

Abstract
We report a family in which two male siblings with Becker muscular dystrophy (BMD) developed severe dilated cardiomyopathy (DCM) and progressive heart failure (HF) at age 11; one died at age 14 years while awaiting heart transplant and the other underwent left ventricular assist device (LVAD) implantation at the same age. Genetic analysis of one sibling showed a novel frameshift mutation in exon 27 of Duchenne muscular dystrophy (DMD) gene (c.3779_3785delCTTTGGAins GG), in which 7 base pairs are deleted and two are inserted. While this predicts an amino acid substitution and premature termination (p.Thr1260Argfs*8), muscle biopsy dystrophin immunostaining instead indicates that the mutation is more likely to alter splicing. Despite relatively preserved skeletal muscular performance, both siblings developed progressive heart failure secondary to early onset DCM. In addition, their 7 year old nephew with delayed gross motor development, mild proximal muscle weakness, and markedly elevated serum creatine kinase (CK) level (> 13,000 IU/L) at 16 months was recently demonstrated to have the familial DMD mutation. Here we report a novel genotype of BMD with early onset DCM and progressive lethal heart failure during early adolescence.
Keywords: Becker muscular dystrophy (BMD), Dilated cardiomyopathy (DCM), Dystrophin, Frameshift mutation, Alternative splicing
Introduction
Among the entities that cause dilated cardiomyopathy in children are dystrophinopathies, including Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), and X-linked dilated cardiomyopathy, allelic conditions caused by defects in dystrophin (1). Dystrophin is an important cytoskeletal protein encoded by the DMD gene located at Xp21.2 (2). DMD is caused by complete loss of the dystrophin protein complicated by progressive skeletal muscle dystrophic pathology, whereas BMD often results from expression of an altered dystrophin protein leading to milder and a more variable clinical presentation than DMD (3). Approximately 70% of BMD patients develop dilated cardiomyopathy (DCM) mostly in the third decade of life or later (4, 5); they rarely develop severe DCM in childhood (6).
Conserving the reading frame of the protein plays a determining factor in disease severity in many dystrophinopathies. Mutations in which the reading frame is conserved resulting in a functional amino- and carboxy-terminus with little effect on the central rod domain yield a milder phenotype consistent with BMD (7), whereas mutations that disrupt the reading frame cause a more severe phenotype consistent with DMD. The reading frame rule was found to be 91–92% consistent in predicting phenotype in simplex cases of young boys (8). However, more recent studies have shown that this rule tends to be better at predicting phenotype for DMD, and there are more exceptions to this rule in BMD (9, 10). Genotype-phenotype correlations for specific mutations in dystrophin related to predisposition to or protection against DCM have been proposed (6, 11), but exactly how this leads to DCM is poorly understood.
Here, we present a family in which two siblings and their nephew are affected by similar skeletal muscle abnormalities consistent with BMD. The brothers developed severe progressive DCM in early adolescence. They have a novel frameshift mutation in the DMD gene responsible for this unique clinical phenotype of BMD and severe early-onset DCM. The importance of genetic screening of the male family members for early detection of lethal congestive heart failure (CHF) will be discussed.
Case Reports
There are three affected males in this family spanning two generations (Figure 1A). The first affected member of the family was diagnosed with BMD at 6 years due to mild skeletal muscle weakness (II-3). His serum CK was extremely elevated (38,000 IU/L). Dystrophin immunostaining in a muscle biopsy was consistent with BMD, and the dystrophin level was quantified as 3–10% of normal by Western blot (Athena Diagnostics, Worcester, MA). He was diagnosed with attention deficit hyperactivity disorder (ADHD), but otherwise remained relatively symptom-free for a long time. At 12 years, however, he developed persistent cough resistant to conventional bronchodilator treatment. Chest X-ray showed cardiomegaly, for which he was referred to cardiology. Initial echocardiogram revealed dilated LV with severely diminished LV systolic function (left ventricle shortening fraction of less than 10%). His clinical status continued to deteriorate despite maximum anti-congestive medications, and he was admitted for further management of intractable CHF. Hemodynamic assessment by cardiac catheterization showed severe LV dysfunction: LV end-diastolic pressure (LVEDP) 35 mmHg and cardiac index (CI) 1.8 L/min/m2 under mechanical ventilation and continuous intravenous inotrope infusion. He died at age 14 due to a cerebral thromboembolic event despite maximum medical treatment while awaiting a heart transplant.
Figure 1. Pedigree and sequencing of individuals II-4 and III-1.
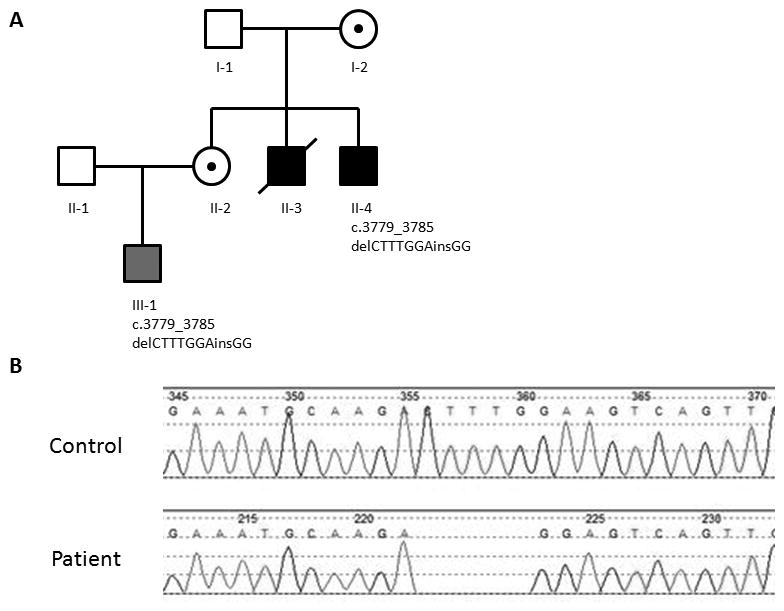
(A) Pedigree of family with BMD. Solid squares indicate individuals with BMD and DCM. Gray square indicates BMD mutation and clinical phenotype without DCM. Circles with a dot represent asymptomatic carrier females. The mutation, c.3779_3785delCTTTGGAinsGG is documented for those who were tested (Emory Genetics Laboratory, Decatur, GA. Figure 1B). (B) Sanger sequences of affected individuals II-4 and III-1.
His younger brother (II-4; 14 years) was diagnosed as BMD at 3 years of age with mild muscle weakness and failure to thrive. He has cognitive impairment with function in the range of 6–8 years old (at 14 years old) and a seizure disorder of unknown etiology. His cardiac status has been followed closely since 6 years of age because of his brother’s history of early-onset of progressive DCM. Sequencing (Emory Genetics Laboratory, Decatur, GA) revealed a novel frameshift mutation in exon 27 of the DMD gene (c.3779_3785delCTTTGGAinsGG) in which 7 base pairs are deleted and two are inserted (Figure 1B). This predicts an amino acid substitution and premature termination in the novel reading frame 8 codons downstream (p.Thr1260Argfs*8). No other potential disease-causing variants were found in the gene. Despite this predicted truncation of protein translation, immunofluorescence studies of a quadriceps muscle biopsy at age 14 instead showed reduced staining for dystrophin and other proteins of the dystrophin-glycoprotein complex and upregulation of utrophin consistent with BMD (Figure 2). The greatest reduction in dystrophin immunofluorescence (perhaps absence of staining) was with an antibody that detects an epitope encoded by exons 27-28 (Figure 2F). Dystrophin epitopes on either side of exons 27-28 were present (Figure 2C, D, E, and G). Dystrophin was also detected in Western blots from the same muscle biopsy using a carboxy terminus antibody. The total amount of the patient’s dystrophin was greatly reduced, and the molecular weight appeared to be slightly reduced (data not shown).
Figure 2. Muscle biopsy evaluation.

Typical histopathologic features of muscular dystrophy are illustrated in this H&E-stained cryosection from the BMD patient II-4 (A). Immunofluorescence studies are shown in panels B through M; staining was performed as previously described (29). A carboxy terminus anti-dystrophin antibody shows the pattern of normal dystrophin staining in control muscle (B) and reduced dystrophin staining in the BMD patient (C). Panels D through H are all sections from the BMD patient stained with various, epitope-specific, anti-dystrophin antibodies: exon 50 (D), exons 38-39 (E), exons 27-28 (F), exons 10-12 (G), and exon 1 (H). They show varying degrees of reduced staining for dystrophin. Utrophin is expressed at the sarcolemma of some muscle fibers in the BMD patient (I); utrophin is commonly upregulated in muscle from dystrophinopathy patients. Beta-dystroglycan [control (J) and BMD (K)] and nNOS [control (L) and BMD (M)] are both reduced in the BMD patient. The scale bar is 100 μm in panel A and 200 μm in panels B through M.
He had normal cardiac function through age 11 years, after which progressive LV systolic dysfunction and dilatation were noted by echocardiogram. His symptoms of CHF continued to worsen over the next 2 years despite escalating medical treatment, and he was admitted to the hospital at 14 years because of severe respiratory distress and cardiac cachexia. Echocardiogram showed markedly dilated LV (LVIDd 7.7 cm: z-score +9.1) with severely diminished LV systolic function (LVSF 15%). Initial hemodynamic assessment by cardiac catheterization was consistent with mild to moderate LV dysfunction under continuous intravenous milrinone infusion (LVEDP 12 mmHg and CI 2.1 L/min/m2). His clinical condition deteriorated despite maximum medical treatment including mechanical ventilation, and eventually he underwent implantation of left ventricular assist device (Heartware HVAD, Framingham, MA). He was discharged home with improved quality of life.
The 7 year old nephew (III-1) of these two affected boys was born to their sister and was diagnosed as BMD in his early childhood. He failed to thrive despite maximizing nutritional efforts (< 1st percentile) and showed generalized muscle weakness and mild developmental delay. At 18 months he was noted to have serum CK above 13,000 IU/L, and he has been followed regularly since that time with a presumed diagnosis of BMD. Molecular analysis revealed identical familial frameshift mutation in DMD to his uncle (II-4). At 18 months he was noted to have serum CK above 13,000 IU/L, and he has been followed regularly since that time with a presumed diagnosis of BMD. A recent echocardiogram was normal (7 years old). Female carriers in the family, the siblings’ mother (I-2) and their sister (II-1), have had no symptoms of muscle weakness or CHF. There is no further family history is available for I-2, as she was adopted in her early childhood.
Clinical manifestations and laboratory data of three BMD patients are summarized in Table 1.
Table 1.
Clinical and Laboratory Findings of the Affected Children
| II-3 | II-4 | III-1 | |
|---|---|---|---|
| Current Age (yrs: In 2013) | 14† | 15 | 7 |
| Diagnosis of BMD (yrs) | 6 | 3 | 3 |
| Diagnosis of DCM (yrs) | 11 2/3 | 11 3/4 | N/A |
|
| |||
| Neurological | Normal | Seizure disorder (resolved) | Mild developmental delay |
| Behavioral & Developmental | ADHD | Mild cognitive delay | ADHD |
|
| |||
| Musculoskeletal | Mild muscle weakness | Mild muscle weakness | Mild muscle weakness |
| Serum CK (IU/L) | 38,256 | >16,000 | 13,860 |
| Muscle Biopsy | (+) | (+) | Not performed |
| Immunohistochemistry | Severely decreased dystrophin protein | Figure 1 | |
| Western blot (protein quantification) | Severely decreased (3 to 10%) | Severely decreased (3 to 10%) | |
|
| |||
| Cardiac | Severe CHF | Severe CHF | None |
| ECG | Sinus tachycardia (130) | T wave inversion in V5, V6 | Normal |
| RVH, RAH, LVH | LVH | ||
| CXR | Cardiomegaly | Cardiomegaly | Not done |
| Echocardiogram | Dilated LV (LVIDd 6.2 cm) | Dilated LV (LVIDd 7.7 cm) | Normal study |
| LVSF < 10% | LVSF 15% | ||
| Cardiac Catheterization | 13 yrs 10 mos | 14 yrs 0 mos | Not performed |
| SvO2: 54%, SaO2 93% | SvO2 70 %, SaO2 99% | ||
| RA (10) mmHg, LDEDP 35 mmHg | RA (3) mmHg, LVEDP 12 mmHg | ||
| C.I. 1.8 L/min/m2, Rp/Rs = 0.43 | C.I. 2.1 L/min/m2, Rp/Rs = 0.28 | ||
| Mechanically ventilated (FiO2 0.5) | On milrinone infusion | ||
| On milrinon infusion | |||
†: Deceased. BMD: Becker muscular dystrophy, DCM: dilated cardiomopathy, ADHD: attention deficit hyperactivity disorder, CK: creatine kinase, CHF: congestive heart failure, RVH: right ventricular hypertrophy, RAH: right atrial hypertrophy, LVH: left ventricular hypertrophy, LVIDd: left ventricular internal diameter in diastole, LVSF: left ventricular shortening fraction, SvO2: mixed venous saturation, SaO2: systemic arterial saturation, RA: right atrial pressure (mean), LVEDP: left ventricular end diastolic pressure, C.I.: cardiac index, Rp/Rs: Pulmonary vascular resistance/systemic vascular resistance.
Discussion
We report a family in which two male siblings with BMD developed severe, early-onset, progressive DCM. Both siblings developed intractable CHF by age 12 that was resistant to conventional anti-congestive medications. BMD patients are known to have milder skeletal muscle involvement than DMD patients and commonly develop DCM in their third decade or later. Cardiac dysfunction is a more frequent primary cause of death in BMD than in DMD (4, 12, 13). There are sporadic case reports of early onset DCM in BMD, but our patients are unique because of their earlier onset, multiple family member involvement with the same clinical phenotype, and a novel genotype.
Dystrophinopathy refers to a clinical spectrum of mutation of DMD gene encompassing BMD, DMD, and X-linked DCM. X-linked DCM can present as a similar clinical course as ours, as reported by others (14–16). Intermediate phenotype between DMD and BMD with variable cardiac involvement is not infrequently encountered. However, all 3 patients here initially presented with primary skeletal muscle weakness with markedly increased serum CK, which is quite different from X-linked DCM where there should be no or very little skeletal muscle involvement (14, 15, 17). Skeletal muscle biopsy demonstrated that two brothers showed markedly reduced but positive dystrophin protein expression consistent with BMD. Collectively, we believe the affected brothers have BMD with severe early-onset DCM rather than X-linked DCM with coincidental skeletal myopathy. Disproportionally severe cardiac phenotype has been reported in limb-girdle type muscular dystrophy with relatively mild skeletal involvement (18).
The X-linked DMD gene has 79 coding exons covering 2.6 million base pairs. The large size of the DMD gene makes it vulnerable to mutation, mostly by deletion of one or more exons. DMD is caused by genetic mutations that lead to disruption of the reading frame of the dystrophin transcript and premature abortion of dystrophin synthesis (19). BMD mutations usually do not disrupt the translational reading frame, and are typically in conjunction with either exon deletions or point mutations that alter splicing such that the resultant mRNA maintains the open reading frame and leads to production of a partially functional but aberrant protein (20–22). Previous reports described patients with BMD phenotype with nonsense mutations as a result of altered exon splicing and exon skipping, excluding the nonsense mutation containing exon (20, 23, 24)
In our BMD patients (II-4 and III-1), we postulate that the mutation in exon 27 (c.3779_3785delCTTTGGAinsGG) results in altered splicing and mRNA that allows production of a truncated dystrophin protein. This interpretation stems from immunohistochemical analysis of a muscle biopsy that demonstrates expression of exons proximal and distal to exon 27 (see Figure 2). Unfortunately, we were unable to recover mRNA of sufficient quality from archived muscle to test this hypothesis directly, but we note that disruption of the splicing enhancer sequence within exon 27 of the DMD gene by nonsense mutation can induce partial exon skipping, and thus results in a BMD phenotype (20, 21). However, this early-onset of severe progressive DCM in BMD patients, as seen in this family, has not been reported and cannot be solely explained by the exon skipping mechanism. Kaspar et al. proposed that the deletions affecting the amino-terminal domain (exons 2-8) are associated with the early-onset of DCM (6), but it does not appear that our cases follow this rule. Double or multiple gene mutations are known to be responsible for more severe clinical phenotype in some hypertrophic cardimyopathy (HCM) (25–27) and rarely in DCM (28). The possibility of second mutation in our cases cannot be ruled out, as we have not performed sequencing of other single gene causes of DCM or whole-exome sequencing.
Genetic predictors of early onset of DCM in BMD patients have been studied extensively (6, 11), but the results are not conclusive as to which genotypes predict a worse natural course. There may be a fundamental difference in regulation of DMD gene transcription between skeletal and cardiac muscle. In this family, both female carriers have been asymptomatic. All three affected children developed a nonspecific central nervous system abnormality, but the clinical hallmark of the two brothers is early-onset lethal DCM. Because early detection of this progressive DCM is essential for optimum management, further effort in investigating genotype-phenotype correlation in BMD should be encouraged.
Acknowledgments
This work was supported by National Institute of Health: 2P20GM103446-12 and Asano/Noguchi Fund (for T.T.) and in part by a Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Grant: U54, NS053672 (for M.O.C. and S.A.M.). Joel Carl assisted with the preparation of Figure 2.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Hermans MC, Pinto YM, Merkies IS, de Die-Smulders CE, Crijns HJ, Faber CG. Hereditary muscular dystrophies and the heart. Neuromuscul Disord. 2010;20(8):479–92. doi: 10.1016/j.nmd.2010.04.008. Epub 2010/07/16. [DOI] [PubMed] [Google Scholar]
- 2.Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53(2):219–28. doi: 10.1016/0092-8674(88)90383-2. Epub 1988/04/22. [DOI] [PubMed] [Google Scholar]
- 3.Connuck DM, Sleeper LA, Colan SD, Cox GF, Towbin JA, Lowe AM, et al. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the Pediatric Cardiomyopathy Registry. Am Heart J. 2008;155(6):998–1005. doi: 10.1016/j.ahj.2008.01.018. Epub 2008/06/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nigro G, Comi LI, Politano L, Limongelli FM, Nigro V, De Rimini ML, et al. Evaluation of the cardiomyopathy in Becker muscular dystrophy. Muscle Nerve. 1995;18(3):283–91. doi: 10.1002/mus.880180304. Epub 1995/03/01. [DOI] [PubMed] [Google Scholar]
- 5.Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, et al. Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation. 1996;94(12):3168–75. doi: 10.1161/01.cir.94.12.3168. Epub 1996/12/15. [DOI] [PubMed] [Google Scholar]
- 6.Kaspar RW, Allen HD, Ray WC, Alvarez CE, Kissel JT, Pestronk A, et al. Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in becker muscular dystrophy. Circulation Cardiovascular genetics. 2009;2(6):544–51. doi: 10.1161/CIRCGENETICS.109.867242. Epub 2009/12/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90–5. doi: 10.1016/0888-7543(88)90113-9. Epub 1988/01/01. [DOI] [PubMed] [Google Scholar]
- 8.Aartsma-Rus A, Janson AA, Heemskerk JA, De Winter CL, Van Ommen GJ, Van Deutekom JC. Therapeutic modulation of DMD splicing by blocking exonic splicing enhancer sites with antisense oligonucleotides. Ann N Y Acad Sci. 2006;1082:74–6. doi: 10.1196/annals.1348.058. Epub 2006/12/06. [DOI] [PubMed] [Google Scholar]
- 9.Kesari A, Pirra LN, Bremadesam L, McIntyre O, Gordon E, Dubrovsky AL, et al. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Hum Mutat. 2008;29(5):728–37. doi: 10.1002/humu.20722. Epub 2008/03/19. [DOI] [PubMed] [Google Scholar]
- 10.Takeshima Y, Yagi M, Okizuka Y, Awano H, Zhang Z, Yamauchi Y, et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet. 2010;55(6):379–88. doi: 10.1038/jhg.2010.49. Epub 2010/05/21. [DOI] [PubMed] [Google Scholar]
- 11.Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, et al. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112(18):2799–804. doi: 10.1161/CIRCULATIONAHA.104.528281. Epub 2005/10/26. [DOI] [PubMed] [Google Scholar]
- 12.Saito M, Kawai H, Akaike M, Adachi K, Nishida Y, Saito S. Cardiac dysfunction with Becker muscular dystrophy. Am Heart J. 1996;132(3):642–7. doi: 10.1016/s0002-8703(96)90250-1. Epub 1996/09/01. [DOI] [PubMed] [Google Scholar]
- 13.Melacini P, Fanin M, Danieli GA, Fasoli G, Villanova C, Angelini C, et al. Cardiac involvement in Becker muscular dystrophy. J Am Coll Cardiol. 1993;22(7):1927–34. doi: 10.1016/0735-1097(93)90781-u. Epub 1993/12/01. [DOI] [PubMed] [Google Scholar]
- 14.Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS, et al. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993;87(6):1854–65. doi: 10.1161/01.cir.87.6.1854. Epub 1993/06/01. [DOI] [PubMed] [Google Scholar]
- 15.Muntoni F, Cau M, Ganau A, Congiu R, Arvedi G, Mateddu A, et al. Brief report: deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy. N Engl J Med. 1993;329(13):921–5. doi: 10.1056/NEJM199309233291304. Epub 1993/09/23. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida K, Ikeda S, Nakamura A, Kagoshima M, Takeda S, Shoji S, et al. Molecular analysis of the Duchenne muscular dystrophy gene in patients with Becker muscular dystrophy presenting with dilated cardiomyopathy. Muscle Nerve. 1993;16(11):1161–6. doi: 10.1002/mus.880161104. Epub 1993/11/01. [DOI] [PubMed] [Google Scholar]
- 17.Diegoli M, Grasso M, Favalli V, Serio A, Gambarin FI, Klersy C, et al. Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. J Am Coll Cardiol. 2011;58(9):925–34. doi: 10.1016/j.jacc.2011.01.072. Epub 2011/08/20. [DOI] [PubMed] [Google Scholar]
- 18.Margeta M, Connolly AM, Winder TL, Pestronk A, Moore SA. Cardiac pathology exceeds skeletal muscle pathology in two cases of limb-girdle muscular dystrophy type 2I. Muscle Nerve. 2009;40(5):883–9. doi: 10.1002/mus.21432. Epub 2009/08/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Deutekom JC, van Ommen GJ. Advances in Duchenne muscular dystrophy gene therapy. Nature reviews Genetics. 2003;4(10):774–83. doi: 10.1038/nrg1180. Epub 2003/10/04. [DOI] [PubMed] [Google Scholar]
- 20.Shiga N, Takeshima Y, Sakamoto H, Inoue K, Yokota Y, Yokoyama M, et al. Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J Clin Invest. 1997;100(9):2204–10. doi: 10.1172/JCI119757. Epub 1997/12/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Howard MT, Sampson JB, et al. Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene. Hum Mutat. 2011;32(3):299–308. doi: 10.1002/humu.21426. Epub 2011/10/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dent KM, Dunn DM, von Niederhausern AC, Aoyagi AT, Kerr L, Bromberg MB, et al. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. American journal of medical genetics Part A. 2005;134(3):295–8. doi: 10.1002/ajmg.a.30617. Epub 2005/02/22. [DOI] [PubMed] [Google Scholar]
- 23.Tuffery-Giraud S, Saquet C, Thorel D, Disset A, Rivier F, Malcolm S, et al. Mutation spectrum leading to an attenuated phenotype in dystrophinopathies. Eur J Hum Genet. 2005;13(12):1254–60. doi: 10.1038/sj.ejhg.5201478. Epub 2005/08/04. [DOI] [PubMed] [Google Scholar]
- 24.Disset A, Bourgeois CF, Benmalek N, Claustres M, Stevenin J, Tuffery-Giraud S. An exon skipping-associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements. Hum Mol Genet. 2006;15(6):999–1013. doi: 10.1093/hmg/ddl015. Epub 2006/02/08. [DOI] [PubMed] [Google Scholar]
- 25.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107(17):2227–32. doi: 10.1161/01.CIR.0000066323.15244.54. Epub 2003/04/23. [DOI] [PubMed] [Google Scholar]
- 26.Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44(9):1903–10. doi: 10.1016/j.jacc.2004.07.045. Epub 2004/11/03. [DOI] [PubMed] [Google Scholar]
- 27.Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005;42(10):e59. doi: 10.1136/jmg.2005.033886. Epub 2005/10/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roncarati R, Viviani Anselmi C, Krawitz P, Lattanzi G, von Kodolitsch Y, Perrot A, et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur J Hum Genet. 2013;21(10):1105–11. doi: 10.1038/ejhg.2013.16. Epub 2013/03/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acsadi G, Moore SA, Cheron A, Delalande O, Bennett L, Kupsky W, et al. Novel mutation in spectrin-like repeat 1 of dystrophin central domain causes protein misfolding and mild Becker muscular dystrophy. J Biol Chem. 2012;287(22):18153–62. doi: 10.1074/jbc.M111.284521. Epub 2012/03/29. [DOI] [PMC free article] [PubMed] [Google Scholar]