

Abstract
Acute intermittent hypoxia (AIH) initiates plasticity in respiratory motor control, including phrenic long term facilitation (pLTF). Since pLTF is enhanced by preconditioning with repetitive exposure to AIH (rAIH), we hypothesized that a rAIH protocol consisting of 3 AIH exposures per week for 10 weeks (3×wAIH; AIH: 10, 5-min episodes of 10.5% O2; 5-min normoxic intervals) would enhance expression of molecules that play key roles in pLTF within the phrenic motor nucleus. Immunohistochemical analyses revealed that 3×wAIH for 10 weeks increased serotonin terminal density in the C4 phrenic motor nucleus and serotonin 2A (5-HT2A) receptor expression in presumptive phrenic motor neurons. Immunoreactive brain derived neurotrophic factor (BDNF) and its high affinity receptor (TrkB) also increased following 3×wAIH. 3×wAIH also increased expression of another hypoxia-sensitive growth factor known to elicit phrenic motor facilitation, vascular endothelial growth factor (VEGF), and its receptor (VEGFR-2). Kinases “downstream” from TrkB and VEGFR-2 were up-regulated in or near presumptive phrenic motor neurons, including phosphorylated extracellular-signal regulated kinase (p-ERK) and protein kinase B (p-AKT). Thus, 3×wAIH up-regulates neurochemicals known to be associated with phrenic motor plasticity. Since 3×wAIH upregulates pro-plasticity molecules without evidence for CNS pathology, it may be a useful therapeutic tool in treating disorders that cause respiratory insufficiency, such as spinal injury or motor neuron disease.
Keywords: Intermittent hypoxia, Phrenic, Plasticity, BDNF, VEGF
Introduction
The neuro-motor system controlling breathing exhibits considerable plasticity in response to physiological challenges (Feldman et al., 2003; Mitchell and Johnson, 2003). For example, acute intermittent hypoxia (AIH; 3 hypoxic episodes) elicits a form of respiratory plasticity known as phrenic long-term facilitation (pLTF; Bach and Mitchell, 1996; Matieka and Sandhu, 2011; Mitchell and Terada, 2011; Mitchell et al., 2001). Our understanding of cellular mechanisms underlying AIH-induced pLTF has advanced considerably in recent years (Dale-Nagle et al., 2010; Feldman et al., 2003; MacFarlane et al., 2009; Mahamed and Mitchell, 2007; Mitchell et al., 2001). For example, pLTF requires spinal serotonin type 2 receptor activation (Baker-Herman and Mitchell, 2002; MacFarlane and Mitchell, 2009; MacFarlane et al., 2011), new synthesis of brain derived neurotrophic factor (BDNF; Baker-Herman et al., 2004), activation of the high affinity BDNF receptor, TrkB (Baker-Herman et al., 2004), with subsequent downstream signaling via extracellular regulated MAP kinases (ERK; Hoffman and Mitchell, unpublished; Wilkerson and Mitchell, 2009).
However, we now know that multiple, distinct cellular mechanisms give rise to long-lasting phrenicmotor facilitation (pMF; a general term that includes pLTF; Dale-Nagle et al., 2010). One alternate pathway to pMF involves activation of adenosine type 2A receptors (A2A), new TrkB synthesis and downstream signaling via protein kinase B/Akt in the region of the phrenic motor nucleus (Golder et al., 2008; Nichols et al., 2012). This novel, adenosine-dependent mechanism assumes predominance when the severity of hypoxic episodes increases, replacing the normal, serotonin-dependent mechanism(Nichols et al., 2012). Another novel pathway involves signaling via the hypoxia-induced growth factor, vascular endothelial growth factor (VEGF; Dale-Nagle et al., 2011). Full expression of VEGF-induced pMF requires activation of both ERKMAP kinases and Akt (Dale-Nagle et al., 2011). The predominance of one pathway versus another in respiratory motor plasticity following intermittent hypoxia may depend on factors such as the intensity and/or duration of hypoxia, or altered conditions during injury or disease (Mitchell and Terada, 2011).
AIH-induced pLTF is enhanced by prior experience with intermittent hypoxia (Ling et al., 2001; Mitchell et al., 2001; Wilkerson et al., 2008). For example, pretreatment with chronic intermittent hypoxia (CIH) enhances pLTF during anesthesia (Ling et al., 2001) and ventilatory LTF in unanesthetized rats (McGuire et al., 2003). It has been suggested that repetitive intermittent hypoxia may be harnessed as a therapeutic approach to increase phrenic motor output in cases of respiratory insufficiency such as following cervical spinal injury or during motor neuron disease (Dale-Nagle et al., 2011; Fuller et al., 2003; Lovett-Barr et al., 2012; Mitchell, 2007; Trumbower et al., 2012).
Although CIH elicits considerable pLTF meta-plasticity (Ling et al., 2001; McGuire et al., 2003), it also elicits morbidity, such as systemic hypertension (Tamisier et al., 2011; Zoccal et al., 2007), sleep fractionation (Perry et al., 2007), hippocampal neuron death and cognitive decline (Gozal et al., 2001; Li et al., 2003; Xu et al., 2004). Thus, less severe protocols of intermittent hypoxia have been suggested as an alternative for therapeutic application (Mitchell, 2007), including daily exposure to acute intermittent hypoxia (dAIH; 10, 5 minute episodes with 5 minute intervals for 7 days; Lovett-Barr et al., 2012; Wilkerson and Mitchell, 2009). Although we know that dAIH increases BDNF and ERK phosphorylation within ventral cervical spinal segments that contain the phrenic motor nucleus (Lovett-Barr et al., 2012; Wilkerson and Mitchell, 2009), other molecules that play key roles in pMF have not been investigated.
Here, we investigate a novel protocol of repetitive acute intermittent hypoxia consisting of AIH (10 episodes per day; 5 min of hypoxia with 5 minute intervals) three times per week for 10 weeks (3×wAIH). Since similar rAIH for four weeks elicits meta-plasticity in pLTF (MacFarlane et al., 2008), we predicted that 3×wAIH for 10 weeks would elicit neurochemical plasticity, but without detectable CNS pathology.
Immunohistochemical techniques were used to localize protein expression in the region of the phrenicmotor nucleus (C4 and C5 ventral segments). In specific, we tested the hypotheses that 3×wAIH (10 weeks): 1) increases the expression of key molecules involved in AIH-induced pLTF, including serotonin, serotonin receptors, BDNF, TrkB and ERK MAP kinases; 2) increases the expression of key molecules that elicit alternate mechanisms of pMF, including TrkB, Akt, VEGF and VEGF receptor 2; and 3) elicits neurochemical plasticity without evidence of non-specific brain pathology, such as hippocampal apoptosis or reactive gliosis. An understanding of 3×wAIH-induced neurochemical plasticity may have important implications as we develop therapeutic strategies to treat clinical disorders involving respiratory insufficiency.
Methods
Twenty adult male Sprague–Dawley rats were randomly assigned to receive either sham normoxia (n=10) or 3×wAIH for 10 weeks (n=10). The day prior to treatment onset, rats were acclimated to the custom-made Plexiglass chambers (1 rat per chamber, dimensions 12 in.×4.5 in. ×4.5 in.) under normoxic conditions (FIO2=0.21). 3×wAIH consisted of 10, 5-min episodes of hypoxia (FIO2=0.105), followed by 5 min normoxic intervals by switching the input gas between O2/N2 mixtures (FIO2=0.105) and medical grade air (FIO2=0.21) 10 times, 3 days per week for 10 weeks. Sham rats were in the chambers for an equivalent period of time, but did not receive hypoxia at any time. Chamber oxygen levels were continuously monitored (AX300-1, Teledyne Analytical Instruments, City of Industry, CA). Both 3×wAIH and normoxia-treated rats rested quietly or slept during exposure periods. All procedures in the present study were carried out in accordance with the National Institute of Health (NIH) guidelines for care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee at University of Wisconsin, Madison.
Immunohistochemistry and immunofluorescence
Immunohistochemistry
All rats were euthanized after treatment and perfused transcardially with ice-cold 0.01 M buffered saline (PBS, pH 7.4) followed by 4% buffered paraformaldehyde. Transverse sections of C4–C5 ventral horn (i.e. the region containing most phrenic motor neurons; Boulenguez et al., 2007; Goshgarian and Rafols, 1981; Mantilla et al., 2009) were processed for immunohistochemistry. The cervical spinal cords were excised, post-fixed overnight, and cryoprotected in 30% sucrose at 4 °C until they sank. Transverse sections (40 μm) were cut using a freezing microtome (Leica SM 200R, Germany). Free-floating sections were washed with 0.1 M Tris-buffered saline with 0.1% Triton-X100 (TBS– Tx; 3×5 min) and incubated in TBS containing 1% H2O2 for 30min. After washing (3×5 min) in TBS–Tx, tissues were blocked with 5% normal goat serum or 5% normal donkey serum at room temperature (RT) for 60 min. Staining was performed by incubating tissue sections with anti 5-HT2A (1/1000, rabbit serum, courtesy of Dr. M. Brownfield, UW-Madison), anti-BDNF (N-20; 1/1,000 Santa Cruz Biotechnology, Santa Cruz, CA), anti-VEGF (A-20; 1/1000 Santa Cruz Biotechnology, Santa Cruz, CA), anti-VEGFR-2 or KDR (Kinase insert Domain Receptor) (V3003; 1/200, Sigma Aldrich), anti-TrkB (1/500, rabbit polyclonal, Santa Cruz Biotechnology, Santa Cruz, CA), or anti-phospho ERK (1/500 rabbit polyclonal; Cell Signaling Technology, Danvers, MA) for 4 °C overnight. The sections were washed and incubated in biotinylated secondary goat anti-rabbit antibody (1:1000, Vector Laboratories, Burlingame, CA). Conjugation with avidin–biotin complex (Vecstatin Elite ABC kit, Vector Laboratories, Burlingame, CA) was followed by visualization with 3,3′-diaminobenzidine-hydrogen peroxidase (Vector Laboratories) according to the manufacturer’s instructions. Sections were then washed in TBS, placed on gelatin-coated slides, dried at RT, dehydrated in a graded alcohol series, and then cleared with xylenes and mounted with Eukitt mounting medium (Electron Microscope Sciences, Hatfield, PA).
All images were captured and analyzed with a digital camera (SPOT II; Diagnostic Instruments, Sterling Heights, MI). Final photomicrographs were created with Adobe Photoshop software (Adobe Systems, San Jose, CA) and all images received equivalent adjustments to tone, scale, gamma and sharpness. Putative phrenic motor neurons were identified based on size (>30 μm) and location within the region of the phrenic motor nucleus in the C4 and C5 ventral horns (Boulenguez et al., 2007; Goshgarian and Rafols, 1981; Mantilla et al., 2009). Sections incubated without primary or secondary antibodies served as negative controls. In addition we preabsorbed the primary BDNF and VEGF antibodies with five fold (by concentration) excess of specific blocking peptides (sc-546 P and sc- 152 P; both from Santa Cruz Biotechnology).
Immunofluorescence
To localize 5-HT, BDNF, VEGF, VEGF-R2, phospho-ERK, Akt and phospho-Akt expression in different cell types or in the cellular compartment of presumptive phrenic motor neurons, tissue sections were incubated at 4 °C overnight with anti-5-HT (1/500, rabbit polyclonal; courtesy of Dr. M. Brownfield, UW-Madison); anti-BDNF (N-20; 1/500, rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA); anti-VEGF (A-20; 1/200, rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA); anti-VEGF (A-20; 1/200, rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA); anti-VEGFR-2 (V3003; 1/200; Sigma Aldrich); anti-phospho ERK (1/200, rabbit polyclonal; Cell Signaling Technology, Danvers, MA); anti-Akt or anti-phospho Akt (1/200, rabbit polyclonal; Cell Signaling Technology, Danvers MA), as well as secondary antibodies targeting markers for neurons (NeuN; 1:500; Chemicon, Temecula, CA), astrocytes (GFAP; 1:1000; Chemicon, Temecula, CA), microglia (OX-42; 1:500, Serotec, Oxford, UK) or synaptic membranes (synaptophysin; 1/200; Neuromic, Edina, MN). After washing with TBS–Tx (3×5 min), tissues were incubated in a mixture of conjugated goat anti-rabbit or donkey anti-goat red fluorescent Alexa 495 and conjugated goat anti-mouse green fluorescent Alexa 488 (1:200, Molecular Probes, Eugene, Oregon) at room temperature for 60min. Stained tissues were mounted in coated glass using anti-fade solution (Prolong Gold anti-fade reagent, Invitrogen, Oregon) and examined using an epifluorescent microscope (Nikon, Japan).
TUNEL assay
In situ detection of apoptotic cells was performed via terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) using the In Situ Cell Death Detection Kit (Roche Molecular Biochemical, Indianapolis, IN) per the manufacturer’s instructions. Briefly, hippocampal sections were permeabilized with 1% Proteinase K (in 50 mM Tris/5 mM EDTA buffer) for 15 min, rinsed with PBS and incubated in the TUNEL reaction mixture for 1 h at 37 °C. Samples were rinsed, mounted in anti-fade solution (Molecular Probes) and analyzed with the fluorescence microscope. Apoptotic cells appeared as red stained cells. TUNEL (Texas Red) stained tissues were subjected to NeuN (green) staining procedures to identify apoptotic hippocampal neurons. Cresyl-violet staining was performed to identify cell death in tissues from the CA1 hippocampal sub-field.
Quantification and statistical analysis
Sections were numbered sequentially, and every 8th section was selected for immunohistochemistry. Thus, ~six sections from each animal at each segmental level were used in this study. The phrenic motor nucleus was identified as a cluster of large, NeuN positive cells in the medio-lateral C4 ventral horn (Boulenguez et al., 2007; Goshgarian and Rafols, 1981; Mantilla et al., 2009). Digital photomicrographs of immunoreactive labeling in the region of phrenic motor neurons were taken (SPOT II; Diagnostic Instruments, Sterling Heights, MI) using a fluorescence microscope (Nikon Eclipse, Japan). Densitometry was performed in a defined, square “area of interest” circumscribing the nucleus (200 μm×200 μm). The number of serotonergic (5-HT) boutons was counted automatically and the intensity of 5-HT2A, BDNF, TrkB, phospho-Akt and phospho-ERK immunostaining was quantified using NIH J Image software (National Institute of Health, Bethesda, MD; http://rsb.info.nih.gov/ij). Data were compared between the 3×wAIH and normoxia treated groups using t-tests. Differences were considered significant if p<0.05. All values are expressed as means±1 SEM.
Results
3×wAIH increases 5-HT terminal density and 5-HT2A receptor expression
NeuN immunofluorescence reveals large neurons in the C4 ventral horn, including presumptive phrenic motor neurons; the region of the phrenic motor nucleus is indicated in Fig. 1A by a white, dashed circle. There was no evidence of cell injury or death following 3×wAIH in the phrenic motor nucleus (Fig. 1D).
Fig. 1.

3×wAIH increases serotonergic (5-HT) terminal density (A–F; red) and 5-HT2A receptor (G–J) immunostaining (brown) in presumptive phrenic motor nucleus in C4 ventral horn (indicated by dotted circle in A–F; solid square in G, I). Presumptive phrenic motor neurons are large, NeuN positive cells (large green cells) in the region of phrenic motor nucleus. The number of serotonergic boutons (K) and 5-HT2A receptor (L) density is significantly increased versus normoxic control rats. Boxes indicate the region surrounding phrenic motor neurons where count/densitometry was performed. Scale bar is 200 μm for lower magnification and 100 μm for higher magnification images (small, white boxes in right hand panels). Data are means±1 SEM. *p<0.001 versus normoxia.
5-HT immunoreactivity in normoxic control rats is shown in Fig. 1B; 5-HT immunostaining revealed nerve fibers with varicosities (i.e. terminals; Fig. 1C, small boxes). 3×wAIH increased the density of serotonergic terminals near phrenic motor neurons (Fig. 1E), expressed as a significant increase in the number of varicosities in the area of interest (3×wAIH: 10,000±1564 boutons; vs. control: 6392±1265; p<0.01, n=5; Figs. 1F–K).
Basal 5-HT2A receptor staining is shown in presumptive phrenic motor neurons in Figs. 1G–H. 3×wAIH increased 5-HT2A receptor immunoreactivity versus normoxic control rats (Figs. 1I–J). Densitometry confirmed that 3×wAIH increased 5-HT2A receptor expression 2.2±0.3 fold vs. controls (p <0.001; n=5; Fig. 1L).
3×wAIH increases BDNF and TrkB immunoreactivity
BDNF expression in the ventral cervical spinal cord is low in control rats (Fig. 2A). 3×wAIH increased BDNF expression in presumptive phrenic motor neurons (Fig. 2B). Densitometry confirmed that 3×wAIH increased BDNF expression in phrenic motor neurons by 1.93±0.28 fold (Fig. 2C; p<0.01; n=5). Immunofluorescence revealed BDNF co-localization with NeuN, and that 3×wAIH enhances BDNF immunoreactivity (Figs. 2G–I) vs. normoxia (Figs. 2D–F).
Fig. 2.

3×wAIH upregulates BDNF and TrkB immunostaining in C4 ventral horn. BDNF immunostaining increases in the phrenic motor nucleus after 3×wAIH (B) versus normoxic controls (A). Boxes indicate the region surrounding phrenic motor neurons in which densitometry was performed. Measurement of optical density (OD) confirmed a significant increase in BDNF immunoreactivity following repetitive AIH (C). Immunofluorescence staining confirmed that BDNF is localized in motor neurons (large NeuN-positive cells in dashed circle), and its immunoreactivity is enhanced following 3×wAIH (D–I). Higher-magnification images from this region are in the bottom right corner. TrkB immunoreactivity (brown color) was Nissl counterstained (blue) to determine expression in presumptive phrenic motor neurons (J–K). Note that 3×wAIH (K) significantly increased TrkB immunostaining versus normoxic controls (J), as confirmed by densitometry (L). Scale bar for A–I is 200 μm; J–K and small boxes are 100 μm. Data are means±1 SEM, *p<0.01; **p<0.001 versus normoxia.
In normoxic rats, TrkB was expressed in presumptive phrenic motor neurons (Fig. 2J), consistent with previous reports of TrkB on the surface of motor neuron cell bodies, axons and dendrites (Yan et al., 1997), including phrenic motor neurons (Golder et al., 2008). 3×wAIH increased TrkB receptor immunoreactivity in presumptive phrenic motor neurons, both in the somata and perikarya (Fig. 2K). Densitometry confirmed significantly increased TrkB receptor expression in motor neurons following 3×wAIH (2.4±0.3 fold; p<0.001; n=4, Fig. 2L).
3×wAIH increases VEGF and VEGFR-2 expression
VEGF protein is expressed in large cells in the region of the phrenic motor nucleus (Fig. 3A), confirming earlier reports in back-labeled phrenic motor neurons (Dale-Nagle et al., 2011). VEGF immunostaining was also apparent in smaller cells, possibly inter-neurons (Figs. 3A–B). After 3×wAIH, VEGF immunostaining was more robust in putative phrenic motor neurons (Fig. 3B). Densitometry confirmed increased VEGF expression in presumptive phrenic motor neurons after 3×wAIH vs. controls (2.9±0.2 vs. 1.0±0.3, respectively; p<0.05; n=5; Fig. 3C). VEGF immunofluorescence was co-localized with NeuN (Fig. 3D–E), but not with GFAP (Fig. 3F) or OX-42 (Fig. 3G), demonstrating that the VEGF isoform (VEGFA) is expressed in presumptive phrenic motor neurons and nearby interneurons.
Fig. 3.

Representative images of VEGF immunostaining in C4 ventral horn. VEGF was expressed in large, NeuN positive cells, including large, presumptive phrenic motor neurons (in black boxes) and interneurons. 3×wAIH significantly increased VEGF protein expression in presumptive phrenic motor neurons versus control, normoxic rats (A–B). Boxes indicate the region in which densitometry was performed; higher-magnification images from this region are in the bottom right corner. Optical density (OD) analysis confirmed significant increase in VEGF immunoreactivity following 3×wAIH (C). Data are means±1 SEM. *p<0.05 versus normoxic controls. Immunofluorescence of large, NeuN positive cells (green) confirmed that VEGF (red) is localized in presumptive phrenic motor neurons (in white circle). VEGF immunoreactivity is enhanced following 3×wAIH (D–E). VEGF was not detectable in GFAP positive astrocytes (F) or in OX-42 positive microglia (G), even after 3×wAIH. Scale bars: 200 μm for lower magnification (A–B, D–E) and 100 μm for higher magnification (small boxes in D–E); F–G is 50 μm.
VEGFR-2 was also expressed in large C4 ventral horn cells (Fig. 4A), and immunostaining was upregulated by 3×wAIH (Fig. 4B). Densitometry revealed increased VEGFR-2 expression versus controls in presumptive phrenic motor neurons (3.8±0.7 vs. 1.0±0.5, respectively; p<0.05; n=5; Fig. 4C). VEGFR-2 immunofluorescence was localized in neurons, including presumptive phrenic motor neurons (Fig. 4D), and 3×wAIH increased its expression (Fig. 4E). VEGFR-2 was not detectable in astrocytes (GFAP positive cells; Fig. 4F) or microglia (OX42 positive cells, Fig. 4G), even after 3×wAIH.
Fig. 4.

Representative images of VEGF receptor-2 (VEGFR-2) immunostaining in C4 ventral horn. VEGFR-2 is expressed in NeuN positive cells, including large, presumptive phrenic motor neurons (small black boxes). 3×wAIH significantly increased VEGFR-2 protein expression in presumptive phrenic motor neurons versus control, normoxic rats (A–B). Optical density (OD) analysis showed significant increase in VEGF-R2 immunoreactivity following 3×wAIH (C). Data are means±1 SEM. *p<0.05 versus normoxic controls. Immunofluorescence for large, NeuN positive cells confirmed that VEGFR-2 is localized in presumptive phrenic motor neurons (white circle). Further, VEGFR-2 immunoreactivity is enhanced following 3×wAIH (D–E). VEGF was not detectable in GFAP positive (green) astrocytes (F) or in OX-42 positive (green) microglia (G), even after 3×wAIH. Scale bars: 200 μm for lower magnification (A–B, D–E) and 100 μm for higher magnification (small boxes in D–E); F–G is 50 μm.
3×wAIH upregulates phosphorylated-ERK
ERK MAP kinases were activated by 3×wAIH in the region of the phrenic motor nucleus. In normoxic rats, p-ERK was only faintly expressed in presumptive phrenic motor neurons (Figs. 5A–B), but was significantly increased following 3×wAIH (Figs. 5C–D). p-ERK co-localized with synaptic or axon-like structures. Densitometry confirmed that 3×wAIH increased p-ERK expression in the phrenic motor nucleus (2.5±0.6 vs. 1.00±0.3, p<0.05; n=5; Fig. 5E).
Fig. 5.

Representative images of p-ERK staining in C4 ventral horn. p-ERK immunoreactivity (brown color) was Nissl counterstained (blue) to assess localization in putative phrenic motor neurons (A–D). 3×wAIH (C–D) significantly increased p-ERK immunoreactivity versus normoxic controls (A–B) as confirmed by optical density (OD) analysis (E). Data are means±1 SEM. *p<0.05 versus normoxia. Higher-magnification images of those cells are in the bottom corner. Immunofluorescence staining showed that p-ERK was found on the membranes of presumptive phrenic motor neurons (arrow head) (F–K) and is colocalized with synaptophysin surrounding the vacant motor neuron somata (stars) (L–Q). Scale bar for A, C is 200 μm; B, D, F–Q is 100 μm.
In control rats, p-ERK was minimally expressed in the soma of large NeuN positive cells of the phrenic motor nucleus, but was absent from their nuclei (Figs. 5F–H). Most p-ERK immunostaining was found in the outer layer of the neuronal membrane (small panel, Fig. 5H). p-ERK increased substantially in the phrenic motor nucleus following 3×wAIH (Figs. 5I–K). This p-ERK immunostaining is characterized by varicosities or synapse-like immunostaining on the surface of motor neurons. However, there was no co-localization of p-ERK with NeuN (small panel, Fig. 5K), suggesting that p-ERK is not located in the nucleus or cytoplasm. On the other hand, p-ERK was co-localized with synaptophysin surrounding presumptive phrenic motor neurons (Figs. 5L–N). p-ERK (colocalized with synaptophysin) increased following 3×wAIH (Figs. 5O–Q).
3×wAIH upregulates Akt and phospho-Akt expression
Akt is predominantly expressed in the cytosol of large ventral horn cells (Figs. 6A–C). Following 3×wAIH, Akt expression is upregulated vs. controls, predominantly in the cell nucleus (small panel, Figs. 6D–F). 3×wAIH also increased cytoplasmic p-AKT expression in presumptive phrenic motor neurons (NeuN positive; Figs. 6J–L) vs. normoxic controls (Figs. 6G–I). Phosphorylated-AKT was not co-localized with synaptophysin in controls (Figs. 6M–O) or following 3×wAIH (Figs. 6P–R).
Fig. 6.

3×wAIH induced Akt (A–F) and p-AKT (G–L) protein expression in presumptive phrenic motor neurons (D–F). Phosphorylated Akt (p-Akt) was dominantly expressed in cytoplasm (small panel, L), but not in neuronal membranes (synaptophysin) of presumptive phrenic motor neurons (M–R). Scale bar is 100 μm.
3×wAIH does not induce hippocampal gliosis, cell death or apoptosis
We found no evidence that 3×wAIH elicits glial activation and/or hippocampal cell death. Hallmarks of glial activation are cellular hypertrophy and increased expression of GFAP (astrocytes) or OX-42 (microglia/macrophages). There were no obvious changes in the morphology of astrocytes or microglia in the hippocampal CA1 subfield following 3×wAIH (Fig. 7). Neither the number of GFAP-positive cells (398±47 vs. 405±38, p=0.84, n=4; Fig. 7C) nor OX42-positive cells (129±32 vs. 132±49, p=0.76, n=4; Fig. 7F) were affected by 3×wAIH. Further, there was no evidence for hippocampal cell death (Figs. 7G–H) or the onset of apoptosis (Figs. 7I–J) after 3×wAIH. Thus, we found no evidence that 3×wAIH causes non-specific brain pathology, similar to our recent report following daily AIH for 7 consecutive days (Lovett-Barr et al., 2012).
Fig. 7.

3×wAIH does not induce gliosis or neuronal death in the CA1 hippocampal sub-field. There was no evidence of reactive astrocytes (A–B) or activated microglia (D–E). Cell counts reveal that the densities of GFAP-positive astrocytes (C) and OX-42 positive microglia (F) were unaffected by 3×wAIH (each n=5). There was no obvious increase of the intensity of either cell marker (A vs. B; D vs. E). Furthermore, there was no evidence that indicates 3×wAIH causes hippocampal cell death (G–H) or apoptosis (I–J) based on cresyl-violet and TUNEL staining, respectively. J′ is a positive control for TUNEL staining taken from the cortex after middle cerebral artery occlusion in a rat. Scale bar for A, B, D, E, 50 μm; G–H, 500 μm; I–J, 100 μm.
Discussion
The concept of plasticity of neuronal properties such as size and shape of somata, density of dendritic spines, formation of new synapses and neurochemical expression has been extensively discussed (Goldin and Segal, 2003; Modney and Hatton, 2006), including altered properties in respiratory neurons (Mitchell and Johnson, 2003). Here, we investigated neurochemical plasticity of respiratory (phrenic) motor neurons caused by repetitive exposure to acute intermittent hypoxia (3×wAIH). We hypothesized that 3×wAIH for 10 weeks would amplify expression of molecules associated with functional plasticity of phrenic motor neurons including AIH-induced pLTF (Feldman et al., 2003; Mitchell and Terada, 2011; Mitchell et al., 2001), as well as adenosine 2A receptor-induced (Golder et al., 2008; Nichols et al., 2012) and VEGF-induced phrenic motor facilitation (Dale-Nagle et al., 2011).
Our emerging understanding of cellular mechanisms giving rise to pLTF (Fig. 8) implicates serotonin (Bach and Mitchell, 1996; Baker-Herman and Mitchell, 2002), 5-HT2 receptors (Fuller et al., 2001; MacFarlane et al., 2011), new synthesis of BDNF and activation of TrkB (Baker-Herman et al., 2004), and downstream signaling via ERK MAP kinases (Hoffman and Mitchell, unpublished; Wilkerson and Mitchell, 2009). Other forms of spinal respiratory motor plasticity linked to hypoxia (Dale-Nagle et al., 2010) include: 1) adenosine receptor induced TrkB transactivation with downstream signaling via Akt (i.e. the S pathway; Dale-Nagle et al., 2010; Golder et al., 2008; Hoffman and Mitchell, 2011; Nichols et al., 2012), and 2) VEGF induced phrenic motor facilitation via ERK and Akt activation (Dale-Nagle et al., 2011). Here we demonstrate that molecules critical for all of these pathways are enhanced by a very modest protocol of repetitive acute intermittent hypoxia, namely 3×wAIH.
Fig. 8.
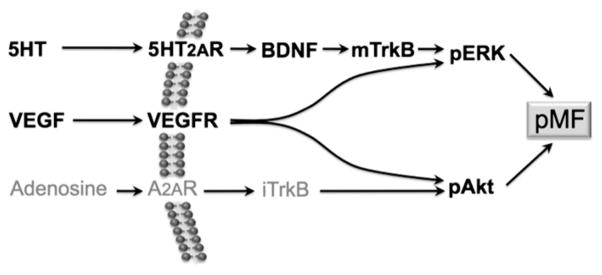
Working model of signaling pathways to hypoxia-induced phrenic motor plasticity. Unique pathways are elicited by 5-HT2A receptors (i.e. the Q pathway; Dale-Nagle et al., 2010), VEGF receptors, and A2A receptors (i.e. the S pathway). These downstream signaling molecules have the potential to play critical roles in enhanced pMF following repetitive AIH. Results from this study indicate that all of these respective molecules are upregulated by 3×wAIH, consistent with the greater capacity for functional plasticity.
Since 3×wAIH enhances the expression of growth/trophic factors within the phrenic motor nucleus (e.g. BDNF and VEGF), as well as their downstream signaling cascades, it may represent a viable therapeutic strategy to initiate preconditioning with a protocol of modest severity, or to restore breathing capacity in patients with respiratory insufficiency (Lovett-Barr et al., 2012; Mitchell, 2007). A key to this therapeutic strategy is the observation that 3×wAIH augments pro-plasticity molecules without evidence for non-specific brain pathology.
Serotonin and serotonin receptors
Millhorn et al. (1980) were the first to demonstrate that pLTF elicited by episodic activation of carotid chemo-afferent neurons is serotonin dependent. Similarly, episodic spinal 5-HT2 receptor activation is both necessary and sufficient for AIH-induced pLTF in rats (Bach and Mitchell, 1996; Baker-Herman and Mitchell, 2002; Fuller et al., 2001; Kinkead and Mitchell, 1999; Lovett-Barr et al., 2006; MacFarlane and Mitchell, 2009; MacFarlane et al., 2011). Meta-plasticity of pLTF is associated with increased serotonin terminal density in the phrenic motor nucleus (Kinkead et al., 1998) and serotonin terminal density below a cervical spinal injury correlates with the pLTF magnitude (Golder and Mitchell, 2005). Here, we demonstrate that 3×wAIH increased the serotonergic terminal density near phrenic motor neurons, and 5-HT2A receptor expression in presumptive phrenic motor neurons. These alterations may confer greater capacity for serotonin-dependent plasticity in phrenic motor neurons.
Altered serotonin receptor expression has been documented in piglet brainstems following intermittent hypercapnia/hypoxia (Say et al., 2007). However, these authors reported reduced 5- HT1A receptor expression without detectable effect on 5-HT2A receptors. Another study of chronic intermittent hypoxia found no change in 5-HT2A–2C binding density in the hypoglossal motor nucleus, or 5-HT2A and 5-HT2C receptor mRNA expression in individual hypoglossal motor neurons (Veasey et al., 2004). This apparent discrepancy with our results may be explained by differences in the respiratory motor nucleus studied (hypoglossal vs. phrenic), age (infant vs. adult), species (piglet vs. rats), or variations in the intermittent hypoxia protocol (e.g. prolonged daily exposures for 12 days vs. brief exposures 3 times per week for 10 weeks; or intermittent hypercapnic hypoxia vs. intermittent poikilocapnic hypoxia).
Alterations in serotonin-dependent respiratory plasticity occur after spinal cord injury. For example, C2 spinal hemisection increases 5-HT2A receptor expression in phrenic motor neurons, perhaps contributing to a strengthened crossed-spinal synaptic pathway to phrenic motor neurons (Fuller et al., 2005). After low-thoracic transection, 5-HT1A/7 and 5- HT2A receptor expressions are both increased in lumbar spinal segments (Giroux et al., 1999; Ung et al., 2005). Another example of plasticity in the serotonergic system that correlates with functional phrenic motor plasticity is the dramatic increase in serotonin terminal density near phrenic motor neurons, accompanied by increased serotonin-dependent pLTF after chronic cervical dorsal rhizotomy (Kinkead et al., 1998). Thus, serotonergic neuron plasticity may contribute to functional plasticity in respiratory (phrenic) motor output following 3×wAIH.
Although our observations are limited to serotonin terminal density and 5-HT2A receptors, we cannot rule out changes in other 5-HT receptor subtypes following 3×wAIH. For example, McGuire et al. (2004) demonstrated that enhanced ventilatory LTF following chronic intermittent hypoxia requires an additional serotonin receptor subtype, most likely 5-HT7 receptors. Possible alterations in other serotonin receptor subtypes following 3×wAIH requires further investigation.
BDNF and TrkB
We recently demonstrated that daily AIH for one week upregulates BDNF and TrkB expressions in the phrenic motor nucleus (Lovett-Barr et al., 2012; Wilkerson and Mitchell, 2009). Similarly, 3×wAIH increases BDNF expression in the phrenic motor nucleus, including the somata of presumptive phrenic motor neurons. BDNF induction following 3×wAIH is associated with TrkB up regulation, suggesting that repetitive AIH may represent a novel and effective means of endogenous BDNF delivery that avoids the difficulties attendant to protein delivery to the CNS (e.g. access across the blood brain barrier, receptor down-regulation, immune responses triggered by protein delivery; Banks and Erickson, 2010; Tóth et al., 2011). As such, repetitive AIH may be an effective treatment for certain forms of injury and/or disease (Mitchell, 2007), including spinal injury (Lovett-Barr et al., 2012).
The association between increased BDNF and TrkB expression is not surprising since TrkB is regulated by the hypoxia-sensitive transcription factor, HIF-1α (Martens et al., 2007). Previous reports demonstrate a requirement for TrkB signaling for 5-HT-induced ERK activation, and for the induction of sensory motor long-term facilitation in Aplysia (Sharma et al., 2006). Collectively, available evidence is consistent with a major role for BDNF and TrkB in respiratory plasticity and meta-plasticity following repetitive AIH.
VEGF and VEGF receptor 2
Hypoxia activates other cellular mechanisms, including increased expression of genes regulated by the transcription factor hypoxia-inducible factor (HIF-1α; Semenza, 2006; Yuan et al., 2011). One example, known to elicit phrenic motor facilitation, is VEGF (Dale-Nagle et al., 2011). Although VEGF was originally thought of as an angiogenic growth factor, it also promotes neurogenesis, neuroprotection and motor plasticity (Chen et al., 2005; Dale-Nagle et al., 2011; Wang et al., 2005). Here we report that 3×wAIH upregulates VEGF and its high affinity receptor, VEGFR-2, in presumptive phrenic motor neurons. The functions of these molecules following 3×wAIH are not yet clear, but they converge on the same, downstream signaling molecules as BDNF/TrkB, including ERK and Akt (Dale-Nagle et al., 2011). Thus, interesting and complex interactions between the BDNF/TrkB and VEGF/VEGFR-2 systems may be expected.
ERK and phosphorylated ERK
ERK MAP kinases play critical roles in the regulation of cell growth and differentiation (Kelly-Spratt et al., 1999; Whittard et al., 2006), as well as neuronal survival and plasticity (Colucci-D’Amato et al., 2003). For example, phosphorylation (and activation) of ERKs is required for hippocampal long-term potentiation (Jones, et al., 1999; Schafe et al., 2008; Xin et al., 2006).
Available evidence suggests a prominent role for ERKs in respiratory plasticity. For example, spinal ERK/MAP kinase activation is necessary for full expression of phrenic motor facilitation elicited by spinal VEGF (Dale-Nagle et al., 2011). ERK activation is also critical for AIH-induced pLTF since: 1) AIH increases phospho-ERK levels near the phrenic motor nucleus (Wilkerson and Mitchell, 2009); and 2) spinal MEK/ERK inhibition blocks AIH-induced pTLF (Hoffman and Mitchell, unpublished).
Hypoxia increases ERK/MAP kinase activity (Conrad et al., 1999; Muller et al., 1997; Wilkerson and Mitchell, 2009). Here, we confirm that 3×wAIH increases phospho-ERK expression in the phrenic motor nucleus; increased expression was observed exclusively in regions immediately surrounding motor neurons (Fig. 5).
Phosphorylated ERK was preferentially increased in the synaptic membrane of presumptive phrenic motor neurons, as revealed by its co-localization with synaptophysin (Fig. 5L–Q). Although pre- versus post-synaptic localization cannot be determined with these methods, this observation with synaptophysin is consistent with the hypothesis that ERK strengthens synaptic inputs following 3×wAIH. As yet, there is no direct evidence to support this hypothesis and additional functional studies are needed.
Akt and phosphorylated Akt
Both Akt and phosphorylated Akt expressions were increased in presumptive phrenic motor neurons following 3×wAIH. However, the distribution of phosphorylated Akt differs strikingly from phosphorylated ERK (Figs. 5 and 6). In contrast to the largely synaptic localization of phospho-ERK following 3×wAIH, phospho-Akt was largely cytosolic/nuclear. The significance of this difference in distribution following 3×wAIH is unknown, but may have important implications concerning the respective roles played by each kinase in respiratory plasticity. The cytosolic/nuclear localization of Akt suggests greater involvement in mechanisms that require nuclear transcriptional activation, whereas the synaptic localization of ERK is consistent with a greater role in synapse-specific mechanisms of plasticity. Thus, these kinases may differ in important respects, such as the time-domain of their respective actions.
Akt regulates cell survival (Hutchinson et al., 2001) and plasticity (Horwood et al., 2006). In its inactive state, Akt is located in the cytosol; however, Akt phosphorylation and activation occur after translocation to the lipid bilayer (Alessi et al., 1996). Activated Akt can be found in the cell nucleus, although its function there is poorly understood. However, once hypoxia up-regulates HIF-1α protein levels, its transcriptional activity is regulated by various growth factors via PI3K/Akt activity (Mazure et al., 1997; Stiehl et al., 2002; Zhong et al., 2000). Thus, Akt-dependent regulation of HIF-1α activity may play a role in the expression of growth factors such as VEGF that are involved in respiratory plasticity.
Significance
Here, we provide evidence that molecules known to play key roles in respiratory plasticity are up-regulated within the phrenic motor nucleus and in presumptive phrenic motor neurons following 3×wAIH, a very modest protocol of repetitive AIH. Specific changes associated with AIH-induced pLTF (i.e. the “Q pathway;” Dale-Nagle et al., 2010) include greater serotonin terminal density, and 5-HT2A receptor, BDNF, TrkB and phospho-ERK expression. We anticipate interesting changes in the expression of other proteins known to play key roles in AIH-induced respiratory plasticity, such as protein kinase C (Devinney and Mitchell, unpublished), serine/threonine protein phosphatases (Wilkerson et al., 2008) and NADPH oxidase (MacFarlane et al., 2009).
Equivalent changes in other signaling cascades leading to phrenic motor facilitation (Dale-Nagle et al., 2010, 2011) include VEGF/VEGFR-2 (ERK and Akt) and the “S pathway” (TrkB/Akt) to phrenic motor facilitation. Although the neurochemical plasticity reported here does not constitute conclusive evidence that any specific change contributes to functional plasticity, they lay a foundation for future studies.
An interesting implication of our findings is that repetitive exposure to “low doses” of intermittent hypoxia may represent an effective means of endogenous growth factor delivery (e.g. BDNF and VEGF). Such growth factor “delivery” may be beneficial in promoting neurogenesis (Gozal et al., 2003), cell survival and/or plasticity as a therapeutic approach to neural injury or disease. Previous attempts to harness growth/trophic factors via exogenous delivery have had poor success (Beck et al., 2005; Borasio et al., 1998; Cedarbaum et al., 1999; Miller et al., 1996; Nefussy et al., 2010), most likely due to complications with protein delivery across the blood brain barrier, immune responses to exogenous proteins and/or down-regulation of relevant receptors. Repetitive acute intermittent hypoxia produces these proteins on site, and is associated with receptor up-regulation, suggesting that this approach has considerable advantages. For example, repetitive acute intermittent hypoxia has been “harnessed” therapeutically in a rodent model of cervical spinal injury; specifically, daily AIH (7 days) restores ventilatory capacity after a cervical spinal hemisection (Lovett-Barr et al., 2012).
One hazard of intermittent hypoxia is the risk of increased blood pressure, CNS inflammation, hippocampal cell death and cognitive impairment (Gozal et al., 2001; Li et al., 2003; Tamisier et al., 2011; Xu et al., 2004; Zoccal et al., 2007) following severe protocols of chronic intermittent hypoxia. However, repetitive AIH is very modest in comparison, and may enable us to capture the benefits without undesirable side effects. Indeed, daily AIH elicits spinal plasticity without evidence for hypertension (Wilkerson and Mitchell, 2009) or hippocampal pathology (Lovett-Barr et al., 2012). We now extend these findings to an even less severe (but prolonged) presentation of intermittent hypoxia, 3×wAIH, which elicits more robust neurochemical changes without evidence of hippocampal pathology. Thus, 3×wAIH may be a useful protocol to treat respiratory insufficiency during lung (e.g. obstructive lung disease), upper airway (e.g. sleep apnea) or neural disorders (e.g. spinal injury or motor neuron disease; Mitchell, 2007).
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL080209, HL69064, HL007654 and NS057778). The authors thank Daniel Harrigan for technical assistance.
References
- Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Banks WA, Erickson MA. The blood–brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37:26–32. doi: 10.1016/j.nbd.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Beck M, Flachenecker P, Magnus T, Giess R, Reiners K, Toyka KV, Naumann M. Autonomic dysfunction in ALS: a preliminary study on the effects of intrathechal BDNF. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:100–103. doi: 10.1080/14660820510028412. [DOI] [PubMed] [Google Scholar]
- Borasio GD, Robberecht W, Leigh PN, Emile J, Giloff RJ, Jerusalem F, Silani V, Vos PE, Wokke JH, Dobbins T. A placebo-controlled trial of insulin-growth factor-1 in amyotrophic lateral sclerosis. European ALS/IGF-1 study group. Neurology. 1998;51:583–586. doi: 10.1212/wnl.51.2.583. [DOI] [PubMed] [Google Scholar]
- Boulenguez P, Gestreau C, Vinit S, Stamegna JC, Kastner A, Gauthier P. Specific and artifactual labeling in the rat spinal cord and medulla after injection of monosynaptic retrograde tracers into the diaphragm. Neurosci Lett. 2007;417 (2):206–211. doi: 10.1016/j.neulet.2007.02.047. [DOI] [PubMed] [Google Scholar]
- Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, Nakanishi A. The ALSFRS_R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III) J Neurol Sci. 1999;169:13–21. doi: 10.1016/s0022-510x(99)00210-5. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang C, Jiang H, Li Y, Zhang L, Robin A, Katakowski M, Lu M, Chopp M. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cereb Blood Flow Metab. 2005;25:281–290. doi: 10.1038/sj.jcbfm.9600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci-D’Amato L, Perrone-Capano C, di Porzio U. Chronic activation of ERK and neurodegenerative diseases. Bioessays. 2003;25:1085–1095. doi: 10.1002/bies.10355. [DOI] [PubMed] [Google Scholar]
- Conrad PW, Rust RT, Han J, Millhorn DE, Beitner-Johnson D. Selective activation of p38alpha and p38gamma by hypoxia. J Biol Chem. 1999;274:23570–23576. doi: 10.1074/jbc.274.33.23570. [DOI] [PubMed] [Google Scholar]
- Dale-Nagle EA, Hoffman MS, MacFarlane PM, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv Exp Med Biol. 2010;669:225–230. doi: 10.1007/978-1-4419-5692-7_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale-Nagle EA, Satriotomo I, Mitchell GS. Spinal vascular endothelial growth factor induces phrenic motor facilitation via extracellular signal-regulated kinase and Akt signaling. J Neurosci. 2011;31:7682–7690. doi: 10.1523/JNEUROSCI.0239-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol. 2001;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Johnson SM, Olson EB, Jr, Mitchell GS. Synaptic pathways to phrenic motoneurons are enhanced by chronic intermittent hypoxia after cervical spinal cord injury. J Neurosci. 2003;23:2993–3000. doi: 10.1523/JNEUROSCI.23-07-02993.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Baker-Herman TL, Golder FJ, Doperalski NJ, Watters JJ, Mitchell GS. Cervical spinal cord injury upregulates ventral spinal 5-HT2A receptors. J Neurotrauma. 2005;22:203–213. doi: 10.1089/neu.2005.22.203. [DOI] [PubMed] [Google Scholar]
- Giroux N, Rossignol S, Reader TA. Autoradiographic study of alpha1- and alpha2-noradrenergic and serotonin1A receptors in the spinal cord of normal and chronically transected cats. J Comp Neurol. 1999;406:402–414. [PubMed] [Google Scholar]
- Golder FJ, Mitchell GS. Spinal synaptic enhancement with acute intermittent hypoxia improves respiratory function after chronic cervical spinal cord injury. J Neurosci. 2005;25:2925–2932. doi: 10.1523/JNEUROSCI.0148-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, Baker-Herman TL, Mitchell GS. Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J Neurosci. 2008;28:2033–2042. doi: 10.1523/JNEUROSCI.3570-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin M, Segal M. Protein kinase C and ERK involvement in dendritic spine plasticity in cultured rodent hippocampal neurons. Eur J Neurosci. 2003;17:2529–2539. doi: 10.1046/j.1460-9568.2003.02694.x. [DOI] [PubMed] [Google Scholar]
- Goshgarian HG, Rafols JA. The phrenic nucleus of the albino rat: a correlative HRP and Golgi study. J Comp Neurol. 1981;201:441–456. doi: 10.1002/cne.902010309. [DOI] [PubMed] [Google Scholar]
- Gozal D, Daniel JM, Dohanich GP. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci. 2001;21:2442–2450. doi: 10.1523/JNEUROSCI.21-07-02442.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, Row BW, Gozal E, Kheirandish L, Neville JJ, Brittian KR, Sachleben LR, Jr, Guo SZ. Temporal aspects of spatial task performance during intermittent hypoxia in the rat: evidence for neurogenesis. Eur J Neurosci. 2003;18:2335–2342. doi: 10.1046/j.1460-9568.2003.02947.x. [DOI] [PubMed] [Google Scholar]
- Hoffman MS, Mitchell GS. Spinal 5-HT7 receptor activation induces long-lasting phrenic motor facilitation. J Physiol. 2011;589:1397–1407. doi: 10.1113/jphysiol.2010.201657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwood JM, Dufour F, Laroche S, Davis S. Signaling mechanisms mediated by phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur J Neurosci. 2006;23:3375–3384. doi: 10.1111/j.1460-9568.2006.04859.x. [DOI] [PubMed] [Google Scholar]
- Hutchinson J, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol Cell Biol. 2001;21:2203–2212. doi: 10.1128/MCB.21.6.2203-2212.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MW, French PJ, Bliss TVP, Rosenblum K. Molecular mechanism of long term potentiation in the insular cortex in vivo. J Neurosci. 1999;19:1–8. doi: 10.1523/JNEUROSCI.19-21-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly-Spratt KS, Klesse LJ, Merenmies J, Parada LF. A TrkB/insulin receptor-related receptor chimeric receptor induces PC12 cell differentiation and exhibits prolonged activation of mitogen-activated protein kinase. Cell Growth Differ. 1999;10:805–812. [PubMed] [Google Scholar]
- Kinkead R, Mitchell GS. Time-dependent hypoxic ventilatory responses in rats: effects of ketanserin and 5-carboxamidotryptamine. Am J Physiol. 1999;277:R658–R666. doi: 10.1152/ajpregu.1999.277.3.R658. [DOI] [PubMed] [Google Scholar]
- Kinkead R, Zhan WZ, Prakash YS, Bach KB, Sieck GC, Mitchell GS. Cervical dorsal rhizotomy enhances serotonergic innervation of phrenic motoneurons and serotonin-dependent long-term facilitation of respiratory motor output in rats. J Neurosci. 1998;18:8436–8443. doi: 10.1523/JNEUROSCI.18-20-08436.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RC, Row BW, Gozal E, Kheirandish L, Fan Q, Brittian KR, et al. Cyclooxygenase 2 and intermittent hypoxia-induced spatial deficits in the rat. Am J Respir Crit Care Med. 2003;168:469–475. doi: 10.1164/rccm.200211-1264OC. [DOI] [PubMed] [Google Scholar]
- Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Jr, Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. J Neurosci. 2001;21:5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barr MR, Mitchell GS, Satriotomo I, Johnson SM. Serotonin-induced in vitro long-term facilitation exhibits differential pattern sensitivity in cervical and thoracic inspiratory motor output. Neuroscience. 2006;142:885–892. doi: 10.1016/j.neuroscience.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Lovett-Barr MR, Satriotomo I, Muir GD, Wilkerson JE, Hoffman MS, Vinit S, Mitchell GS. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic spinal injury. J Neurosci. 2012;32:3591–3600. doi: 10.1523/JNEUROSCI.2908-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J Physiol. 2009;587:5469–5481. doi: 10.1113/jphysiol.2009.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Wilkerson JE, Lovett-Barr MR, Mitchell GS. Reactive oxygen species and respiratory plasticity following intermittent hypoxia. Respir Physiol Neurobiol. 2008;587:1931–1942. doi: 10.1016/j.resp.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Satriotomo I, Windelborn JA, Mitchell GS. NADPH oxidase activity is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. J Physiol. 2009;164:263–271. doi: 10.1113/jphysiol.2008.165597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Vinit S, Mitchell GS. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neuroscience. 2011;178:45–55. doi: 10.1016/j.neuroscience.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol. 2007;92:27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods. 2009;182:244–249. doi: 10.1016/j.jneumeth.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens LK, Kirschner KM, Warnecke C, Scholz H. Hypoxia-inducible factor is a transcriptional activator of the TrkB neurotrophin receptor gene. J Biol Chem. 2007;282 (19):14379–14388. doi: 10.1074/jbc.M609857200. [DOI] [PubMed] [Google Scholar]
- Matieka JH, Sandhu KS. Experimental protocols and preparations to study respiratory long term facilitation. Respir Physiol Neurobiol. 2011;176:1–11. doi: 10.1016/j.resp.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazure NM, Chen EY, Laderoute KRs, Giaccia AJ. Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood. 1997;90:3322–3331. [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Chronic intermittent hypoxia enhances ventilatory long-term facilitation in awake rats. J Appl Physiol. 2003;95:1499–1508. doi: 10.1152/japplphysiol.00044.2003. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Serotonin receptor subtypes required for ventilatory long-term facilitation and its enhancement after chronic intermittent hypoxia in awake rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R334–R341. doi: 10.1152/ajpregu.00463.2003. [DOI] [PubMed] [Google Scholar]
- Miller RG, Bryan WW, Dietz MA, Munsat TL, Petajan JH, Smith SA, Goodpasture JC. Toxicity and tolerability of recombinant human ciliary neurotrophic factors in patients with amyotrophic lateral sclerosis. Neurology. 1996;47:1329–1331. doi: 10.1212/wnl.47.5.1329. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by endogenous central serotonin. Respir Physiol. 1980;42:171–198. doi: 10.1016/0034-5687(80)90113-9. [DOI] [PubMed] [Google Scholar]
- Mitchell GS. Respiratory plasticity following intermittent hypoxia: a guide for novel therapeutic approaches to ventilatory control disorders. In: Gaultier C, editor. Genetic Basis for Respiratory Control Disorders. Springer Publishing Company; New York: 2007. [Google Scholar]
- Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol. 2003;94:358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Terada J. Should we standardize protocols and preparations used to study respiratory plasticity? Respir Physiol Neurobiol. 2011;177:92–97. doi: 10.1016/j.resp.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Modney BK, Hatton GI. Multiple synapse formation: a possible compensatory mechanism for increased cell size in rat supraoptic nucleus. J Neuroendocrinol. 2006;1:21–27. doi: 10.1111/j.1365-2826.1989.tb00072.x. [DOI] [PubMed] [Google Scholar]
- Muller J, Krauss B, Kaltschmidt C, Baeuerle P, Rupec R. Hypoxia induces c-fos transcription via a mitogen-activated protein kinase-dependent pathway. J Biol Chem. 1997;272:23435–23439. doi: 10.1074/jbc.272.37.23435. [DOI] [PubMed] [Google Scholar]
- Nefussy B, Artamonov I, Deutsch V, Naparstek E, Nagler A, Drory VE. Recombinant human granulocyte-colony stimulating factor administration for treating amyotrophic lateral sclerosis: a pilot study. Amyotroph Lateral Scler. 2010;11:187–193. doi: 10.3109/17482960902933809. [DOI] [PubMed] [Google Scholar]
- Nichols NL, Dale EA, Mitchell GS. Severe acute intermittent hypoxia elicits phrenic long-term facilitation by a novel adenosine-dependent mechanism. J Appl Physiol. 2012;112:1678–1688. doi: 10.1152/japplphysiol.00060.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JC, D’Almeida V, Souza FG, Schoorlemmer GH, Colombari E, Tufik S. Consequences of subchronic and chronic exposure to intermittent hypoxia and sleep deprivation on cardiovascular risk factors in rats. Respir Physiol Neurobiol. 2007;156:250–258. doi: 10.1016/j.resp.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Say M, Machaalani R, Waters KA. Changes in serotoninergic receptors 1A and 2A in the piglet brainstem after intermittent hypercapnic hypoxia (IHH) and nicotine. Brain Res. 2007;1152:17–26. doi: 10.1016/j.brainres.2007.03.037. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Swank MW, Rodrigues SM, Debiec J, Doyère V. Phosphorylation of ERK/MAP kinase is required for long-term potentiation in anatomically restricted regions of the lateral amygdala in vivo. Learn Mem. 2008;15:55–62. doi: 10.1101/lm.746808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Regulation of physiological responses to continuous and intermittent hypoxia by hypoxia-inducible factor 1. Exp Physiol. 2006;91:803–806. doi: 10.1113/expphysiol.2006.033498. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Sherff CM, Stough S, Hsuan V, Carew TJ. A tropomyosin-related kinase B ligand is required for ERK activation, long-term synaptic facilitation, and long-term memory in Aplysia. Proc Natl Acad Sci U S A. 2006;103:14206–14210. doi: 10.1073/pnas.0603412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiehl DP, Jelkmann W, Wenger RH, Hellwig-Burgel T. Normoxic induction of the hypoxia-inducible factor 1α by insulin and interleukin-1β involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002;512:157–162. doi: 10.1016/s0014-5793(02)02247-0. [DOI] [PubMed] [Google Scholar]
- Tamisier R, Pépin JL, Rémy J, Baguet JP, Taylor JA, Weiss JW, Lévy P. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J. 2011;13:119–128. doi: 10.1183/09031936.00204209. [DOI] [PubMed] [Google Scholar]
- Tóth A, Veszelka S, Nakagawa S, Niwa M, Deli MA. Patented in vitro blood–brain barrier model in CNS drug discovery. Recent Pat CNS Drug Discov. 2011;6:107–118. doi: 10.2174/157488911795933910. [DOI] [PubMed] [Google Scholar]
- Trumbower RD, Jayaraman A, Mitchell GS, Rymer WZ. Exposure to acute intermittent hypoxia augments somatic motor function in human with incomplete spinal cord injury. Neurorehabil Neural Repair. 2012;26:163–172. doi: 10.1177/1545968311412055. [DOI] [PubMed] [Google Scholar]
- Ung RV, Landry ES, Lapointe N, Rouillard C, Levesque D, Guertin P. Expression, activation, and function of 5-HT2A receptor in the lumbar spinal cord of adult paraplegic mice. Soc Neurosci Abstr 516.7 2005 [Google Scholar]
- Veasey SC, Zhan G, Fenik P, Practico D. Long term intermittent hypoxia reduced excitatory hypoglossal nerve output. Am J Respir Crit Care Med. 2004;170:665–672. doi: 10.1164/rccm.200403-261OC. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kilic E, Kilic Ü, Weber B, Bassetti CL, Marti HH, Hermann DM. VEGF overexpression induces post-ischaemic neuroprotection, but facilitates haemodynamic steal phenomena. Brain. 2005;128:52–63. doi: 10.1093/brain/awh325. [DOI] [PubMed] [Google Scholar]
- Whittard JD, Sakurai T, Cassella MR, Gazdoiu M, Felsenfeld DP. MAP kinase pathway-dependent phosphorylation of the L1-CAM ankyrin-binding site regulates neuronal growth. Mol Biol Cell. 2006;17:2696–2706. doi: 10.1091/mbc.E06-01-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JE, Mitchell GS. Daily intermittent hypoxia augments spinal BDNF levels, ERK phosphorylation and respiratory long-term facilitation. Exp Neurol. 2009;217:116–123. doi: 10.1016/j.expneurol.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JE, Satriotomo I, Baker-Herman TL, Watters JJ, Mitchell GS. Okadaic acid-sensitive protein phosphatases constrain phrenic long-term facilitation after sustained hypoxia. J Neurosci. 2008;28:2949–2958. doi: 10.1523/JNEUROSCI.5539-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin WJ, Gong QJ, Xu JT, Yang HW, Zang Y, Zhang T, Li YY, Liu XG. Role of phosphorylation of ERK in induction and maintenance of LTP of the C-fiber evoked field potentials in spinal dorsal horn. J Neurosci Res. 2006;84:934–943. doi: 10.1002/jnr.21013. [DOI] [PubMed] [Google Scholar]
- Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B, et al. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126:313–323. doi: 10.1016/j.neuroscience.2004.03.055. [DOI] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997;378:135–157. [PubMed] [Google Scholar]
- Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol. 2011;226:2925–2933. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1 alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- Zoccal DB, Bonagamba LG, Antunes-Rodrigues J, Machado BH. Plasma corticosterone levels is elevated in rats submitted to chronic intermittent hypoxia. Auton Neurosci. 2007;134:115–117. doi: 10.1016/j.autneu.2007.01.004. [DOI] [PubMed] [Google Scholar]