

Summary
Fatty acids (FAs) provide cellular energy under starvation, yet how they mobilize and move into mitochondria in starved cells, driving oxidative respiration, is unclear. Here, we clarify this process by visualizing FA trafficking with a fluorescent FA probe. The labeled FA accumulated in lipid droplets (LDs) in well-fed cells, but moved from LDs into mitochondria when cells were starved. Autophagy in starved cells replenished LDs with FAs, increasing LD number over time. Cytoplasmic lipases removed FAs from LDs, enabling their transfer into mitochondria. This required mitochondria to be highly fused and localized near LDs. When mitochondrial fusion was prevented in starved cells, FAs neither homogeneously distributed within mitochondria nor became efficiently metabolized. Instead, FAs re-associated with LDs and fluxed into neighboring cells. FAs thus engage in complex trafficking itineraries regulated by cytoplasmic lipases, autophagy and mitochondrial fusion dynamics, ensuring maximum oxidative metabolism and avoidance of FA toxicity in starved cells.
Introduction
Cells adapt to nutrient starvation by shifting their metabolism from reliance on glucose metabolism to dependence on mitochondrial fatty acid (FA) oxidation. The biochemical basis for this metabolic reprogramming under starvation is well established (Eaton, 2002; Finn and Dice, 2006; Kerner and Hoppel, 2000; O’Neill et al., 2012). However, how FAs become mobilized and delivered into mitochondria for driving FA oxidation under starvation is far from clear.
FAs are stored within cells as energy-rich triacylglycerols in lipid droplets (LDs) in addition to being found on cellular membranes. Excess free FAs in the cytoplasm are harmful to cells: they can generate damaging bioactive lipids or disrupt mitochondrial membrane integrity (Unger et al., 2010). When mobilizing FAs from stores under starvation conditions, therefore, cells need to adjust FA trafficking pathways to avoid FA toxicity caused by overabundance of free FAs in the cytoplasm or within mitochondria.
Cells use two primary mechanisms for mobilizing FAs during nutrient stress. One is through autophagic digestion of membrane-bound organelles (i.e., the ER) or LDs (Axe et al., 2008; Hayashi-Nishino et al., 2009; Kristensen et al., 2008; Singh et al., 2009a; Yla-Anttila et al., 2009). This involves autophagosomal engulfment of the organelle/LD and fusion with the lysosome, where, hydrolytic enzymes digest the organelle/LD, releasing free FAs that quickly move into the cytoplasm (Singh et al., 2009a). When LDs are the substrate, this process is called lipophagy. While effective for bulk release of FAs into the cytoplasm in starved cells, FA mobilization by autophagy requires ways to avoid FA toxicity due to its potential to cause overabundance of free FAs in the cytoplasm. This could entail FAs either being immediately taken up into mitochondria or first moved to some storage compartment. Clearly, other FA trafficking pathways must function in conjunction with autophagy to manage released FAs in this mode of FA mobilization.
A second mechanism for mobilizing FAs during starvation is by lipolytic consumption of LDs. Here, cytoplasmic neutral lipases directly hydrolyze triacylglycerols on the LD surface. An advantage of this mechanism is that it can be regulated at the level of lipase activity, fine-tuned by the cell (Wang et al., 2008; Zechner et al., 2012). But the fate of FAs released by this mechanism remains an issue. Do the FAs move directly from LDs into mitochondria (possible if LDs and mitochondria are in close proximity), or do the FAs first mix with cytoplasmic pools? If the former, how do cells ensure that all mitochondria obtain adequate levels of FAs to drive β-oxidation-based metabolism? If the latter, how do cells avoid FA toxicity? Given these unanswered questions, it is not surprising that the respective roles of autophagy and lipolysis (i.e., lipase digestion of LDs) in mobilizing FAs are ambiguous (Kim et al., 2013; Smirnova et al., 2006; Wang et al., 2008).
Mitochondria represent the primary site for β-oxidation where FAs are enzymatically broken down to sustain cellular energy levels during nutrient stress. This requires mitochondria to import FAs to yield the metabolic intermediates driving respiration (Eaton, 2002; Kerner and Hoppel, 2000; O’Neill et al., 2012). Upon starvation, cells up-regulate enzymes required for mitochondrial FA import and β-oxidation (Eaton, 2002; Kerner and Hoppel, 2000). Interestingly, cells also remodel mitochondria into highly connected networks (Gomes et al., 2011; Rambold et al., 2011), by modulating mitochondrial fission/fusion dynamics, regulated by proteins including fusion proteins, mitofusin 1 and 2 (Mfn1 and Mfn2) (on the outer mitochondrial membrane) and optic atrophy protein 1 (Opa1) (on the inner mitochondrial membrane), and the fission protein dynamin related protein 1 (Drp1) (Hoppins et al., 2007; Hoppins and Nunnari, 2009). It remains to be tested, however, whether mitochondrial fusion occurring during starvation facilitates FA trafficking and oxidation during nutrient stress.
Here, we investigate how cells coordinate FA mobilization, trafficking and mitochondrial β-oxidation. Using a pulse-chase labeling method to visualize movement of FAs in live cells, we demonstrate that starved cells use primarily LDs as a conduit for supplying mitochondria with FAs for β-oxidation. This involves cytoplasmic, neutral lipase-mediated FA mobilization rather than lipophagy. Autophagy promoted lipid buildup in LDs, replenishing LDs with new FAs that then fluxed into mitochondria. Surprisingly, FA flux into mitochondria required LDs to be in close proximity with mitochondria and mitochondria to be highly tubulated/fused. This enabled FAs to homogeneously distribute inside mitochondria after import from LDs, ensuring FA oxidation and downstream oxidative phosphorylation reactions were optimized. Defects in mitochondrial fusion caused massive alterations in cellular FA routing. Not only were non-metabolized FAs redirected to and stored in LDs, they became excessively expulsed from cells. Given the role of FAs as signaling molecules, FA re-routing under these conditions could alter the function of whole cell populations, relevant to studies on obesity, diabetes and mitochondrial disorders.
Results
Nutrient stress increases FA transfer from LDs to mitochondria
Under starvation, cells shift to mitochondrial FA-driven oxidative phosphorylation for ATP production. This requires transfer of FAs, including those stored in LDs, into mitochondria (Finn and Dice, 2006; Kerner and Hoppel, 2000). To understand how this process is regulated, we developed a pulse-chase assay to track FAs in relation to LDs and mitochondria in starved mouse embryonic fibroblasts (MEFs) (Figure 1A). For this assay we utilized BODIPY558/568 C12 (Red C12), a saturated FA analog with a tail composed of 12 carbons and a BODIPY 558/568 fluorophore covalently bound at the hydrophobic end, with an overall length approximately equivalent to that of an 18-carbon FA. BODIPY-FAs have been used in studies of lipid trafficking, and have been shown to incorporate into LD-specific neutral lipids (Herms et al., 2013; Kassan et al., 2013; Thumser and Storch, 2007; Wang et al., 2011). In our pulse-chase assay, cells were first labeled overnight with trace amounts (1 μM) of Red C12, which accumulated in neutral lipids within LDs (Figure 1B, 0h). They were then placed in complete or nutrient-depleted medium in the absence of Red C12 for 6h or 24h before labeling LDs or mitochondria and visualizing fluorescence in the cells with spinning-disk confocal microscopy.
Figure 1. Fatty acid trafficking can be visualized using a fluorescent fatty acid pulse-chase assay.

(A) Schematic representation of the fluorescent FA pulse-chase assay: cells were pulsed with Red C12 overnight, washed, and incubated with CM for 1 h in order to allow the Red C12 to accumulate in LDs. Cells were then chased in CM or HBSS for the indicated periods of time and imaged to determine the subcellular localization of the FA. (B–D) WT MEFs were assayed as described in Figure 1A and chased in CM or HBSS for 0 h, 6 h, or 24 h. LDs were labeled using (B) BODIPY 493/503 and (C) mitochondria were labeled using MitoTracker Green. Scale bar, 10μm. (D) Relative cellular localization of Red C12 was quantified by Pearson’s coefficient analysis. Data were expressed as means ± SEM. (E) TLC resolving Red C12 isolated from WT MEFs assayed as described above and chased for 6 h or 24 h with HBSS in the absence or presence of etomoxir, see also Figure S1.
Cells chased in nutrient-rich medium (i.e., complete medium, CM) showed nearly all Red C12 signal localized to LDs throughout the pulse-chase labeling period (Figure 1B, CM). Little, if any, Red C12 signal was transported into mitochondria (Figure 1C, CM). By contrast, cells in nutrient-depleted medium showed a dramatic loss of Red C12 signal from LDs between 6–24 h of chase (Figure 1B, HBSS), with the signal redistributing into mitochondria (Figure 1C, HBSS). LD-to-mitochondria transfer of FAs in starved cells was already detectable within 6 h (Figure 1C, HBSS, and Figure S2A, B) and was most prominent after 24 h (Figure 1C, D), with overall reduction of cellular Red C12 levels (Figure S1A). Near complete overlap of Red C12 with MitoTracker-positive structures occurred after 24 h of starvation, with no apparent labeling of other organelles (Figure 1C, HBSS).
Thin layer chromatography (TLC) revealed that the amount of esterified Red C12 in cells decreased with time after nutrient deprivation while the amount of free Red C12 increased (Figure 1E), consistent with our imaging results above. In addition, we observed slower migrating breakdown products of Red C12 with time after starvation. Inhibition of the mitochondrial FA importer CPT-I with etomoxir inhibited the translocation of Red C12 into mitochondria and the appearance of Red C12 breakdown products (Figure 1E, Figure S1B and Figure S6E), indicating Red C12 as a substrate for β-oxidation. Thus, starvation initiates a process whereby cells selectively and efficiently transfer FAs from LDs to mitochondria for oxidation.
Cytoplasmic lipase activity liberates FAs from LDs for transfer to and metabolism within mitochondria
FAs can be released from LDs by two distinct mechanisms: lipolysis and lipophagy (Singh et al., 2009a; Zechner et al., 2012). To determine which of these two mechanisms is responsible for FA delivery to mitochondria under starvation, we employed the Red C12 pulse-chase labeling protocol, examining the effect of inhibiting either lipase activity or autophagy.
To test the role of lipases in FA transfer to mitochondria, we depleted cells of adipose triglyceride lipase (ATGL) (Figure S2C), a ubiquitously expressed neutral lipase that can digest LDs (Smirnova et al., 2006), and then followed the fate of Red C12 during pulse-chase labeling. Unlike MEFs treated with noncoding control siRNA, which showed a dramatic increase in co-localization between Red C12 and MitoTracker after 24 h starvation, ATGL-silenced MEFs showed only minimal redistribution of Red C12 from LDs into mitochondria (Figure 2A, B). Instead, the Red C12 signal remained localized to LDs (Figure 2A). Pharmacological inhibition of lipase function with DEUP also significantly reduced starvation-induced mitochondrial accumulation of Red C12 in the pulse-chase assay (Figure S2A, B, DEUP). Therefore, lipase activity is necessary for FA delivery from LDs into mitochondria in starved MEFs.
Figure 2. Cytoplasmic lipase activity, but not lipophagy are essential to liberate FAs from LDs for transfer to mitochondria.
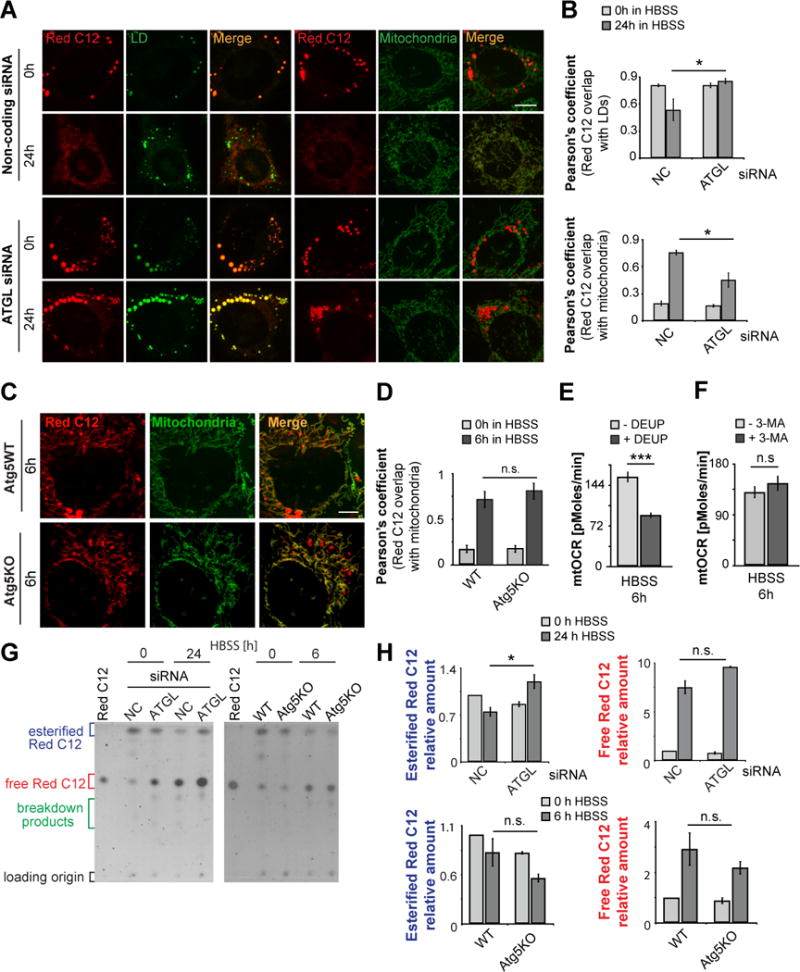
(A) WT MEFs were assayed as described in Figure 1A after RNA interference using non-coding or ATGL siRNA. Scale bar, 10μm. (B) Quantification of the correlation between Red C12 signal and LDs (upper graph) or mitochondria (lower graph) in the experiment shown in Figure 2A. (C, D) Atg5-WT and Atg5-deficient cells were assayed as described in Figure 1A. Scale bar, 10μm. (D) Quantification of the correlation between Red C12 signal and mitochondria in the experiment described in Figure 2C. (E, F) Mitochondrial respiration is dependent on cytoplasmic lipase activity. Mitochondrial respiratory activity of 6 h starved WT MEFs was measured in the presence of (E) the lipase inhibitor DEUP and (F) 3-MA to inhibit autophagy. (G) TLC resolving Red C12 isolated from cells assayed as described in Figure 2A (left panel) or 2C (right panel). (H) Relative amounts of esterified and free Red C12 were quantified from images of TLC plates, normalized to Red C12 levels of WT cells at the 0 h time point. Data were expressed as means ± SEM, see also Figure S2.
We next tested the role of autophagic activity in FA trafficking to mitochondria. Cells were pulse-chase labeled with Red C12 under starvation conditions in the presence of the autophagy inhibitor 3-methyladenine (3-MA). Notably, Red C12 trafficking into mitochondria was not significantly altered compared to vehicle-treated cells (Figure S2A, B, 3-MA). To further test autophagy’s role in FA delivery to mitochondria, we examined whether Red C12 transfer to mitochondria was affected in starved Atg5-deficient MEFs (Atg5 KO), in which autophagosome biogenesis is blocked (Mizushima et al., 2001). No impairment in Red C12 transfer to mitochondria was observed (Figure 2C, D). TLC data corroborated the imaging results, with levels of esterified Red C12 decreasing in response to starvation in WT and Atg5 KO, but not ATGL-depleted, MEFs (Figure 2G, H). Interestingly, in ATGL-depleted MEFs levels of free Red C12 still increased in response to starvation, indicating that compensatory mechanisms such as up-regulation of phospholipase activity may exist (Cabodevilla et al., 2013).
That lipolysis (i.e., lipase digestion of LDs) was necessary for mitochondrial respiration under starvation was demonstrated by measuring mitochondrial oxygen consumption rate (mtOCR). Using DEUP treatment to inactivate lipases, we found that in 6h starved cells, but not fed cells, mtOCR levels were significantly reduced relative to untreated cells (Figure 2E and Figure S2G). By contrast, in similarly starved cells treated with 3-MA to inhibit autophagy, no reduction in mtOCR levels was observed (Figure 2F). However, autophagy inhibition at times of prolonged starvation significantly reduced mitochondrial respiration (Figure S2H), as reported before (Singh et al., 2009a). Therefore, FA substrates used for mitochondrial respiration in acutely nutrient stressed cells are primarily derived from lipolysis of LDs rather than through autophagy.
To address directly whether lipophagy is active in HBSS-starved MEFs, we performed co-localization studies between the autophagosomal marker LC3 and LDs. Although we detected lipophagy in individual MEFs (Figure S2E), autophagic LD degradation did not increase in 6h or 24h HBSS relative to fed cells (Figure S2D, E and F). In contrast to HBSS, under milder starvation conditions where only serum but not amino acids and glucose was limited, lipophagy was significantly induced (Figure S2E 6h and 24h), as reported previously (Ouimet et al., 2011; Schulze et al., 2013; Singh et al., 2009a). Together, these data suggest that under HBSS starvation conditions, MEFs are not reliant on lipophagy to supply mitochondria with LD-derived FAs. Rather, cytoplasmic neutral lipase activity, which releases FAs from LDs, appears to be the primary mechanism.
Autophagy drives LD growth during starvation
Given that lipophagy is not induced in HBBS-starved MEFs, we next explored whether other autophagic pathways that are distinct from lipophagy play roles in regulating FA flux into mitochondria and sustaining mitochondrial metabolism in starved cells. Bulk autophagosomal degradation dramatically increases during starvation (Mizushima, 2007), so we investigated whether this type of autophagic activity helps recycle FAs to maintain LD stores for continual delivery of FAs to mitochondria. Supporting this possibility, we observed that BODIPY493/503-labeled LDs grew continually in starved cells (Figure 3A), despite efficient transfer of pulsed Red C12 pools into mitochondria (Figure 1B). LD number almost doubled upon 6h starvation and reached its maximum after 24 h of starvation, with five times more LDs per cell (Figure 3B). The average LD size per cell increased by 2-fold after 24 h starvation (Figure 3B), shifting toward larger LDs in the heterogeneously sized LD population (Figure S3A). Finally, total LD volume per cell showed a significant increase (Figure S3B) and triglyceride storage, measured enzymatically, was boosted (Figure S3C).
Figure 3. Autophagy drives LD growth during starvation.
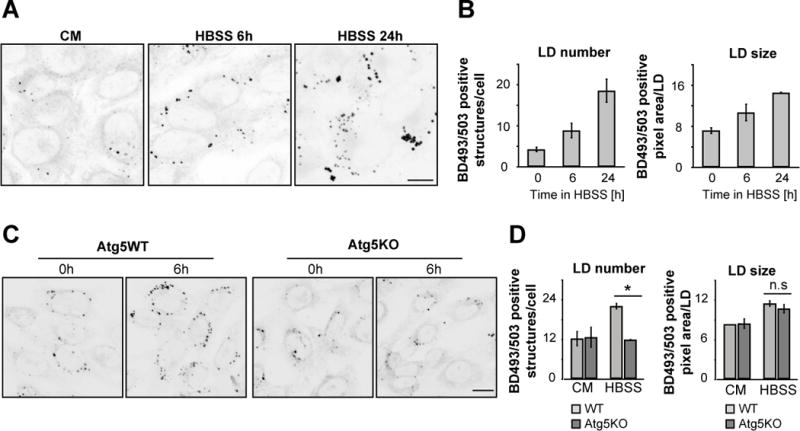
(A–D) WT or Atg5KO MEFs were incubated in CM or HBSS for the time periods indicated, BODIPY 493/503 was added to visualize LDs and images of live cells were captured and presented as inverted greyscale micrographs (A and C). To determine LD growth during starvation LD size and number were measured (B and D). Scale bars, 50 μm. Data were expressed as means ± SEM, see also Figure S3.
As a further test to the hypothesis that autophagic activity during starvation helps maintain LD stores, we examined the effect of inhibiting autophagy on LD growth during starvation. No up-regulation of LD abundance occurred in Atg5KO cells compared to wild type (WT) cells under starvation conditions (Figure 3C, D). LD number-increase during starvation was also prevented by pharmacological inhibition of autophagy initiation using 3-MA (Figure S3D) or lysosomal degradation of autophagosomes using bafilomycin A1 (Figure S3E).
To further confirm autophagy-derived FAs drive LD growth in starved cells, we performed a FA pulse-chase assay using phosphatidylcholine that was fluorescent-tagged at its FA tail (FL HPC) (Figure 4A). Cells were first labeled overnight with trace amounts of FL HPC, which integrated into various cellular membranes, including the ER, mitochondria and Golgi apparatus under fed conditions (Figure 4B, 0h CM and Figure S4A). Cells chased in CM retained the FL HPC signal in cellular membrane structures (Figure 4B, 6h CM). In contrast in starved MEFs, FL HPC signal on membranes was decreased due to it redistributing into LDs (Figure 4B, 6h HBSS, Figure S4B). Inhibition of autophagy using 3-MA, bafilomycin A1 or Atg5-deficient MEFs reduced this transfer (Figure 4B, C).
Figure 4. Autophagy mobilizes phospholipids from cellular membranes during starvation.
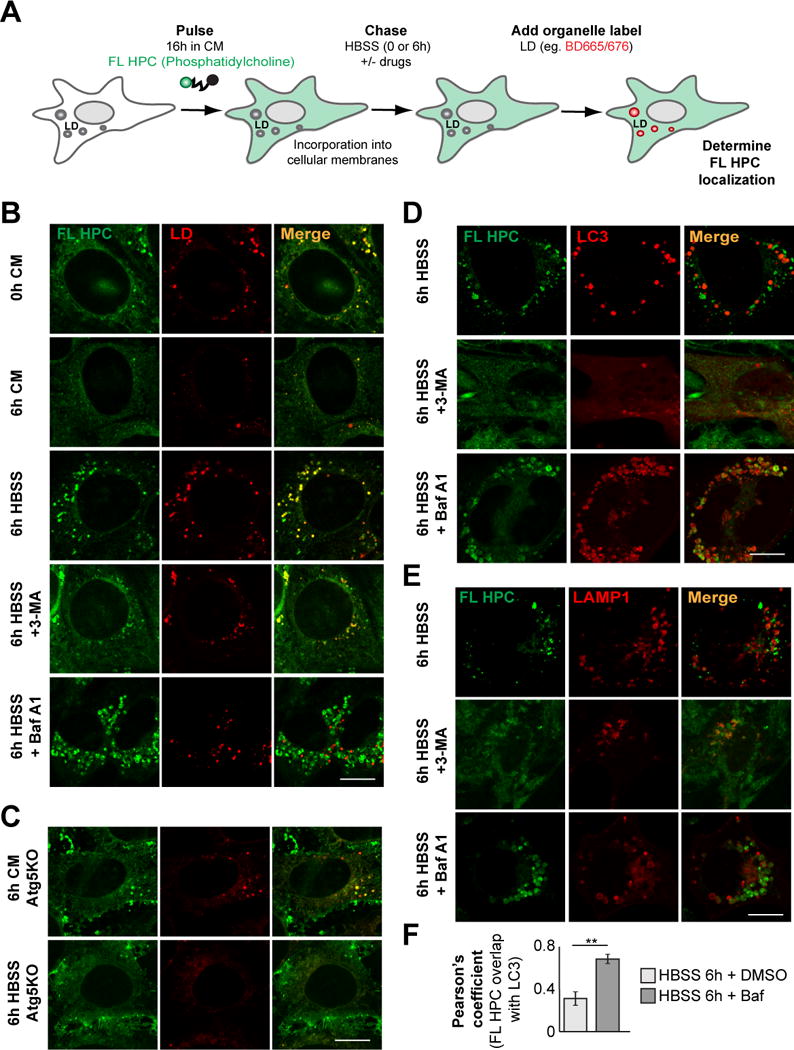
(A, B) FL HPC loaded WT MEFs were chased in HBSS for 6 h in the absence or presence of 3-MA or Bafilomycin A1 (Baf A1) and the subcellular localization of FL HPC was determined. LDs were stained with BODIPY665/676. (C) Atg5KO MEFs were assayed as described in Figure 4B. (D, E) WT MEFs expressing mCherry-LC3 or LAMP1-mCherry were assayed as described in Figure 2B. (F) Quantification of the correlation between FL HPC signal and autophagosomes or lysosomes in the experiments described in Figure 2D and E. Data were expressed as means ± SEM, see also Figure S4.
Figure 6. Mitochondrial fusion deficiencies result in increased FA storage.
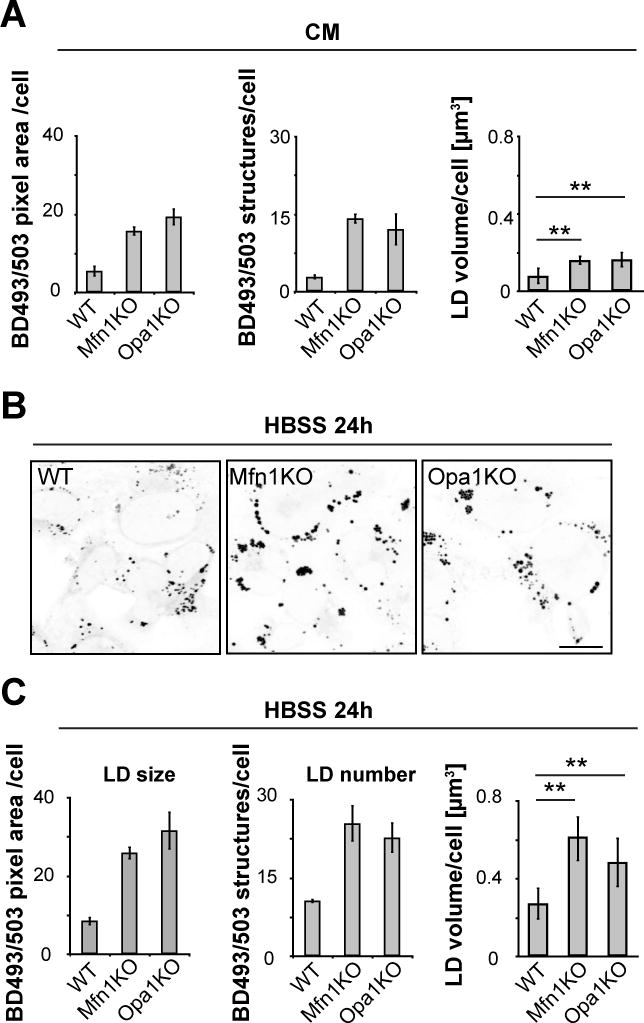
WT, Mfn1KO and Opa1KO cells were grown in complete (A) or starvation (B, C) medium for 24h, BODIPY493/503 was added to visualize LDs, and LD size, number and volume were determined. Scale bar, 50 μm. Data were expressed as means ± SEM, see also Figure S6.
That autophagy is actively involved in FL HPC recycling from cellular membranes was further confirmed by co-localization studies in MEFs transfected with the autophagosomal marker mCherry-LC3 or lysosomal marker LAMP1-mCherry. Upon starvation, FL HPC could be partially found in autophagosomes/lysosomes (Figure 4D and E) in addition to LDs (Figure 4B). Autophagosome/lysosome localization could be prevented by 3-MA (Figure 4D and E, 3-MA). When autophagosome-lysosome fusion was reduced with high concentrations of bafilomycin A1 (Bjorkoy et al., 2005), significant trapping of FL HPC in autophagosomes occurred (Figure 4D, F). In addition, there was less localization of FL HPC with lysosomes labeled with LAMP1 (Figure 4E, BafA1) and with late endosomes labeled with Rab5 (Figure S4, C). Together, these data suggest that autophagy is involved in shuttling FL HPC from cellular membranes into lysosomes. FAs hydrolyzed from the phospholipids would then be released into the cytoplasm and associate with LDs.
Fused mitochondria ensure FAs distribute throughout mitochondria for maximal β-oxidation
When starved MEFs were imaged after expression of the mitochondrial marker mito-RFP and labeling with the LD marker BODIPY493/503, we found that virtually all LDs were closely associated with mitochondria (Figure 5A). Monitoring mitochondria and the distribution of FAs delivered into them in starved cells further revealed that mitochondria were highly fused and FAs were homogenously distributed throughout the mitochondria (Figure 5A, D) (Gomes et al., 2011; Rambold et al., 2011). One interpretation of these results is that close association between LDs and mitochondria allows FAs to traffic directly from LDs into mitochondria. The fused state of mitochondria, in turn, would ensure that FAs are equilibrated throughout the mitochondrial system (assuming limited numbers of LDs per cell), maximizing FA availability for β-oxidation reactions.
Figure 5. Mitochondrial fusion is required for mitochondrial FA distribution and oxidation.
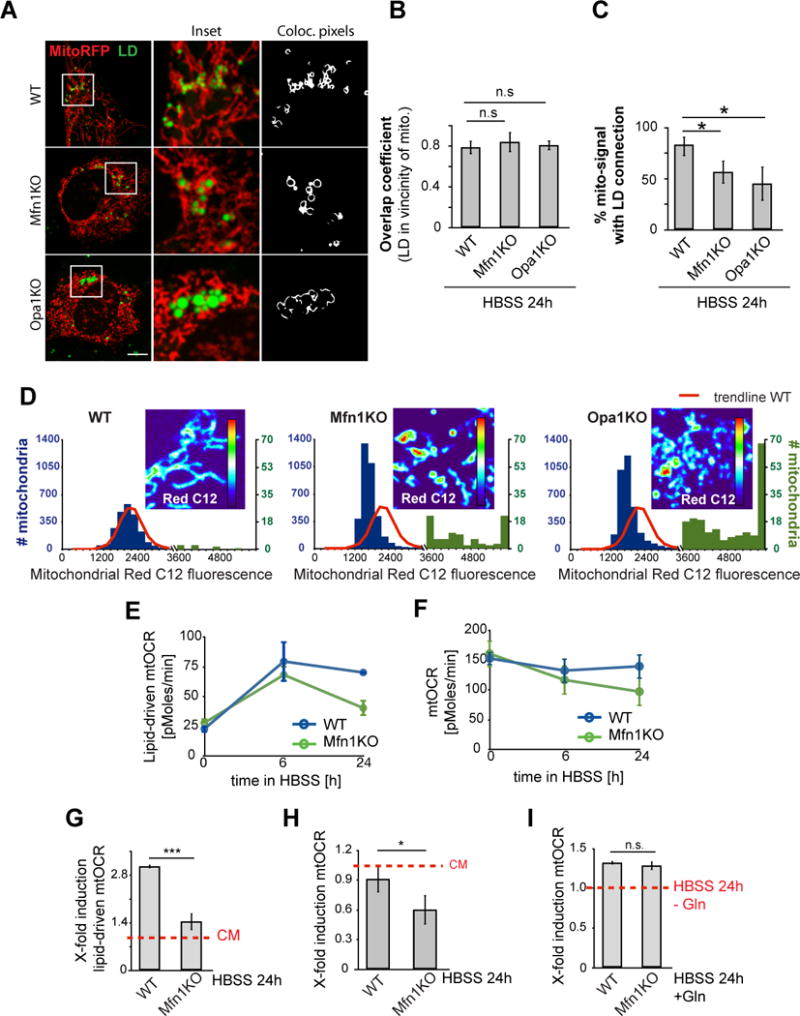
(A–C) WT, Mfn1 KO or Opa1 KO MEFs were transfected with the mitochondrial marker mito-RFP and labeled with BODIPY493/503 to visualize LDs. (A) Live images were acquired, (B) the ability of LDs to gain states of high proximity to mitochondria was measured using the overlap coefficient, (C) and overall mitochondrial content with direct contact to LDs was measured. (D) Red C12 localization in WT, Mfn1 KO or Opa1 KO MEFs was assayed as described in Figure 1A (24 h HBSS). Red C12 intensities in individual mitochondria were plotted as histograms, with blue and green bars representing higher and lower ranges of intensities, respectively. Red lines: WT trendline. Representative images of mitochondrial Red C12 in each cell line were presented as heatmaps, blue: lowest, and red: highest mitochondrial Red C12 levels. Scale bars, 10 μm. (E–F) Mitochondrial respiratory activity was measured in WT and Mfn1KO MEFs, incubated in CM or HBSS for the time points indicated. (E) Lipid-driven mtOCR and (F) total mtOCR were determined. Lipid-specific respiration was analyzed by acute CPT-I inhibition with etomoxir. (G, H) Respiratory induction levels for lipid-driven mtOCR and total mtOCR levels were determined, using mtOCR levels in CM as baseline levels for each cell line. (I) Glutamine (Gln)-driven respiration was determined by acute injection of glutamine to 24 h starved cells and induction levels were determined. Data were expressed as means ± SEM, see also Figure S5.
To test this hypothesis, we first examined whether the fused state of mitochondria was necessary for the homogenous dissemination of FAs within mitochondria. Red C12 pulse-chase labeling was performed in MEFs deficient in the major mitochondrial fusion proteins Mfn1 or Opa1. In the Mfn1- or Opa1- deficient cells, mitochondria did not form networks and were fragmented (Figure 5A) (Chen et al., 2005; Gomes et al., 2011; Rambold et al., 2011). Despite this, LDs were still in close proximity with mitochondria and there was transfer of Red C12 into mitochondrial elements (Figure 5A, B and D). However, many mitochondrial elements were not in close proximity to LDs (Figure 5C) and Red C12 did not become homogenously distributed across the mitochondrial population (Figure 5D), resulting in either very low or exceedingly high Red C12 signals relative to that detected in WT cells (Figure 5D). In contrast to Red C12, MitoTracker signal was uniform throughout the mitochondria of WT, Mfn1KO and Opa1 KO cells (data not shown). Therefore, mitochondrial fusion is required to homogenously distribute LD-derived FAs throughout the mitochondrial network in starved cells.
We next investigated whether a homogenous distribution of FAs throughout mitochondria is necessary for optimal β-oxidation within mitochondria in starved cells. To address this, we measured FA-driven mtOCR and its contribution to total mtOCR in control or Mfn1KO cells under starvation. Mfn1KO cells were used as a primary model since, unlike Opa1KO and Mfn1/2KO MEFs (Chen et al., 2005; Cogliati et al., 2014; Frezza et al., 2006) (Figure S5A), their respiration levels under fed conditions (Figure 5E, F) and mitophagy levels under starvation conditions (Figure S5, B) were comparable to WT cells. In starved WT cells, there was an expected rapid increase in FA oxidation over 6 h, followed by sustained oxidation for at least 24h (Figure 5E, G). The boosted β-oxidation enabled these cells to nearly maintain equivalent total mitochondrial respiration levels over the entire starvation period (Figure 5F, H). Mfn1-deficient cells up-regulated initial levels of β-oxidation comparable to control cells, however the levels were not maintained over 24h, with FA oxidation becoming significantly reduced (Figure 5E, G). Consequently, total mitochondrial respiration levels in Mfn1KO cells dropped significantly over time (Figure 5H), accompanied by excessive accumulation of mitochondrial FA breakdown intermediates (Figure S6E).
We next determined whether the requirement of having fused mitochondria was specific for FA metabolism and not for metabolism of other metabolic substrates, such as glutamine. Unlike FAs, which are concentrated in LDs, glutamine is diffusely distributed in the cytoplasm and is taken up into mitochondria via transporters distributed throughout the organelle. The fused state of mitochondria may therefore be irrelevant for glutamine to be effectively metabolized. To address this possibility, we measured mtOCR levels in starved cells cultured for 24h with and without glutamine addition in WT and Mfn1KO cells. A 1.3-fold induction of mitochondrial respiration in response to glutamine addition occurred for both control and Mfn1KO cells (Figure 5I). This revealed that mitochondrial fragmentation does not affect glutamine oxidation, unlike its effect on oxidation of FAs. Therefore, β-oxidation requires mitochondria to be highly fused, in contrast to other mitochondrial reactions such as glutamine oxidation, which are unaffected by mitochondrial morphology. This could be explained if delivery of FAs to mitochondria occurs primarily at limited sites (i.e., LDs in close proximity to mitochondria), thereby requiring the mitochondrial system to be highly fused to equilibrate newly arriving FAs.
Defects in mitochondrial fusion lead to lipid accumulation in LDs
Previous work has shown that FA buildup can be toxic to mitochondria (Unger et al., 2010), and that cells from patients with inherited β-oxidation defects can re-export FAs out of mitochondria (Nada et al., 1995; Schaefer et al., 1995). Given the observed heterogeneous buildup of mitochondrial FAs in starved Mfn1KO or Opa1KO cells, we explored how mitochondria cope with an excessive load of non-metabolized FAs. One possibility is that the cells redirect FAs back to LDs to be esterified to inert triacylglycerols. This predicts a buildup of LDs in these cells. Consistent with this, live cell imaging of BODIPY493/503 in Mfn1KO and Opa1KO cells in CM showed slightly higher cellular LD load than WT cells as measured by increased LD size, number and volume (Figure 6A). The increase in LD number and size was more pronounced when fusion-deficient KO cells were starved, with three times higher LD volume per cell occurring compared to fusion-competent control cells (Figure 6B, C). Pulse-chase Red C12 labeling experiments further revealed that although net transfer of Red C12 into mitochondria in starved cells deficient in mitochondrial fusion was not significantly affected (Figure S6A, D), there was greater buildup of the Red C12 in LDs (Figure S6A, B, C). Greater accumulation of Red C12 in LDs was also seen in non-starved cells deficient in mitochondrial fusion compared to WT cells, with Opa1KO cells showing a more extreme phenotype than Mfn1KO cells, most likely due to their increased bioenergetic deficiency (Figure S6A, B, C). Despite this, lack of Mfn1 or Opa1 did not affect overall FA release from LDs or the abundance of the mitochondrial FA importer CPT-I under starvation conditions (Figure S6C, F). Together, these results suggest that to compensate for their inability to effectively metabolize FAs, mitochondrial fusion-deficient cells reroute FAs to re-associate with LDs.
Defects in mitochondrial fusion lead to increased FA expulsion from cells
Given LD storage capacity is thought to be limited in cells of non-adipose origin, we wondered if cells deficient in mitochondrial fusion used further coping mechanisms to limit free FA levels in their cytoplasm. One mechanism would be to transfer FAs into the extracellular space (Herms et al., 2013). To test this possibility, we combined Red C12 labeling of FAs with a cellular co-culture system to serve as optical readout for FA release (Figure 7A). WT MEFs were pulsed with Red C12, and the resulting “donor” cells were then co-cultured for 24h in nutrient-depleted medium with “acceptor” WT MEFs labeled with a green cellular dye. A small amount of Red C12 was transferred from WT donor cells to acceptor cells (Figure 7B, C). These results demonstrate that FAs are exported at low levels in MEFs during starvation. Inhibiting FA release from LDs by ATGL depletion drastically reduced FA transfer into acceptor cells (Figure S7A), thus verifying LD stores as a major source for the FA expulsion. Performing co-culture experiments with Mfn1KO donor cells revealed both higher levels of donor cell labeling and significant transfer of Red C12 into the WT acceptor cells, as shown both by fluorescence imaging and quantitative flow cytometry (Figure 7B, C). Similar high levels of starvation-induced Red C12 release was also observed in fusion-deficient Opa1KO cells (Figure 7B, C). Since cell survival rates in Mfn1KO and Opa1KO cells were comparable to WT cells (Figure S7B), we can exclude the possibility that transferred FAs were released from dying cells. Together these data suggest that reduction in β-oxidation rates, induced by mitochondria fusion-deficiency, increases rates of FA efflux out of cells. This could be an important compensatory mechanism to avoid toxic FA buildup in the cytoplasm.
Figure 7. Mitochondrial fusion deficiencies result in increased cellular export of FAs.
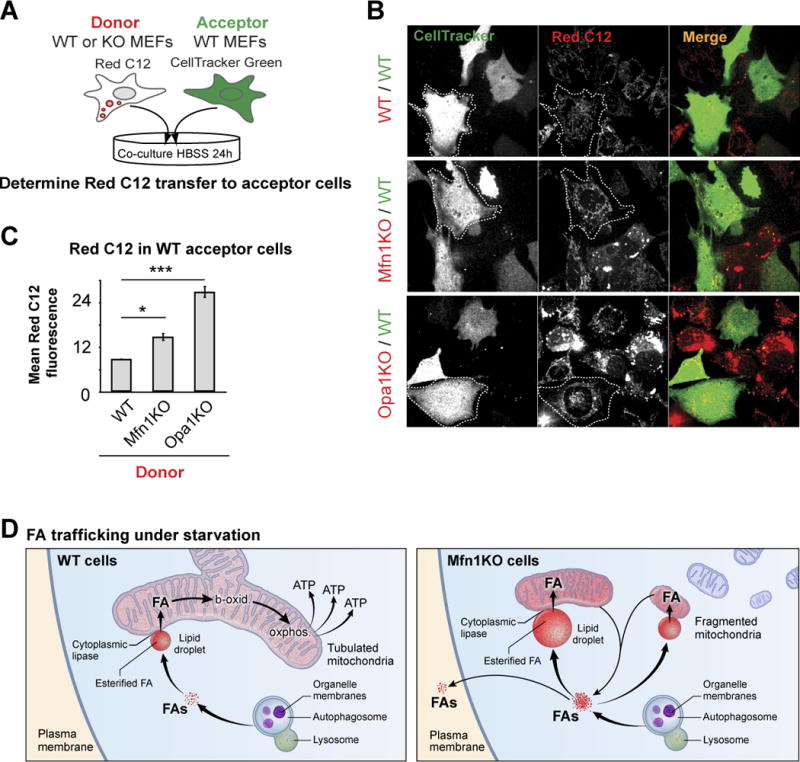
(A) Schematic representation of the co-culture assay to visualize cellular FA export. Red C12 pre-labeled donor cells (WT, Mfn1KO, or Opa1KO MEFs) were washed excessively and then co-cultured for 24 h in HBSS with CellTracker green-labeled acceptor cells (WT MEFs). (B) Live cell images of donor and acceptor cells were acquired and (C) levels of Red C12 transfer into acceptor cells measured by flow cytometry analysis. Data were expressed as means ± SEM see also Figure S7. (D) Model of FA trafficking in starved cells. In WT cells, autophagy releases FAs from phospholipids within organelle membranes; these FAs flux through LDs into tubulated mitochondria, where they become homogenously distributed throughout the mitochondrial network and are efficiently metabolized to produce ATP. In fusion-deficient Mfn1KO cells, fragmented mitochondria receive highly variable amounts of FA. These FAs are not efficiently metabolized, resulting in re-direction of FA flow, resulting in increased FA storage within LDs and efflux of FAs from the cell.
Discussion
In this study, we define the pathways regulating FA flux into mitochondria during nutrient stress, crucial for enabling cells to shift their metabolism to FA oxidation and thereby survive starvation. Using pulse-chase labeling assays, we found that FA flux into mitochondria was co-dependent on LD lipolysis, autophagy and mitochondrial fusion dynamics (Figure 7D). Unless all three systems contributed, FAs did not successfully redistribute into and throughout mitochondria to maximize oxidative phosphorylation, and instead accumulated within LDs and/or were released from cells. As discussed below, our findings are relevant to the broader question of how cells and organisms adapt cellular FA flow and storage to changing nutrient availability and metabolic need.
In exploring how FAs move into mitochondria in starved cells, we found that rather than being delivered from a free cytoplasmic pool (as occurs for other metabolites), FAs used LDs as their conduit. This may explain why LDs and mitochondria are so closely associated; it would enable FAs to move efficiently between these two organelles. Close spatial positioning of LDs and mitochondria has been reported before in various cell types (Shaw et al., 2008; Sturmey et al., 2006; Tarnopolsky et al., 2007; Vock et al., 1996). Indeed, electron microscopic studies of skeletal muscle showed chains of alternating mitochondria-LD structures (Tarnopolsky et al., 2007), with increased interactions in response to energy-intensive exercise (Shaw et al., 2008). It remains to be established what mechanisms underlie the close positioning of LDs and mitochondria, but roles for microtubules (Valetti et al., 1999; Welte et al., 2005), SNAP23 (Jagerstrom et al., 2009) and perilipin 5 (Wang et al., 2011) have been proposed.
FA release from LDs has been linked to cytoplasmic lipases and lipophagy (i.e., autophagic digestion of LDs), with their relative contributions still unclear. Recent studies identified pleiotropic roles for autophagy in lipid metabolism that may be tissue- or condition-specific (Dupont et al., 2014; Kim et al., 2013; Shibata et al., 2009; Shibata et al., 2010; Singh et al., 2009a; Singh et al., 2009b). Here, we demonstrated that during acute starvation in mammalian cells, autophagy is dispensable for supplying mitochondria with FAs to sustain oxidative respiration. This is because inhibiting lipase activity (by silencing ATGL or treating with DEUP), which blocked FA flux into mitochondria, reduced mitochondrial oxidative metabolism and resulted in enlarged LDs in starved cells. By contrast, we found that conditions of serum depletion in the presence of amino acids and glucose led to upregulation of lipophagy, consistent with previous studies (Ouimet et al., 2011; Schulze et al., 2013; Singh et al., 2009a). How the availability of different nutrients dictates autophagosomal specificity remains to be addressed. One possibility is that the signaling pathways that control autophagy are sensitive to different nutrient stress and this determines what substrate is targeted. For example, the major autophagy regulator mammalian target of rapamycin (mTOR) is rapidly inactivated during HBSS but not during serum starvation alone (Peterson et al., 2011). Additionally, specialized cell types might differentially utilize lipases versus lipophagy for their response to starvation, for example, release of FAs from LDs by autophagy might be of particular importance in cell types with low lipase activity, such as hepatocytes (Singh et al., 2009a; Walther and Farese, 2012).
Why do starved mammalian cells prefer lipolysis to lipophagy in trafficking FAs into mitochondria? We speculate this arises from several factors. One factor relates to avoidance of FA overload and toxicity. When FA levels become too high in the cytoplasm, FA toxicity occurs, leading to membrane permeability and mitochondrial dysfunction (Unger et al., 2010). Digestion of LDs by autophagy would likely release the stored FAs in large quantities from lysosomes into the cytoplasm, requiring coordination of FA flux into mitochondria with storage of excess FAs in LDs. By contrast, FA flux into mitochondria via lipolysis would only require regulation of lipases and LD-associated proteins, such as perilipin 5 (Dalen et al., 2007; Granneman et al., 2011; Wolins et al., 2006; Yamaguchi et al., 2006). Close positioning of LDs with mitochondria, which we observed, could provide a further mechanism whereby high concentrations of FAs flow directly from LDs into mitochondria, avoiding buildup in the cytoplasm. This transfer of FAs is likely to occur in association with FA-binding proteins (FABPs), a family of small cytoplasmic proteins capable of reversibly binding FAs with high affinity. The ubiquitously expressed H-FABP in particular has been implicated in transfer of FAs to mitochondria (Furuhashi and Hotamisligil, 2008). Once locally delivered into mitochondria, the FAs could then readily distribute throughout the mitochondrial system, given the highly fused state of mitochondria in starved cells (Gomes et al., 2011; Rambold et al., 2011).
A second factor for why starved mammalian cells might prefer lipolysis to lipophagy for trafficking of FAs into mitochondria could be related to the FA species released by lipolysis versus lipophagy. The FA forms generated by lipolysis may be more readily imported into mitochondria (or metabolized there) compared to FAs released during lipophagy. In line with this, a recent report demonstrated that lipid-dependent PPAR signaling specifically requires cytoplasmic lipase-catalyzed hydrolysis of intracellular triglyceride stores, but not FAs from other sources, to build a functional signaling complex (Haemmerle et al., 2011). Given that lipase-activated PPAR signaling drives mitochondrial biogenesis and oxidative phosphorylation, FA release from LDs could contribute to the coordination between FA transfer to mitochondria and regulation of mitochondrial activity. Finally, it is even conceivable that components of LDs, such as LD-associated proteins, dissociate during lipolysis and regulate FA trafficking or import.
Given that FAs flux into mitochondria from LDs via lipolysis in starved cells, what supplies LDs with FAs so they can be continuously delivered into mitochondria? We found this function is mediated by autophagy. Autophagic digestion of cellular membranes released FAs into the cytoplasm from where they were at least partially incorporated into LDs. Therefore, inhibiting autophagy did not prevent FAs from moving into mitochondria and driving β-oxidation as long as LDs were abundant.
Interestingly, we found that effective FA delivery into mitochondria in starved cells involved a role for mitochondrial fusion dynamics. Unless mitochondria were highly fused, FAs did not distribute homogenously throughout the mitochondrial system and energy production within mitochondria by oxidative phosphorylation was reduced. In cells with fragmented mitochondria, FAs excessively accumulated in some mitochondrial elements and diminished in others, presumably due to only some mitochondrial elements being associated close to a LD. The consequence of these effects on FA trafficking and metabolism were major, with levels of FA metabolism reduced, more FAs built up in LDs, and more FAs fluxed out the cell to neighboring cells.
Changes in mitochondrial shape and ultrastructure are known to accompany lipid-associated diseases and cellular lipid excess (Galloway and Yoon, 2012, 2013; Yoon et al., 2011). Our results showing the functional interrelation of mitochondrial fusion with cellular FA trafficking inside and in-between cells provide conclusive evidence for alterations in mitochondrial structural integrity can actively contributing to changes in cellular lipid homeostasis. Our data are consistent with a model in which mitochondrial fusion-deficient cells respond to reduced β-oxidation levels by re-routing FAs from mitochondria into LDs and the extracellular medium, similar to studies showing that mitochondrial import enzymes reverse FA transport when β-oxidation is impaired (Eaton et al., 1996; Nada et al., 1995; Schaefer et al., 1995; Watmough et al., 1988). The level of FA re-routing was dosage-dependent, with higher FA flux occurring in respiratory defective Opa1KO than Mfn1KO cells. The correlation between respiratory deficiency and FA export might make FA-rerouting also relevant for metabolic defects resulting from mechanisms other than altered mitochondrial fission/fusion dynamics.
The system we have described for mobilizing FAs during starvation may play a role in other cellular contexts. For example, FA oxidation is critical in highly oxidative cells, like skeletal muscle specifically during exercise or the heart (Fritz et al., 1958; Grynberg and Demaison, 1996). Emerging evidence is also correlating the presence of fused mitochondria with many cellular states of high-energy demand, including G1/S cell cycle transition (Mitra et al., 2009) and chemical stress (Tondera et al., 2009). Moreover, recent studies showed that fusion is required to sustain mitochondrial activity during excitation-contraction coupling in skeletal muscle and the activity of Agrp-neurons in lipid fed mice (Dietrich et al., 2013; Eisner et al., 2014). In addition, because FAs and their derivatives play important roles in regulating cellular signaling cascades and contribute to FA-driven modulations in metabolism, inflammatory responses and cell death programs (Calder, 2013; Wahli and Michalik, 2012; Zechner et al., 2012) alterations in their levels likely have huge health risks (Currie et al., 2013; Krahmer et al., 2013). Therefore, future work studying the FA flux system described here (involving autophagosomes, LDs and mitochondria,) has great potential for contributing to our understanding of the pathophysiology of lipid-associated diseases, such as obesity, diabetes, cancer and inflammatory disorders.
Experimental Procedures
Fluorescent FA pulse-chase and co-culture experiments
MEFs were incubated with complete medium (DMEM with 10% FBS and 4 mM glutamine, CM) containing 1 μM BODIPY 558/568 C12 (Red C12, Life Technologies) or 2 μM β-BODIPY FL C12-HPC (FL HPC, Life Technologies) for 16 h. Cells were then washed three times with CM, incubated for 1 h in order to allow the fluorescent lipids to incorporate into LDs or cellular membranes, and then chased for the time indicated in CM or Hank’s buffered saline solution (HBSS) in the absence or presence of various drugs. Mitochondria were labeled with 100 nM MitoTracker Green FM (Life Technologies) for 30 minutes prior to imaging. To label LDs, BODIPY 493/503 (Life Technologies) or BODIPY665/676 (Life Technologies) was added to cells at 200 ng/ml immediately prior to imaging and was present during imaging.
For FA transfer co-culture assays, donor cells were incubated with 2 μM Red C12 for 16 h in CM. Cells were then washed three times with CM followed by 1 h incubation. In parallel, acceptor cells were labeled for 30 min at 37 °C with 5 μM CellTracker Green (Life Technologies), according to the manufacturer’s instructions. Acceptor cells were trypsinized and co-plated with donor cells. After 24h in HBSS cells were either imaged, or BODIPY 558/568 C12 fluorescence determined in CellTracker Green-positive acceptor cells using a FACSCalibur cell analyzer (BD Biosciences).
Image processing, analysis and statistics
Images were analyzed using Slidebook Imaging Software (3i) and ImageJ (National Institutes of Health). Image brightness and contrast were adjusted in Adobe Photoshop CS.
For fluorescent FA pulse-chase assays, Z-stacks were analyzed using Slidebook to determine Pearson’s coefficients and mean fluorescence intensity. To quantify fluorescence intensity of Red C12 in mitochondria, a mask was made using the MitoTracker channel and then the mean fluorescence intensity in the Red C12 channel was calculated either across the entire mask or for individual objects larger than 10 pixels.
For LD-mitochondria proximity binary Z-stack images were analyzed using ImageJ ‘JACoP’ software to determine the overlap coefficient. For connectivity between mitochondria and LDs, a mask was created from segmented mitochondrial images and the percentage of mitochondrial signal connected to a site of LD-mitochondria proximity versus total mitochondrial signal was calculated.
LD size and number were quantified with the Image J ‘analyze particles’ function in thresholded images with size (pixel2) settings from 0.1 to 100 and circularity from 0 to 1. To determine LD volume/cell thresholded Z-stacks were subjected to the Volume function of the Slidebook (3i) software.
Data were expressed as means ± SEM. Statistical analysis among groups was performed using Student’s t test. * p< 0.05, ** p< 0.001, *** p< 0.0001.
Lipid extraction and TLC
Fluorescent pulse-chase assays were performed as described above, except that 1×107 cells were grown in 10 cm dishes and harvested by trypsinization. Lipids were extracted by homogenizing cell pellets in chloroform at 3×107 cells/ml and then spotted onto silica gel preparative TLC plates (Sigma-Aldrich). Separation of lipids was performed by developing the plates in a solvent system of cyclohexane/ethyl acetate, 1:2 (vol/vol), and fluorescent lipids were visualized using a Typhoon 9410 Molecular Imager (GE Amersham).
Mitochondrial activity measurements
Mitochondrial activity was determined using the Seahorse Flux Analyzer XF96, according to the manufacturer’s instructions. Briefly, 2×104 cells were seeded on 10μg/ml fibronectin-coated Seahorse 96 well plates. After 24 h, cells were incubated in CM or HBSS for the time points indicated. Sixteen replicates were performed per cell line and the average value was taken per experiment. To determine mitochondrial OCR (mtOCR), rotenone and antimycin were injected to acutely inhibit mitochondrial-driven oxygen consumption. mtOCR was determined by subtracting non-mitochondrial OCR from total OCR levels. Lipid oxidation-driven mtOCR was determined by inhibition of the mitochondrial FA importer CPT-I using etomoxir. OCR-data were normalized by cell number.
Supplementary Material
Acknowledgments
We thank N. Mizushima and D. C. Chan for contributing the Atg5KO and Mfn-deficient cell lines, respectively. We thank S. van Engelenburg, T. Lämmermann and G. Diering for helpful discussions and technical assistance. We are thankful to C. Stratakis for providing access to the Seahorse Flux Analyzer. We thank Alan Hoofring for help with illustrations. A.S.R. was supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
A.S.R., S.C. and J.L.-S. designed research; A.S.R. and S.C. performed research; A.S.R. and S.C. analyzed data, and A.S.R. and J.L.-S. wrote the paper.
References
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. The Journal of cell biology. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. The Journal of cell biology. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder PC. Long chain fatty acids and gene expression in inflammation and immunity. Current opinion in clinical nutrition and metabolic care. 2013;16:425–433. doi: 10.1097/MCO.0b013e3283620616. [DOI] [PubMed] [Google Scholar]
- Cabodevilla AG, Sanchez-Caballero L, Nintou E, Boiadijieva VG, Picatoste F, Gubern A, Claro E. Cell survival during complete nutrient deprivation depends on lipid droplet-fueled b-oxidation of fatty acids. The Journal of biological chemistry. 2013;288:27777–27788. doi: 10.1074/jbc.M113.466656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. The Journal of biological chemistry. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie E, Schulze A, Zechner R, Walther TC, Farese RV., Jr Cellular fatty acid metabolism and cancer. Cell metabolism. 2013;18:153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalen KT, Dahl T, Holter E, Arntsen B, Londos C, Sztalryd C, Nebb HI. LSDP5 is a PAT protein specifically expressed in fatty acid oxidizing tissues. Biochim Biophys Acta. 2007;1771:210–227. doi: 10.1016/j.bbalip.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Dietrich MO, Liu ZW, Horvath TL. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell. 155:188–199. doi: 10.1016/j.cell.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, Robenek H, Proikas-Cezanne T, Deretic V. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Current biology: CB. 2014;24:609–620. doi: 10.1016/j.cub.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton S. Control of mitochondrial beta-oxidation flux. Prog Lipid Res. 2002;41:197–239. doi: 10.1016/s0163-7827(01)00024-8. [DOI] [PubMed] [Google Scholar]
- Eaton S, Pourfarzam M, Bartlett K. The effect of respiratory chain impairment of beta-oxidation in rat heart mitochondria. Biochem J. 1996;319(Pt 2):633–640. doi: 10.1042/bj3190633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner V, Lenaers G, Hajnoczky G. Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. The Journal of cell biology. 205:179–195. doi: 10.1083/jcb.201312066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn PF, Dice JF. Proteolytic and lipolytic responses to starvation. Nutrition. 2006;22:830–844. doi: 10.1016/j.nut.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Fritz IB, Davis DG, Holtrop RH, Dundee H. Fatty acid oxidation by skeletal muscle during rest and activity. The American journal of physiology. 1958;194:379–386. doi: 10.1152/ajplegacy.1958.194.2.379. [DOI] [PubMed] [Google Scholar]
- Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nature reviews Drug discovery. 2008;7:489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway CA, Yoon Y. Perspectives on: SGP symposium on mitochondrial physiology and medicine: what comes first, misshape or dysfunction? The view from metabolic excess. J Gen Physiol. 2012;139:455–463. doi: 10.1085/jgp.201210771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway CA, Yoon Y. Mitochondrial morphology in metabolic diseases. Antioxidants & redox signaling. 2013;19:415–430. doi: 10.1089/ars.2012.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman JG, Moore HP, Mottillo EP, Zhu Z, Zou L. Interactions of perilipin-5 (Plin5) with adipose triglyceride lipase. The Journal o fbiological chemistry. 2011;286:5126–5135. doi: 10.1074/jbc.M110.180711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynberg A, Demaison L. Fatty acid oxidation in the heart. Journal of cardiovascular pharmacology. 1996;28(Suppl 1):S11–17. doi: 10.1097/00005344-199600003-00003. [DOI] [PubMed] [Google Scholar]
- Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nature medicine. 2011;17:1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- Herms A, Bosch M, Ariotti N, Reddy BJ, Fajardo A, Fernandez-Vidal A, Alvarez-Guaita A, Fernandez-Rojo MA, Rentero C, Tebar F, et al. Cell-to-cell heterogeneity in lipid droplets suggests a mechanism to reduce lipotoxicity. Current biology: CB. 23:1489–1496. doi: 10.1016/j.cub.2013.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annual review of biochemistry. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Hoppins S, Nunnari J. The molecular mechanism of mitochondrial fusion. Biochim Biophys Acta. 2009;1793:20–26. doi: 10.1016/j.bbamcr.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Jagerstrom S, Polesie S, Wickstrom Y, Johansson BR, Schroder HD, Hojlund K, Bostrom P. Lipid droplets interact with mitochondria using SNAP23. Cell Biol Int. 2009;33:934–940. doi: 10.1016/j.cellbi.2009.06.011. [DOI] [PubMed] [Google Scholar]
- Kassan A, Herms A, Fernandez-Vidal A, Bosch M, Schieber NL, Reddy BJ, Fajardo A, Gelabert-Baldrich M, Tebar F, Enrich C, et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. The Journal of cell biology. 2013;203:985–1001. doi: 10.1083/jcb.201305142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerner J, Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000;1486:1–17. doi: 10.1016/s1388-1981(00)00044-5. [DOI] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim do H, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nature medicine. 19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- Krahmer N, Farese RV, Jr, Walther TC. Balancing the fat: lipid droplets and human disease. EMBO Mol Med. 5:905–915. doi: 10.1002/emmm.201100671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AR, Schandorff S, Hoyer-Hansen M, Nielsen MO, Jaattela M, Dengjel J, Andersen JS. Ordered organelle degradation during starvation-induced autophagy. Molecular & cellular proteomics: MCP. 2008;7:2419–2428. doi: 10.1074/mcp.M800184-MCP200. [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. The Journal of cell biology. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nada MA, Rhead WJ, Sprecher H, Schulz H, Roe CR. Evidence for intermediate channeling in mitochondrial beta-oxidation. The Journal of biological chemistry. 1995;270:530–535. doi: 10.1074/jbc.270.2.530. [DOI] [PubMed] [Google Scholar]
- O’Neill HM, Holloway GP, Steinberg GR. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: implications for obesity. Mol Cell Endocrinol. 366:135–151. doi: 10.1016/j.mce.2012.06.019. [DOI] [PubMed] [Google Scholar]
- Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell metabolism. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A. 108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer J, Pourfarzam M, Bartlett K, Jackson S, Turnbull DM. Fatty acid oxidation in peripheral blood cells: characterization and use for the diagnosis of defects of fatty acid oxidation. Pediatr Res. 1995;37:354–360. doi: 10.1203/00006450-199503000-00017. [DOI] [PubMed] [Google Scholar]
- Schulze RJ, Weller SG, Schroeder B, Krueger EW, Chi S, Casey CA, McNiven MA. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. The Journal of cell biology. 2013;203:315–326. doi: 10.1083/jcb.201306140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CS, Jones DA, Wagenmakers AJ. Network distribution of mitochondria and lipid droplets in human muscle fibres. Histochem Cell Biol. 2008;129:65–72. doi: 10.1007/s00418-007-0349-8. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, Arai H, Tanaka K, Kominami E, Uchiyama Y. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun. 2009;382:419–423. doi: 10.1016/j.bbrc.2009.03.039. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yoshimura K, Tamura H, Ueno T, Nishimura T, Inoue T, Sasaki M, Koike M, Arai H, Kominami E, et al. LC3, a microtubule-associated protein1A/B light chain3, is involved in cytoplasmic lipid droplet formation. Biochem Biophys Res Commun. 393:274–279. doi: 10.1016/j.bbrc.2010.01.121. [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009a;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009b;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Goldberg EB, Makarova KS, Lin L, Brown WJ, Jackson CL. ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO reports. 2006;7:106–113. doi: 10.1038/sj.embor.7400559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturmey RG, O’Toole PJ, Leese HJ. Fluorescence resonance energy transfer analysis of mitochondrial:lipid association in the porcine oocyte. Reproduction. 2006;132:829–837. doi: 10.1530/REP-06-0073. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Rennie CD, Robertshaw HA, Fedak-Tarnopolsky SN, Devries MC, Hamadeh MJ. Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1271–1278. doi: 10.1152/ajpregu.00472.2006. [DOI] [PubMed] [Google Scholar]
- Thumser AE, Storch J. Characterization of a BODIPY-labeled fluorescent fatty acid analogue. Binding to fatty acid-binding proteins, intracellular localization, and metabolism. Molecular and cellular biochemistry. 2007;299:67–73. doi: 10.1007/s11010-005-9041-2. [DOI] [PubMed] [Google Scholar]
- Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. Embo J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, Clark GO, Scherer PE, Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta. 1801:209–214. doi: 10.1016/j.bbalip.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Valetti C, Wetzel DM, Schrader M, Hasbani MJ, Gill SR, Kreis TE, Schroer TA. Role of dynactin in endocytic traffic: effects of dynamitin overexpression and colocalization with CLIP-170. Mol Biol Cell. 1999;10:4107–4120. doi: 10.1091/mbc.10.12.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vock R, Hoppeler H, Claassen H, Wu DX, Billeter R, Weber JM, Taylor CR, Weibel ER. Design of the oxygen and substrate pathways. VI. structural basis of intracellular substrate supply to mitochondria in muscle cells. J Exp Biol. 1996;199:1689–1697. doi: 10.1242/jeb.199.8.1689. [DOI] [PubMed] [Google Scholar]
- Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends in endocrinology and metabolism: TEM. 2012;23:351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annual review of biochemistry. 81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, Sztalryd C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. Journal of lipid research. 52:2159–2168. doi: 10.1194/jlr.M017939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, Sztalryd C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. Journal of lipid research. 2011;52:2159–2168. doi: 10.1194/jlr.M017939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Soni KG, Semache M, Casavant S, Fortier M, Pan L, Mitchell GA. Lipolysis and the integrated physiology of lipid energy metabolism. Mol Genet Metab. 2008;95:117–126. doi: 10.1016/j.ymgme.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Watmough NJ, Bhuiyan AK, Bartlett K, Sherratt HS, Turnbull DM. Skeletal muscle mitochondrial beta-oxidation. A study of the products of oxidation of [U-14C]hexadecanoate by h.p.l.c. using continuous on-line radiochemical detection. Biochem J. 1988;253:541–547. doi: 10.1042/bj2530541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte MA, Cermelli S, Griner J, Viera A, Guo Y, Kim DH, Gindhart JG, Gross SP. Regulation of lipid-droplet transport by the perilipin homolog LSD2. Current biology: CB. 2005;15:1266–1275. doi: 10.1016/j.cub.2005.06.062. [DOI] [PubMed] [Google Scholar]
- Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Corce MA, Gropler MC, Yamaguchi T, Matsushita S, Motojima K, Hirose F, Osumi T. MLDP, a novel PAT family protein localized to lipid droplets and enriched in the heart, is regulated by peroxisome proliferator-activated receptor alpha. The Journal of biological chemistry. 2006;281:14232–14240. doi: 10.1074/jbc.M601682200. [DOI] [PubMed] [Google Scholar]
- Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–1185. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
- Yoon Y, Galloway CA, Jhun BS, Yu T. Mitochondrial dynamics in diabetes. Antioxidants & redox signaling. 2011;14:439–457. doi: 10.1089/ars.2010.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F. FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell metabolism. 2012;15:279–291. doi: 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.