) |
| 15–19 | −3.68 +/− 3.82 (11) | 0.330 +/− 0.087 (11) | 1461 +/− 108 (11) | 1830 +/− 121 (11) |
Structural Analysis
Light microscope images of 124 cleared individual A/J temporal bones (99 right temporal bones, 25 left temporal bones) were scored to quantify the opacity of the otoconial membrane, the area of otoconial distribution, and estimate of the percent of the utricular and saccular surfaces covered by otoconia (integer coverage scores). Images were also used to document the presence of unusually large or giant otoconia. A total of 245 sensory maculae (utricles and saccules) were evaluated.
Otoconial dysgenesis was a prominent feature in macular sensors of A/J mice. In the normal mouse (e.g., C57BL/6J), saccular and utricular maculae are completely covered by an otoconial membrane fully laden with otoconia (figure 4A, 4C & 4I) and would receive a coverage score of 4. As can be seen in figure 5A, otoconial coverage scores for A/J mice were variable and took on values between 0 (absent otoconia) and 4 (normal) across all end organs. The distributions of coverage scores for utricles and saccules are summarized in Table 2. There were no normal otoconial coverage scores for the A/J utricle. Most utricles had a coverage scored of 1 (64.5%) and about a third (32.3 %) were scored as 2. Only a few animals (n = 3) were given a score of 3. In contrast there were 37 saccules with normal otoconial scores in the A/J mice. However, the majority (69%) had lower scores of 3 (31.1%) or 2 (30.3%). A few saccules were scored as 1 (n = 9; 7.6%). No saccules were scored as 0. Otoconia were completely absent in only one case involving only the left utricle (the right side was not measured). The saccule was normal in this case. Coverage scores for the left utricle versus right utricle and the left saccule versus right saccule were not significantly different (rmMANOVA F4,13 = 2.181, P = 0.128; MANOVA F2,116 = 1.42, P = 0.246). The distributions of the utricle and saccule coverage scores were significantly different (related-Wilcoxin SRT = 8.046, P < 0.001) and this was reflected by a significantly higher mean score for the saccule (2.85 +/− 0.93) compared to the utricle (1.35 +/− 0.53) as shown in figure 5A (rmMANOVA F3,91 = 74.55, P = 1.99 × 10−24). Table 3 summarizes mean coverage scores in terms of individual end organs harvested (utricle versus saccule). Mean coverage scores were well below 4 for both end organs indicating reduced otoconial coverage for both macular sensors. Moreover, the significantly lower mean utricular coverage score suggested that otoconial dysgenesis was more severe for utricular epithelia. These characteristics can also be seen in the general summary data for individual end organs in Table 3. Mean coverage score across all A/J animals for the combined end organ values (left and right utricle and saccule) was 2.06 +/− 0.6.
Figure 4.

SEM micrographs of utricles and saccules from C57 and A/J mice. Note co-existence of giant and normal otoconia in the A/J saccule. Otoconial dysgenesis was present in neonates (B, F, D, H) as well as adults (J, K). Overall, A/J otoconia (B, D, J & K) are less numerous than C57 controls (A, C), and the abnormality is more severe in the utricle. The saccular otoconia can appear normal but often evidence major defects (B, F). A/J utricles often have a few, large or giant crystals (D (arrows), J and K). The giant otoconia appear to be composed of numerous fused crystals under higher magnification (F, H vs. E, G). The middle panels (E, F, G, H) are higher magnification of the lower regions in the respective upper panels. Calibration bars are 50 microns for A, B, C & D;; 5 microns for E, F, G & H, 300 microns for I and 200 microns for J & K. Red arrows in lower right of C & D indicate directions for anterior and lateral.
Figure 5.

A/J coverage (A) and opacity (B) scores. Values for individual utricles (filled circles) and saccules (open triangles) demonstrate the distributions for the data. Summary mean and standard deviations are represented for the two end organs to the right by large symbols with error bars.
Table 2.
Distribution of otoconia coverage scores for A/J strain. 4 = normal. 3 = 50% > to 75%. 2 = 25% > to 50%. 1 = 0% > to 25%. 0 = no otoconia.
| Utricle (n = 124) | Saccule (n = 119) | ||||
|---|---|---|---|---|---|
| Score | n | % | Score | n | % |
| 4 | 0 | 0.0 | 4 | 37 | 31.1 |
| 3 | 3 | 2.4 | 3 | 37 | 31.1 |
| 2 | 40 | 32.3 | 2 | 36 | 30.2 |
| 1 | 80 | 64.5 | 1 | 9 | 7.6 |
| 0 | 1 | 0.8 | 0 | 0 | 0.0 |
Table 3.
Summary of coverage and opacity measures for A/J otoconia. Measures for each ear independently as well as means for both ears are represented. Data include all end organs given as mean +/− SD (n), where n= number of end organs sampled. All ages, 1 to 19 months. Limits for opacity are minimum = 0 and maximum = 255. Mean opacity for the 7 normal C57 mice of the present study was: 139.2 +/− 6.8 (7). Note that values are slightly different here than those represented in the text. Those in the text reflect means and variance calculated across animals rather than individual end organs.
| Right Ear | Left Ear | Average | |
|---|---|---|---|
| Utricle | |||
| Coverage | 1.33 +/− 0.53 (99) | 1.48 +/− 0.59 (25) | 1.36 +/− 0.55 (124) |
| Opacity | 58.02 +/− 16.85 (96) | 59.93 +/− 15.81 (23) | 58.39 +/− 16.61 (119) |
| Saccule | |||
| Coverage | 2.79 +/− 0.94 (95) | 3.13 +/− 0.95 (24) | 2.86 +/− 0.95 (119) |
| Opacity | 97.83 +/− 33.62 (94) | 117.81 +/− 38.7 (21) | 101.48 +/− 35.28 (115) |
| All End Organs (Utricle & Saccule) | |||
| Coverage | 2.05 +/− 1.05 (194) | 2.29 +/− 1.14 (49) | 2.09 +/− 1.07 (243) |
| Opacity | 77.71 +/− 33.12 (190) | 87.55 +/− 40.98 (44) | 79.56 +/− 34.86 (234) |
Similar results were obtained with the measurement of the OM opacity in A/J mice (figure 5B). No differences in opacity were found with comparisons between left and right ears (rmMANOVA F4,13 = 2.181, P = 0.128). Opacity levels for the saccule (100.8 +/− 34.3) were significantly higher than the utricle (58.5 +/− 16.4, rmMANOVA F3,91 = 74.55, P = 1.99 × 10−24) and thus were also consistent with a more severe otoconial loss in utricular versus saccular sensors. The distribution of utricle and saccule opacity scores was also significantly different (related-Wilcoxin SRT = 8.311, P < 0.001). The A/J opacity data are illustrated in figure 5B and summarized in Table 3 for individual end organs. A/J opacity levels were significantly lower than those for C57BL/6J control animals (A/J: 80.99 +/− 23.2; C57: 139.2 +/− 6.8; ANOVA F1,107 = 43.5, P = 1.64 × 10−9).
As noted, A/J OM coverage scores varied widely especially for saccular elements. Although there was no overall difference in the mean or distribution of values for left and right end organs, the question arises as to whether in a given animal the coverage in one ear was predictive of scores in the other ear? In the small number of A/J animals where both ears were examined (n = 20), combined utricle and saccule coverage scores for one ear failed to show a positive relationship with scores for the other ear (Regression F1,14 = 2.591, P = 0.130; R = −0.395 and R2 = 0.156). On the other hand, left and right coverage scores for the saccule alone were well correlated and provided a positive predictor for the other ear (Table 4, F1,18 = 19.653, P = 3.21 × 10−4, R = 0.722, R2 = 0.522). Left and right scores for the utricle alone, although consistently very low, were not correlated (Regression F1,18 = 0.018, P = 0.895, R = 0.032, R2 = 0.001). This finding is not surprising in the case of the A/J utricle since there were essentially only two possible scores in these animals (1 or 2, only one animal had a score of 0) and the difference in coverage between any two of them was only 25% and difficult to distinguish. The average opacity for a given ear (average of both end organs: utricle and saccule for each ear) was well correlated with the average opacity for the opposite ear (Regression F1,17 = 16.856, P = 0.001, R = 0.706, R2 = 0.498, slope = 0.812, intercept = 9.352). This correlation was largely due to the strong correlation between left and right saccules that was evident over a wide range of saccular opacity values (~30 to 170). Similar to the low utricular coverage scores noted above for A/J, the low utricular opacities (Table 3) showed a relatively small range of values (~28 to 105) and a poor correlation between ears (R = 0.161, R2 = 0.026, P = 0.51). These and other findings are summarized in Table 4. The findings of similar levels of dysgenesis and correlated otoconial metrics between the two ears of A/J mice suggest a pathological process that is bilaterally symmetrical with similar magnitudes for the respective end organs of the two ears.
Table 4.
Summary correlations for A/J left and right epithelia. Pairwise sampling of data was made in animals where both left and right ears were measured. Left versus right correlations were evaluated for utricle, saccule and summed end organs.
| Coverage Score | Opacity | |||||
|---|---|---|---|---|---|---|
| Epithelium | R | R2 | Significance | R | R2 | Significance |
| Utricle | 0.032 | 0.001 | P = 0.895 | 0.161 | 0.026 | P = 0.510 |
| Saccule | 0.722 | 0.522 | P = 3.21 × 10−4 | 0.81 | 0.657 | P = 4.5 × 10−5 |
| Utricle + Saccule | −0.395 | 0.156 | P = 0.130 | 0.706 | 0.498 | P = 0.001 |
There were notable abnormalities in otoconial size in A/J mice. In about 22% of A/J inner ears (n = 27) the otoconial membranes were laden with unusually large or giant otoconia, which could often be identified using light microscopy in cleared material. Rather than a diffuse thick bed of hundreds of small otoconia covering the epithelium, several giant otoconia of varying sizes laid on the surface of the OM as illustrated by SEM micrographs of figure 4B, 4D (neonates, P4) and figure 4J, 4K (adults). Using light microscopy, large and giant otoconia were identified only in the utricle. However, unusually large or giant otoconia were also seen in the saccule when viewed under the higher resolution of the SEM (see below, figure 4B & 4F).
SEM images and/or high magnification light microscopy examination of utricles and saccules from 70 A/J mice at various ages (E18.5, P0, P4, P11, P22, 1.5M, 3M, 5M, 7M, 10M. M = months) consistently showed the presence of several giant otoconia in the utricle (figures 4D, 4H, 4J and 4K) and to a lesser extent multiple large to giant otoconia in the saccule that often co-existed with normal crystals (Figure 4B & 4F). Generally, A/J otoconia were less numerous than in C57BL/6J controls, and the giant crystals were loosely anchored. The giant otoconia appeared to be composed of numerous fused crystals (crystallites, figures 4F, 4H, 6A, 6B). Large otoconia in A/J mice actually had fewer crystallites in each otoconium compared to control, where each control crystal evidenced up to 20 crystallites arranged in a rosette-like pattern (figures 4F, 4H, 4K, 6A, 6B). Compared to controls, A/J crystallites were not so cylindrical and hexagonal in shape when viewed in cross-section. In addition, the ratio of organic matrix to inorganic crystallites appeared to be greater in the A/J otoconium (asterisk in figure 6B). There were no other apparent gross structural defects in the vestibule of the A/J mice (e.g., Figure 6E, 6F).
Figure 6.

Abnormal morphology of crystallites in the A/J otoconium. (A, B) The large A/J otoconia actually have fewer inorganic crystallites in each crystal (arrows in B vs. A). Compared to controls, A/J crystallites are flat and not so hexagonal cylinder-like in the cross-sectional view, and the relative portion of the organic matrix is increased in the A/J otoconium (asterisk in B). (C, D) Toluidine Blue-staining shows overall normal epithelial cellular structures but large to giant otoconia. (E, F) TEM shows overall normal ultrastructures of hair and supporting cells at this stage (P4). HC, hair cells; O, Otoconia; SC, supporting cells.
Given no significant differences for otoconial metrics for the left and right ears (reported above), scores were averaged between sides and evaluated for age effects. There was no significant relationship between mean otoconial coverage scores and age for the utricle (Regression F1,102 = 0.350, P = 0.555, R2 =0.003) or the saccule (Regression F1,97 = 3.193, P = 0.077, R2 = 0.032). Mean OM opacity for each A/J animal is plotted as a function of age in figure 5B. There was virtually no effect of age on utricle (F1,98 = 9.964, P = 0.002, R2 = 0.092, slope =1.083 opacity units/month, intercept = 50.302) or saccule (F1,95 = 7.952, P = 0.006, R2 = 0.077, slope = 2.078 opacity units/month, intercept = 85.073) opacity. Although, the regressions indicate significance, the effect sizes and estimated slopes for the two end organs are negligible and this can be appreciated from figure 5B. Thus, otoconial dysgenesis in A/J mice does not appear to be linked to a major aging process. Abnormal crystals were present in neonates as well as mature animals (figure 4).
Ultrastructural analysis of the utricle and saccule showed normal cellular structures generally at postnatal stages (figure 6C, 6D, 6E & 6F). However, in some cases A/J stereocilia had a disheveled appearance (figure 7B, 7D, 7F & 7G), and some had extra numbers of tall stereocilia but lacked the intermediate rows (lacked the typical staircase formation). The abnormal bundle organization and bundle loss were much more frequent and more severe in the A/J cochlea, especially in outer hair cells (figure 8) whose bundles were often not arranged in a “V” shape and did not have the shorter rows (figure 8B & 8F). Cochlear stereociliary bundle loss was observed at less than 1 month of age, and was much more severe than in older C57BL/6J mice. For example, A/J bundle loss was worse in 1.5 month-old mice compared to 3 month-old C57BL/6J mice (figure 8A & 8B) and 5 month-old A/J worse than 7 month-old C57BL/6J mice (figure 8G & 8H). Inner hair cell bundle loss and disorganization (figure 8C & 8D) were less severe than that of outer hair cell bundles, and there was only occasional bundle loss in the vestibule at 7 months (data not shown). Since bundle disorganization in the cochlea was observed at neonatal stages, it is possibly a precursor to hair cell loss reported in the A/J cochlea by Zheng and coworkers (Zheng et al., 2009).
Figure 7.

Disheveled and disorganized stereocilia in the A/J vestibule (the utricle is shown). (A, B) Phalloidin-staining and (C–G) SEM show that some A/J bundles had extra numbers of tall stereocilia but lacked the intermediate rows (lacked the typical staircase formation, exemplified by the arrow in B, also seen in D & F).
Figure 8.

SEM of the apical epithelial surface of the middle turn of the cochlea. Disheveled, disorganized and degenerating stereocilia in the A/J cochlea. The bundle abnormality is more frequent and more severe in the cochlea than the vestibule of A/J mice, especially of outer hair cells whose bundles are often not arranged in a “V” shape and did not have the shorter rows (B, F). Bundle loss is seen as early as less than 1 month of age in the cochlea, and is much more severe than older C57 mice. e.g. Bundle loss is worse in 1.5M A/J than 3M C57 (A, B), or worse in 5M A/J than 7M C57 (G, H). Inner hair bundle loss and disorganization (C, D) are less severe than that of outer bundles.
To test the hypothesis that the abnormal matrix-crystallite arrangement in A/J otoconia may have arisen from altered incorporation of otoconial proteins, we performed semi-quantitative and quantitative expression studies of important otoconins including Oc90, otolin and keratin sulfate proteoglycan (KSPG, figures 9 & 10). There was indeed an increase in the epithelial expression and otoconial deposition of Oc90 and otolin compared to that in C57. The increase in Oc90 incorporation in A/J otoconia was most evident on the Western blot (figure 10A). Two groups of otoconial protein extracts were compared between C57 and A/J. In one group, an equal amount of total protein was loaded for the C57 and A/J comparison (data not shown). In the second group, total otoconial extract from one A/J and one C57 mouse was loaded and compared in figure 10A. Both groups showed a higher level of Oc90 in A/J otoconia.
Figure 9.

Deposition of otoconial proteins in A/J and C57 otoconia as detected by fluorescent immunostaining (the saccule is shown). (A, B) Abundant presence of Oc90 protein in the giant crystals of A/J mice. The open arrow shows large to giant otoconia attached to the squamous epithelium of the A/J mice. (C, D) At a dilution of 1:200, the otolin antibody stains weakly the C57 sensory epithelium and otoconia (C), whereas A/J crystals have intense fluorescent signals (D and inset). The giant crystal pointed by the arrow is magnified in the inset. (E, F) KSPG antibody also shows an intense staining pattern in both A/J and C57 mice.
Figure 10.
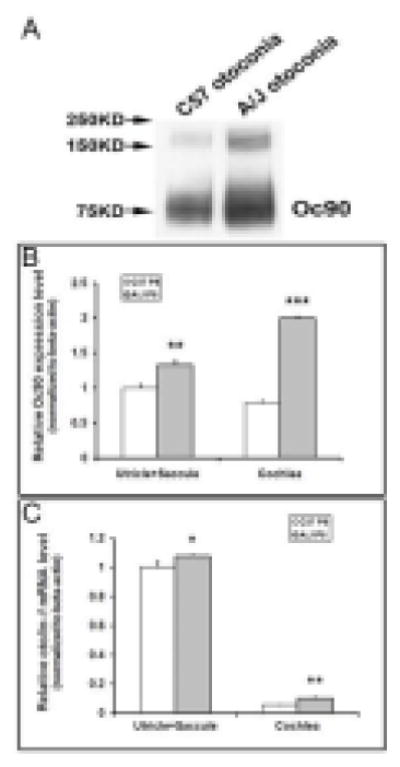
Otoconial deposition and epithelial expression of otoconial proteins in A/J vs. C57 inner ears as detected by Western blotting (A) and real-time quantitative RT-PCR (B, C). In (A), total otoconia extract from one C57 and one A/J mouse at age P4 was loaded in each lane (labeled as “O”). This increase of Oc90 in A/J otoconia is confirmed by loading an equal amount of total protein in each lane (not shown). (B, C) Real-time PCR shows a significant increase in both Oc90 and Otol1 mRNA in the A/J inner ear epithelia as compared to C57 tissues at age P8 (*, P < 0.05; **, P < 0.01; ***, P < 0.001, n=3).
The increase in otolin incorporation was also evident in immunostained otoconia sections (figure 9D & 10C). At 1:200 dilution of the primary antibody against a C-terminal unique region of otolin (Zhao et al., 2007), control crystals and epithelium were only weakly stained, whereas A/J crystals had intense fluorescent signals (figure 9D and inset). Because Western blotting of otolin showed several bands due to possible oligomerization, it was difficult to compare the protein quantity on the blot (data not shown). Nevertheless, real-time quantitative RT-PCR showed a significant increase in Otol1, as well as Oc90, mRNA in the A/J inner ear epithelia as compared to C57 tissues at age P8 (figure 10B, 10C).
The pattern of otoconin deposition associated with the otoconial membrane was evaluated using fluorescent immunostaining. Figure 9 illustrates such patterns in the saccule for C57 and A/J mice. Oc90, otolin and KSPG were all well labeled in A/J maculae (Figure 9B, 9D & 9F) suggesting the presence of abundant protein in each case. (Figure 9F, 9E). Staining patterns were qualitatively different in C57 versus A/J material in that for C57 maculae the labeling of the otoconial layer appeared smooth and continuous with a relatively uniform intensity throughout, thus reflecting the comparatively uniform, compact distribution of abundant small normal otoconia (figure 9A, 9C & 9E). In contrast, the labeling pattern in A/J material appears rough, discontinuous with non-uniform intensity and abundant labeling associated with irregularly sized and distributed otoconia including abnormally large to giant crystals (Figure 9B, Oc90 open arrow and 9D, solid arrow and inset). There were numerous, normal to giant otoconia on the squamous epithelium of the A/J utricle and saccule (denoted by open arrow in Figure 9B, as well as unmarked in 9D) that appeared to be formed there because of the tight adherence to the epithelium.
In summary, all three otoconins examined (Oc90, otolin & KSPG) were present in otoconial assemblies of A/J mice. They were present at higher levels compared to normal animals and the deposition of each constituent reflected a highly disordered otoconial distribution that included non-uniform irregular assemblies of normal and abnormal otoconia, many of which assembled as excessively large or giant crystals.
Evaluation of Consomic Mice
An initial attempt was made to evaluate whether genes having known variants that produce otoconial dysgenesis in other strains were also responsible for the otoconial defects reported here for A/J mice. To this end, we evaluated vestibular function and morphology in the C57BL/6J-Chr 17 A/J/NaJ consomic mouse, which has a C57BL/6J genome where chromosome 17 of the A/J mouse (Chr 17A/J) is substituted for the natural Chr 17C57. Chr 17 contains two genes, Nox3 (NADPH oxidase 3, Bergstrom et al., 1998; Paffenholz et al., 2004) and Noxo1 (Nox organizer 1, Kiss et al., 2006), that when null, lead to complete otoconial agenesis. Thus, we hypothesized that Chr 17 contains an unknown gene variant (or variants) responsible for the A/J vestibular phenotype. If true, then one expects that substitution of Chr 17A/J for Chr 17C57 will produce otoconia dysgenesis and vestibular deficits in the otherwise normal C57BL/6J genome.
Both A/J and C57 strains share the same gene mutation Cdh23ahl on Chr 10 (Johnson et al., 1997; 2000; Noben-Trauth et al., 2003) and exhibit age-related hearing loss (AHL). In addition, C57 normally carries the ahl3 variant allele, which reportedly enhances the susceptibility to AHL (Ahl3C57BL/6J, locus available only, Nemoto et al., 2004, Chr 17) and the A/J strain manifests the “susceptibility allele” ahl4 (Csahl4-A/J, citrate synthase, Chr 10, Zheng et al., 2009; Johnson et al., 2012), which reportedly exacerbates AHL. The consomic mouse (C57BL/6J-Chr 17A/J/NaJ), hereafter referred to as C17 mouse, therefore is thought to lack the ahl3 variant normally found in C57BL/6J and also clearly lacks the ahl4 allele that normally is found in A/J mice on Chr 10. Therefore, the differences in AHL one would expect in the consomic mouse are those effects conferred by eliminating the ahl3 allele.
The C57BL/6J strain normally retains robust vestibular function over the entire life span (up to 24 months, Mock 2008). Thus, the combined presence of ahl and ahl3 in C57 normally (as noted above), does not lead to age-related vestibular dysfunction. As noted, the hypothesis was that gene variants within Chr 17A/J contribute to the A/J vestibular phenotype. Since normal C57 mice show no vestibular deficits, the appearance of vestibular dysfunction in the consomic mouse could only be due to the presence of Chr 17A/J.
The results showed that C57 mice consomic for Chr 17A/J had altered VsEP responses. Table 5 summarizes mean values for consomic C17, A/J, and C57 strains of animals at ages between 1 and 13 months-old. Mean values for AJ VsEP thresholds, amplitudes and latencies shown in Table 5 were all significantly different than those for C57 (P < 0.001). Distributions and means for VsEP thresholds were significantly elevated in consomic C17 mice compared to age-matched normal C57 controls (MANOVA, F2,223 = 15.88, P = 3.56 × 10−7, Post Hoc LSD P = 1.85 × 10−3; Mann-Whitney U = −2.382, P = 0.017). P1 latencies were also significantly different (MANOVA F2,223 = 19.1, P = 2.2 × 10−8, Post Hoc LSD P = 2.48 × 10−7; Mann-Whitney U = −4.902, P < 0.001). Response amplitudes were the same for control C57 and C17 consomic mice.
Table 5.
Comparison of VsEP response metrics for A/J, C17 consomic and C57 mouse strains. Only P1 latency was available for C17 consomic animals as metric for peripheral activation times.
| Strain | Threshold | P1N1 | P1latency |
|---|---|---|---|
| A/J | −7.86 +/− 2.66 (101) ### | 0.53 +/− 0.19 (103) ### | 1422.04 +/− 97.04 (103) |
| C17 | −8.83 +/− 4 (52)* | 0.67 +/− 0.3 (50) | 1437.57 +/− 121.68 (49)** |
| C57 | −10.5 +/− 2.79 (75) | 0.71 +/− 0.25 (77) | 1322.34 +/− 135.32 (77) |
Age-matched animals 1 to 13 months-old. Mean +/− SD (n). Comparison of C17 to control C57:
P < 0.02.
P < 0.001.
Comparison of C17 to A/J:
P = 0.003. All metrics shown here for AJ were significantly different than those for C57 (P < 0.001).
Interestingly, although the mean thresholds were the same for A/J and C17 mice, threshold distributions were significantly different (Post Hoc Mann-Whitney U = −3.013, P = 0.003). Moreover, P1-N1 mean amplitudes for C17 were significantly larger, and their distribution different, than those for A/J mice (MANOVA F2,223 = 14.5, P =1.0 × 10−6, Post Hoc LSD P = 0.001; Post Hoc Mann-Whitney U = 2.913, P = 0.003). Latencies for C17 and A/J mice were the same. Thus, although vestibular function in C17 consomic mice was significantly degraded compared to C57 laboratory controls, it was not equivalent to the A/J functional deficit.
An otoconial deficit was present in C17 consomic animals where over 80% of the animals had an appreciable otoconial loss of 25% or more based on coverage scores. Coverage scores were obtained in 46 C17 mice, where both ears were examined in 43 and one ear in 3 animals. There was no difference in coverage scores between ears (Saccule: related -Wilcoxin SRT = −1.606, P = 0.108; Utricle: related-Wilcoxin SRT = 0.626, P = 0.532; rmMANOVA F2,40 = 1.374, P = 0.265; ANOVA P > 0.100). Mean combined otoconial score for utricle and saccule across all C17 animals was 2.99 +/− 0.30 (46), a value that is three standard deviations below the normal value of 4.0 (T test: t45 = −22.6, P = 3.05 × 10−26, 2-tailed). Summary findings for individual end organs in C17 mice are shown in Table 6. Loss of otoconial coverage was more pronounced in the utricle (mean 2.62 +/− 0.45 (46)) compared to saccule (mean: 3.38 +/− 0.42 (46)) in C17 mice (rmMANOVA F3,43 = 52.404, P = 2.06 × 10−14; Post Hoc ANOVA = 65.2, P = 2.66 × 10−10; related-Wilcoxin SRT = 5.179 xx, P < 0.001). The utricle was normal (score of 4) in only three (3.3%) of the utricles examined, whereas 35 (39.8%) of the saccules were normal. All control C57 mice (n = 7) had full OM coverage and received coverage scores of 4. In every case, the mean combined coverage score for C17 mice (average of left, right, saccule & utricle) was below 4. On the other hand, C17 mice had higher mean coverage scores and a different distribution than the age-matched A/J animals (A/J: 2.06 +/− 0.6; C17: 2.99 +/− 0.30; ANOVA, F1,139 = 107.5, P = 5.16 × 10−19; Mann-Whitney U = 8.324, P < 0.001).
Table 6.
Summary of otoconial measurements in consomic C17 mice ages 1 to 12.7 months-old. Values are calculated across individual end organs rather than across animals. Mean +/− SD (n). n= number of end organs sampled.
| C17 | Right Ear | Left Ear | Average |
|---|---|---|---|
| End Organ: | |||
| Utricle | |||
| Coverage | 2.60 +/− 0.58 (45) | 2.68 +/− 0.56 (44) | 2.64 +/− 0.57 (89) |
| Opacity | 152.45 +/− 9.96 (43) | 152.21 +/− 11.46 (40) | 152.33 +/− 10.65 (83) |
| Saccule | |||
| Coverage | 3.44 +/− 0.55 (45) | 3.33 +/− 0.47 (43) | 3.39 +/− 0.51 (88) |
| Opacity | 153.17 +/− 14.41 (44) | 151.03 +/− 15.43 (41) | 152.14 +/− 14.86 (85) |
| All End Organs (Utricle & Saccule) | |||
| Coverage | 3.02 +/− 0.7 (90) | 3.00 +/− 0.61 (87) | 3.01 +/− 0.66 (177) |
| Opacity | 152.81 +/− 12.35 (87) | 151.61 +/− 13.54 (81) | 152.23 +/− 12.91 (168) |
Opacity scores for the utricle versus the saccule were not significantly different in C17 mice (rmMANOVA: F3,43 =52.4; ANOVA, F1,45 = 0.014, P = 0.967) unlike those in A/J mice. The opacity scores for C17 were more comparable to and slightly higher than those obtained in control C57 mice (C17: 151.9 +/−9.34; C57: 139.2 +/− 6.8; rmANOVA F1,50 = 11.95, P = 0.001). This contrasts with the very low opacity levels found in A/J mice that were significantly lower than both C17 and C57 mice (A/J vs C17: ANOVA: F1,145 = 392.3, P = 4.4 × 10−43; A/J vs C57 is reported above).
We see from above that despite a clear degradation of vestibular sensitivity and abnormal distribution of otoconia coverage scores, the phenotype of the C17 mice did not duplicate the A/J phenotype precisely. Thresholds, although higher than controls, were not distributed as high as A/J mice and the otoconial deficit was not as severe as in A/J (Mann-Whitney U = −3.013, P = 0.003; C17: −8.83 +/− 4.0 (52); range: maximum = +4.5 dB, minimum = −13.5; A/J: −7.88 +/− 2.66 (102); maximum > +6dB, minimum = −13.5 dB). Moreover, there was no evidence of giant otoconia in the C17 mouse. Thus, A/J Chr17 alone does not appear to account fully for all otoconial and neurosensory components of the A/J phenotype. Clearly, gene variants on A/J Chr17 do contribute to the A/J phenotype; however, the evidence suggests further that gene variants located on other chromosomes may also contribute.
Identifying other candidate chromosomes
To explore the possibility of other gene variants contributing to the A/J phenotype, we established an N2 backcross between the vestibular dysfunction-sensitive A/J strain and the vestibular dysfunction-resistant C57BL/6J. Briefly, A/J females were bred to C57BL/6J males to create A/J X C57BL/6J F1 hybrid progeny. Among the F1 hybrid mice, females were backcrossed to A/J males to generate approximately 400 N2 backcross progeny. According to a convention that lists females first, the cross can be described as [A/J X C57BL/6J] X A/J or abbreviated as [A X B] X A (where A=A/J and B=C57BL/6J). N2 backcross mice of both sexes were subjected to VsEP testing and scored for the following parameters: VsEP threshold, P1-N1 amplitude, P1 latency, saccular otoconial anatomy and utricular otoconial anatomy. Otoconia were scored by two methods: semi-quantitatively, from 0 to 4 representing a spectrum of dysmorphology from unaffected to severely affected phenotypes; and qualitatively, as 0 or 1 representing a binary distinction between unaffected and affected phenotypes. Each N2 mouse was also scored for A/J or C57BL/6J genotype at 150 single nucleotide polymorphisms (SNPs) spaced at 10 cM intervals across the genome. Associations were assessed using R/qtl software (version 1.23–16) (Broman et al., 2003) at The Jackson Laboratory’s Computational Sciences/Statistics and Analysis group.
The strongest and most interesting association involved P1 latency. P1 latency among 250 backcross mice ranged from 0.9 to 1.6 msec and showed a normal bell-shaped distribution. The main effect of sex was determined to be significant (ANOVA: F1,1 = 5.13, P < 0.0243). Further, three QTL mapping models were fit to the P1 latency data: Model 1, No Covariates (Y ~ Q); Model 2, Additive Covariate (Y ~ Q + Sex); and Model 3, Full Model (Y ~ Q + Sex + Q × Sex). For each of the three models, 1000 permutations were used to obtain LOD significance thresholds for P < 0.01, P < 0.05, P < 0.10 and P < 0.63 respectively (Doerge and Churchill, 1996). The X Chr was permuted separately with slightly different LOD significance thresholds. From these analyses, the full model identified a strong QTL (with QTL*Sex interaction) on proximal Chr 9 (LOD = 4.577, P < 9.08 × 10-7) and with suggestive QTLs on Chr 1 and Chr 4. A genome wide two-dimensional scan identified a significant pair of epistatic loci on proximal Chr 9 (the same locus) and Chr X (p < 0.05). Multiple regression analyses substantiated the epistatic interaction between the loci on Chr 9 and Chr X and further implicated the loci on Chr 1 and Chr 4. The Chr 9 QTL interaction with sex shows a shortening of P1 latency in males with AB genotype at the Chr 9 QTL. Reciprocally, and perhaps relatedly, the Chr 9 QTL interaction with the locus on Chr X shows a prolongation of P1 latency among females with both the AB genotype at the QTL on Chr 9 and the AA genotype on the interacting locus on Chr X. The localization of the Chr 9 QTL (defined here as the union of QTL Confidence Intervals defined by 1.5 LOD drop off and Bayes credible interval approaches) identified a 95% confidence interval for the Chr 9 QTL as the region from 0–30 cM (approximately 0–60 Mbp). In summary, QTL analysis revealed a highly significant association between VsEP P1 latency and Chr 9 and a significant sex by Chr 9 interaction.
Discussion
Our results show that the A/J inbred mouse strain has a vestibular phenotype consisting of vestibular sensory loss accompanied by otoconial dysgenesis and limited structural changes in sensory hair cells. There was also evidence of a limited age-related vestibular impairment. We have characterized the phenotype of A/J in some detail.
VsEP waveforms and thresholds varied widely and responses were absent in two animals. Vestibular functional loss ranged from mild to profound. Such wide variation is not characteristic of normal vestibular function in the control mouse (e.g., C57BL/6J). On average, macular sensors in A/J mice were less sensitive to vestibular stimulation at all ages and exhibited lower amplitudes and prolonged latencies compared to normal age-matched C57 mice. Varying amplitudes, elevated thresholds and/or absent VsEP responses have previously been shown to be associated with otoconial dysgenesis and/or agenesis (Jones et al., 1999, 2004) and the findings here for A/J mice are consistent with such observations.
There was a limited aging component to macular functional changes in A/J mice, however this effect accounted for a small proportion of the variation in threshold and latencies. Decreases in amplitudes in A/J mice were more closely linked to age (R2 = 0.211). In general, the influence of age (effect size) was small in A/J compared to C57 animals presumably due to a much larger influence of age-independent otoconia dysgenesis and perhaps other processes associated with the A/J phenotype.
The findings of elevated VsEP thresholds and prolonged latencies in Chr 17A/J consomic mice compared to C57 controls demonstrates that Chr 17 harbors one or more genetic variants that likely contribute substantially to the A/J phenotype. The fact that some VsEP characteristics were significantly different in A/J versus C17 mice suggests that there may be other gene variants located on other chromosomes that contribute to the A/J phenotype.
In support of this idea, QTL analysis evaluating VsEP P1 latency indicated a strong linkage to Chr 9 with suggestive associations for Chr 1 and Chr 4 and additional epistatic influences by Chr X. These observations support the hypothesis that the A/J phenotype derives in part from gene variation(s) on Chr 17 (based on C17 mice) with potential modifying influences located on Chrs 1, 4, 9 and X. These chromosomes will be targets of future investigation.
It is of considerable interest that a genetic basis for human benign positional paroxysmal vertigo (BPPV) has been recently identified (Gizzi et al., 2014). BPPV is thought to result, in many cases, from cupulolithiasis (i.e., an abnormal attachment of otoconial crystals to cupular structures). The ectopic otoconia are thought to be free otoconial crystals that are dislodged from degenerating utricles. Gizzi and colleagues further identified a gene locus for the BPPV trait on human chromosome 15 (LOD score of ~2.8). The homologous locus for the mouse is found on chromosome 9, which is identified here with a link to otoconial dysgenesis. This raises the possibility that a gene variant on chromosome 9 in the A/J mouse may be related to the locus associated with BPPV on human chromosome 15.
There was little evidence of extensive morphological abnormalities in the vestibular sensory epithelium itself. Analysis of the utricle and saccule material in A/J provided some evidence of abnormal formation in vestibular stereociliary bundles suggesting that functional deficits may involve more than just otoconial dysgenesis. However, alone such observations do not appear to explain the vestibular dysfunction observed. Nonetheless, there may be a subtle underlying process associated with the hair cell bundle abnormalities, one that contributes to vestibular functional deficits in A/J mice. Perhaps these processes contribute to the underlying aging effects on VsEP parameters. Presently it is not clear whether the stereocilia and otoconia abnormalities are molecularly/mechanistically linked. In zebrafish, cilia are critical to normal otolith formation and anchoring, as deficiency in any one of 16 cilia-related genes, including dyneins, centrosomal proteins, and tubulin-modifying enzymes, causes defective otoliths (Colantonio et al., 2009; Lee et al., 2012). However, the observed otolith defects could have been caused by other defective cellular functions as well, since many of these genes have overlapping roles in cilium biogenesis, vesicle (protein) transport, and cell-cell signaling.
Our primary focus was to characterize vestibular function. However, there arose an opportunity to provide new insight regarding cochlear structure for the A/J mouse strain. Ultrastructural data presented here confirmed accelerated structural damage in A/J mice compared to C57 controls as reported by Zheng et al., (2009) and Johnson et al., (2012). The disorganized A/J hair bundles somewhat resemble those of other mouse mutants carrying null mutations in the cadherin 23 (Cdh23), myosin VIIa and myosin VI genes (Boeda et al., 2002; Holme and Steel 2002). As noted above, C57 and A/J both carry a G to A transition at nucleotide position 753 of the Cdh23 gene, known as the ahl allele (Cdh23ahl) (Di Palma et al., 2001; Johnson et al., 1997; 2000; 2006; Noben-Trauth et al., 2003). This hypomorphic allele causes in-frame skipping of exon 7 and reduces message stability. Besides being a tip-link component, Cdh23 also interacts with the above myosins, and these features become disrupted in other Cdh23 mouse mutants.
Otoconial defects in A/J mice were striking. The appearance of giant otoconia, altered crystalline structures, reduced otoconial coverage and low OM opacity suggest the primary bases for the vestibular functional deficits observed. The general features of the otoconial dysgenesis point to a highly variable yet bilaterally symmetrical process that affects the utricle to a much greater extent than the saccule. Otoconial dysgenesis appeared to be independent of age in as much as metrics used to quantify otoconial status evidence no systematic trend with age and large or giant crystals were identified in young and old mice alike.
It is not clear at the present time whether altered ionic compositions exist in A/J endolymph (e.g., for Ca2+ and CO32−), however such ionic changes could have caused the abnormal CaCO3 crystallite morphology noted here in A/J mice. On the other hand, altered incorporation of organic components of otoconia alone (e.g., Oc90 and otolin) could also cause such problems inasmuch as organic matrix has been shown to regulate the growth, morphology and stability of each otoconium (Sollner et al., 2003; Murayama et al., 2005; Zhao et al., 2007; Kang et al., 2008).
Clearly, all three major organic matrix constituents examined here (Oc90, otolin and KSPG) were present over saccular and utricular sensory patches in A/J mice. This finding tends to rule out the possibility that low or absent levels of these proteins (otoconins) led to the abnormal matrix-crystalline arrangement in A/J otoconia. The significance of having higher levels of these protein constituents in otoconial assemblies in terms of mechanisms of formation is not clear at this juncture. However, it is clear that the incorporation of these proteins and the distribution of their deposition is abnormal in A/J mice.
The A/J otoconial deficit is clearly not a total agenesis like that found in Nox3, p22phox and Noxo1 null mutations (Bergstrom et al., 1998; Paffenholtz et al., 2004; Kiss et al., 2006; Nakano et al., 2008). Rather, the extent of dysgenesis is highly variable producing a corresponding variable functional deficit. One feature, abnormally large or giant otoconia, has been reported in a number of mouse models. For example aberrant crystals have been described: 1) following targeted deletion of Oc90 (Zhao et al., 2007); 2) in the absence of pendrin (Nakaya et al., 2007; Li et al., 2013); 3) associated with the human Pierre Robin Triad (Gruen et al., 2005); and 4) in otopetrin1 (Otop1) null mice (Besson et al., 2005; Hurle et al., 2003; Ornitz et al., 1998). In these cases, crystallization is not completely eliminated, but rather mineralization and assembly of otoconia are dramatically altered, producing abnormal crystals (e.g., giant otoconia) in addition to the absence of or substantially reduced amounts of normal otoconia. Interestingly, preliminary evaluation of Otop1 levels in A/J mice showed a 30% reduction (unpublished data). In the A/J mouse, mineralization clearly occurs and the phenotype includes both abnormal crystallization and reduced amounts of apparently normal otoconia.
Findings in consomic C17 mice suggest that the A/J chromosome 17 alone produced an otoconial dysgenesis which was similar to, but attenuated compared to, the A/J phenotype. This clearly indicates first, that one or more unknown gene variants are present on Chr 17, which contributes to the A/J phenotype. This variant cannot be a null mutation of either known candidate genes present on Chr 17 (i.e., Nox3 or Noxo1) because that would presumably result in otoconial agenesis. The gene variant on Chr 17 presumably must be either distinct from Nox3/Noxo1 or conceivably be a variant of Nox3 or Noxo1 that reduces but does not abolish the function of the protein product(s) entirely. Sequence data for Nox3 and Noxo1 in the C57 and AJ strains are available. A list of described variants between the C57BL/6J and A/J strains within and around (± 10 kbp) the Nox3 and Noxo1 genes was compiled from the Wellcome Trust Sanger Institute Mouse Genomes Project Query page (https://www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1410). For Nox3, 154 single nucleotide polymorphisms (SNPs), 49 indels and 0 structural variants were described. For Noxo1, 152 SNPs, 38 indels and 2 structural variants were listed. The vast majority of these variants were designated as noncoding (upstream, downstream, intergenic or intronic) variants each with, presumably, a small likelihood of causation of the vestibular phenotype seen in A/J mice. There was, however, one exception. This SNP (designated rs50679113) was described as a splice region variant (a G to A transition) occurring on Chromosome 17 at position 24,697,122 (build GRCm38/mm10) in an alternate noncoding 5′ exon of Noxo1. This variant represents an interesting candidate for further study. Closer examination revealed that many other strains also carry this SNP in Noxo1 but have no known otoconial phenotype. Thus, it may be the case that another Chr 17 gene variant, one distinct from Nox3 and Noxo1, is responsible for the phenotype in the consomic Chr17 mouse.
What then is responsible for the full A/J phenotype if the phenotype cannot be fully explained by a candidate gene or genes on Chr 17? The results of QTL analysis pointed to other chromosomes in the A/J mouse that may be responsible for the additional deficits presented by A/J mice. These included interacting epistatic loci on Chr 9 and Chr X with possible weaker influences located on Chrs 1 and 4. These possibilities will be examined in future research.
In summary, the findings of the present study point to the existence of novel genes involved in the formation and/or maintenance of otoconia in macular sensors of the mouse; genes that are important for normal vestibular function in the A/J mouse strain. The A/J phenotype represents a complex trait involving multiple gene variants. Vestibular dysfunction in A/J mice was signaled by mild to profound loss of vestibular function manifested generally as elevated VsEP thresholds, diminished amplitudes and prolonged latencies. This was accompanied by structural defects including remarkable otoconial dysgenesis and limited defects in macular stereociliary bundles. It is unclear whether the otoconial and stereociliary abnormalities are molecularly or mechanistically related. The phenotype of A/J was partially replicated in normal mice by substituting the A/J chromosome 17 (Chr 17A/J) for the chromosome 17 in C57 mice. The consomic mouse (C57BL/6J-Chr 17A/J/NaJ) exhibited loss of vestibular function and otoconial dysgenesis similar to, but less severe than, A/J mice. An important locus on the A/J chromosome 17 therefore likely contributes to the A/J otoconial phenotype. QTL analysis identified additional potential epistatic modifiers on Chr 1, 4, 9 and X. Findings also confirmed accelerated cochlear ultrastructural changes in A/J mice. Given the marked otoconial pathology, the A/J strain provides a new opportunity to better understand the mechanisms and genes responsible for otoconial formation and maintenance. Understanding the biosynthesis of otoconia in this animal model may help us to understand human conditions of otoconial pathology. Identifying genes critical for normal vestibular structure and function is important and can lead to the discovery of new and potentially valuable genetic markers for human and animal inner ear disease. Such markers can be used to identify individuals at risk for developing inner ear dysfunction, improve accuracy of diagnosis and, with time, could suggest treatment strategies offering significant benefits to patients.
Acknowledgments
This work was supported by grants from the National Institute on Deafness and Other Communication Disorders (DC006443 to SMJ and DC008603 to YWL). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Deafness and Other Communication Disorders or the National Institutes of Health. Special thanks to Kristal Mills for assistance with data collection, and Greg Tipton, Choongheon Lee and David Raybine for assistance with data analysis.
References
- Ahituv N, Avraham KB. Mouse models for human deafness: Current tools for new fashions. Trends in Molecular Medicine. 2002;8:447–451. doi: 10.1016/s1471-4914(02)02388-2. [DOI] [PubMed] [Google Scholar]
- Beisel KW, Lundberg YW, Maklad A, Fritzsch B. Development and evolution of the vestibular sensory apparatus of the mammalian ear. J Vestib Res. 2005;15:225–241. [PMC free article] [PubMed] [Google Scholar]
- Bergstrom RA, You Y, Erway LC, Lyon MF, Schimenti JC. Deletion mapping of the head tilt (het) gene in mice: A vestibular mutation causing specific absence of otoliths. Genetics. 1998;150:815–822. doi: 10.1093/genetics/150.2.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson V, Nalesso V, Herpin A, Bizot JC, Messaddeq N, Romand R, Puech A, Blanquet V, Hérault Y. Training and aging modulate the loss-of-balance phenotype observed in a new ENU-induced allele of Otopetrin1. Biol Cell. 2005:787–98. doi: 10.1042/BC20040525. [DOI] [PubMed] [Google Scholar]
- Boeda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Petit C. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21(24):6689–99. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen SS, Churchill GA. R/qtl: QTL mapping in experimental crosses. Informatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- Colantonio JR, Vermot J, Wu D, Langenbacher AD, Fraser S, Chen JN, Hill KL. The dynein regulatory complex is required for ciliary motility and otolith biogenesis in the inner ear. Nature. 2009;457:205–209. doi: 10.1038/nature07520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns K, van Alphen AM, van Spaendonck MP, van de Heyning PH, Timmermans JP, de Zeeuw CI, van Camp G. Circling behavior in the Ecl mouse is caused by lateral semicircular canal defects. J Comp Neurol. 2004;468(4):587–595. doi: 10.1002/cne.10975. [DOI] [PubMed] [Google Scholar]
- Dickie MM, Deol MS. Jackson waltzer, jv. Mouse News Letter. 1966;35:31. [Google Scholar]
- Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27(1):103–7. doi: 10.1038/83660. [DOI] [PubMed] [Google Scholar]
- Doerge RW, Churchill GA. Permutation tests for multiple loci affecting a quantitative character. Genetics. 1996;142:285–294. doi: 10.1093/genetics/142.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizzi MS, Peddareddygari LR, Grewal RP. A familial form of benign paroxysmal positional vertigo maps to chromosome 15. International Journal of Neuroscience. 2014;2014:1–4. doi: 10.3109/00207454.2014.953157. Early online. [DOI] [PubMed] [Google Scholar]
- Goodyear RJ, Jones SM, Sharifi L, Forge A, Richardson G. Hair-bundle defects and loss of function in the vestibular end organs of mice lacking the receptor-like inositol lipid phosphatase, PTPRQ. Journal of Neuroscienleece. 2012;32(8):2762–2772. doi: 10.1523/JNEUROSCI.3635-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruen PM, Carranza A, Karmody CS, Bachor E. Anomalies of the Ear in the Pierre Robin Triad. Annals of Otology, Rhinology & Laryngology. 2005;114:605–613. doi: 10.1177/000348940511400805. [DOI] [PubMed] [Google Scholar]
- Henry KR. Age-related auditory loss and genetics: an electrocochleographic comparison of six inbred strains of mice. J Gerontol. 1982;37(3):275–82. doi: 10.1093/geronj/37.3.275. [DOI] [PubMed] [Google Scholar]
- Holme RH, Steel KP. Stereocilia defects in waltzer (Cdh23), shaker1 (Myo7a) and double waltzer/shaker1 mutant mice. Hear Res. 2002;169(1–2):13–23. doi: 10.1016/s0378-5955(02)00334-9. [DOI] [PubMed] [Google Scholar]
- Hurle B, OIgnatova E, Massironi SM, Mashimo T, Riso X, Thalmann I, Thalmann R, Ornitz DM. Non- syndromic vestibular disorder with otconial agenesis in tilted/mergulhador mice caused by mutations in otopetrin1. Hum Mol Genet. 2003;12:777–789. doi: 10.1093/hmg/ddg087. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Erway LC, Cook SA, Willott J, Zheng QY. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear Res. 1997;114(1–2):83–92. doi: 10.1016/s0378-5955(97)00155-x. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Bykhovskaya Y, Spirina O, Fischel-Ghodsian N. A nuclear-mitochondrial DNA interaction affecting hearing impairment in mice. Nat Genet. 2001;27(2):191–4. doi: 10.1038/84831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Erway LC. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics. 2000;70(2):171–80. doi: 10.1006/geno.2000.6377. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Noben-Trauth K. Strain background effects and genetic modifiers of hearing in mice. Brain Res. 2006;1091(1):79–88. doi: 10.1016/j.brainres.2006.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Erway LC, Cook SA, Willott JF, Zheng QY. A major gene affecting age-related hearing loss in C57BL/6J mice. Hearing Research. 1997;114:83–92. doi: 10.1016/s0378-5955(97)00155-x. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Gagnon LH, Longo-Guess C, Kane KL. Association of a citrate synthase missense mutation with age-related hearing loss in A/J mice. Neurobiology of Aging. 2012;33:1720–1729. doi: 10.1016/j.neurobiolaging.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Jones TA. Vestibular Sensory Evoked Potentials. In: Jacobson GP, Shepard NT, editors. Balance Function Assessment and Management. 2. Chp 20. Plural Publishing; SanDiego: 2014. [Google Scholar]
- Jones SM, Erway LC, Bergstrom RA, Schimenti JC, Jones TA. Vestibular responses to linear acceleration are absent in otoconia-deficient C57BL/6JEi-het mice. Hear Res. 1999;135(1–2):56–60. doi: 10.1016/s0378-5955(99)00090-8. [DOI] [PubMed] [Google Scholar]
- Jones SM, Erway LC, Johnson KR, Yu H, Jones TA. Gravity receptor function in mice with graded otoconial deficiencies. Hear Res. 2004;191(1–2):34–40. doi: 10.1016/j.heares.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Jones SM, Jones TA, Johnson KR, Yu H, Erway LC, Zheng QY. A comparison of vestibular and auditory phenotypes in inbred mouse strains. Brain Res. 2006;1091(1):40–6. doi: 10.1016/j.brainres.2006.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Mock BE, Bergstrom DE. Inner ear function in fourteen inbred strains identifies A/J as an otoconia-deficient strain. Assoc Res Otolaryngol Abst 2008 [Google Scholar]
- Jones TA, Jones SM. Short latency compound action potentials from mammalian gravity receptor organs. Hear Res. 1999;136(1–2):75–85. doi: 10.1016/s0378-5955(99)00110-0. [DOI] [PubMed] [Google Scholar]
- Kang YJ, Stevenson AK, Yau PM, Kollmar R. Sparc protein is required for normal growth of zebrafish otoliths. J Assoc Res Otolaryngol. 2008;9(4):436–51. doi: 10.1007/s10162-008-0137-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss PJ, Knisz J, Zhang Y, Baltrusaitis J, Sigmund CD, Thalmann R, Smith RJ, Verpy E, Banfi B. Inactivation of NADPH oxidase organizer 1 results in severe imbalance. Curr Biol. 2006;16(2):208–213. doi: 10.1016/j.cub.2005.12.025. [DOI] [PubMed] [Google Scholar]
- Lane PW. Tilted (tlt) Mouse News Lett. 1986;75:28. [Google Scholar]
- Lane PW. New mutants and linkages: Tilted (tlt) Mouse News Lett. 1987;77:129. [Google Scholar]
- Lee JE, Silhavy JL, Zaki MS, Schroth J, Bielas SL, Marsh SE, Olvera J, Brancati F, Iannicelli M, Ikegami K, Schlossman AM, Merriman B, Attie-Bitach T, Logan CV, Glass IA, Cluckey A, Louie CM, Lee JH, Raynes HR, Rapin I, Castroviejo IP, Setou M, Barbot C, Boltshauser E, Nelson SF, Hildebrandt F, Johnson CA, Doherty DA, Valente EM, Gleeson JG. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat Genet. 2012;44:193–199. doi: 10.1038/ng.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Conrad T, Jones SM, Lagziel A, Starost AF, Belyantseva I, Friedman TB, Morell RJ. A null mutation of mouse Kcna10 causes significant vestibular and mild hearing dysfunction. Hearing Research. 2013;300:1–9. doi: 10.1016/j.heares.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Sanneman JD, Harbidge DG, Zhou F, Taku I, Nelson R, Picard N, Chambrey R, Eladri D, Miesner T, Griffith AJ, Marcus DC, Wangemann P. SLC26A4 targeted to the endolymphatic sac rescues hearing and balance in Slc26a4 mutant mice. PLoS Genet. 9(7):e1003641. doi: 10.1371/journal.pgen.1003641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mock BE. PhD Dissertation. East Carolina University; 2008. Functional aging of th inner ear sensory systems in mouse models of age-related hearing loss. [Google Scholar]
- Murayama E, Herbomel P, Kawakami A, Takeda H, Nagasawa H. Otolith matrix proteins OMP-1 and Otolin-1 are necessary for normal otolith growth and their correct anchoring onto the sensory maculae. Mech Dev. 2005;122(6):791–803. doi: 10.1016/j.mod.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Longo-Guess CM, Bergstrom DE, Nauseef WM, Jones SM, Banfi B. Immuno-vestibular syndrome caused by the inactivation of p22phox. J Clin Invest. 2008;118:1176–1185. doi: 10.1172/JCI33835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya K, Harbidge DG, Wangemann P, Schultz BD, Green ED, Wall SM, Marcus DC. Lack of pendrin-HCO3 transport elevates vestibular endolymphatic [Ca2+] by inhibition of acid- sensitive TRPV5 and TRPV6 channels. Am J Physiol Renal Physiol. 2007;292:F1314–F1321. doi: 10.1152/ajprenal.00432.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazareth AM, Jones TA. Central and peripheral components of short latency vestibular responses in the chicken. J Vestib Res. 1998;8(3):233–52. [PubMed] [Google Scholar]
- Nemoto M, Morita Y, Mishima Y, Takahashi S, Nomura T, Ushiki T, Shiroishi T, Kikkawa Y, Yonekawa H, Kominami R. Ahl3, a third locus on mouse chromosome 17 affecting age-related hearing loss. Biochemical and Biophysical Research Communications. 2004;324(2004):1283–1288. doi: 10.1016/j.bbrc.2004.09.186. [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nature Genetics. 2003;35(1):21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noben-Trauth J, Johnson KR. Inheritance patterns of progressive hearing loss in laboratory strains of mice. Brain Research. 2009;1277:42–51. doi: 10.1016/j.brainres.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Bohne BA, Thalmann I, Harding GW, Thalmann R. Otoconial agenesis in tilted mutant mice. Hear Res. 1998;112:60–70. doi: 10.1016/s0378-5955(98)00080-x. [DOI] [PubMed] [Google Scholar]
- Paffenholz R, Bergstrom RA, Pasutto F, Wabnitz P, Munroe RJ, Jagla W, Heinzmann U, Marquardt A, Bareiss A, Laufs J, Russ A, Stumm G, Schimenti JC, Bergstrom DE. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev. 2004;1:1–6. doi: 10.1101/gad.1172504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner C, Burghammer M, Busch-Nentwich E, Berger J, Schwarz H, Riekel C, Nicolson T. Control of crystal size and lattice formation by starmaker in otolith biomineralization. Science. 2003;302(5643):282–6. doi: 10.1126/science.1088443. [DOI] [PubMed] [Google Scholar]
- Sweet H. Head tilt. Mouse news letter. 1980;63:19–21. [Google Scholar]
- Zhao X, Yang H, Yamoah EN, Lundberg YW. Gene targeting reveals the role of Oc90 as the essential organizer of the otoconial organic matrix. Dev Biol. 2007;304(2):508–24. doi: 10.1016/j.ydbio.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Ding D, Yu H, Salvi RJ, Johnson KR. A locus on distal chromosome 10 (ahl4) affecting age-related hearing loss in A/J mice. Neurobiol Aging. 2009;30(10):1693–705. doi: 10.1016/j.neurobiolaging.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]