

Abstract
Recovery from encephalomyelitis induced by infection with mosquito-borne alphaviruses is associated with a high risk of lifelong debilitating neurological deficits. Infection of mice with the prototypic alphavirus, Sindbis virus, provides an animal model with which to study disease mechanisms and examine potential therapeutics. Infectious virus is cleared from the brain within a week after infection, but viral RNA is cleared slowly and persists for the life of the animal. However, no studies have examined the effect of infection on neurocognitive function over time. In the present study, we examined neurocognitive function at different phases of infection in five-week-old C57BL/6 mice intranasally inoculated with Sindbis virus. At the peak of active virus infection, mice demonstrated hyperactivity, decreased anxiety, and marked hippocampal-dependent memory deficits, the latter of which persisted beyond clearance of infectious virus and resolution of clinical signs of disease. Previous studies indicate that neuronal damage during alphavirus encephalomyelitis is primarily due to inflammatory cell infiltration and glutamate excitotoxicity rather than directly by virus infection. Therefore, mice were treated with 6-diazo-5-oxo-l-norleucine (DON), a glutamine antagonist that can suppress both the immune response and excitotoxicity. Treatment with DON decreased inflammatory cell infiltration and cell death in the hippocampus and partially prevented development of clinical signs and neurocognitive impairment despite the presence of infectious virus and high viral RNA levels. This study presents the first report of neurocognitive sequelae in mice with alphavirus encephalomyelitis and provides a model system for further elucidation of the pathogenesis of virus infection and assessment of potential therapies.
Keywords: Sindbis virus, alphavirus, encephalomyelitis, fear conditioning, hippocampus, 6-diazo-5-oxo-L-norleucine (DON)
Introduction
Arthropod-borne alphaviruses and flaviviruses are plus-strand enveloped RNA viruses that pose an increasing worldwide threat to human populations as disease vectors expand into new geographic locations (Gubler 2002; van den Hurk et al. 2009; Griffin 2010; Lambrechts et al. 2010; Weaver and Reisen 2010). The New World alphaviruses, which include eastern equine encephalitis virus (EEEV), western equine encephalitis virus (WEEV), and Venezuelan equine encephalitis virus (VEEV), cause encephalomyelitis in humans and horses with varying rates of mortality (Steele et al. 2007; Griffin 2013). Many patients that recover from the acute clinical disease, especially infants and children, are left with life-long debilitating neurological defects, such as cognitive deficits, impaired motor control, and emotional and behavioral disturbances (Bruyn and Lennette 1953; Finley et al. 1955; Earnest et al. 1971; Villari et al. 1995). Currently, no treatments beyond symptomatic care are available, and no licensed human vaccines exist (Griffin 2010). All three viruses are endemic in the Americas, and encephalomyelitis outbreaks caused by EEEV and VEEV have increased over the last few decades (Weaver et al. 1996; Silverman et al. 2013). Therefore, it is increasingly important to understand the mechanisms responsible for the long-term consequences of alphavirus infection and to develop therapeutic interventions.
Sindbis virus (SINV), the prototypic alphavirus, produces rash and arthritis in humans but is neurotropic in mice and provides a valuable model for studying alphavirus-induced encephalomyelitis. In susceptible mice, nonlethal SINV infection consists of three phases in the brain: 1) presence of high levels of both infectious virus and viral RNA until about 7–8 days post infection (DPI); 2) undetectable infectious virus with significant yet declining viral RNA levels from about 10 to 60 DPI; and 3) chronic low-but-detectable steady state viral RNA levels from 60 DPI on, presumably for the remaining life of the animal (Metcalf and Griffin 2011). However, it is currently unknown whether SINV infection results in cognitive dysfunction in mice. Therefore, the aim of this study was to use this mouse model of alphavirus encephalomyelitis to determine the impact of viral infection on cognitive function and relate that to changes in brain structure and function. Motor, anxiety, and neurocognitive function were tested at each of the three different phases of SINV infection in mice. These data were correlated with the presence of infectious virus and viral RNA in the brain, along with severity of inflammation and cell death.
Neuronal damage resulting from alphavirus infection is due to both the immune response and glutamate excitotoxicity, and previous studies have shown that inhibition of these mechanisms can protect mice from fatal viral encephalomyelitis (Greene et al. 2008). However, treatment has not been evaluated for prevention of sequelae in nonfatal infection. Because the glutamine antagonist, 6-diazo-5-oxo-L-norleucine (DON), affects both of these pathologic mechanisms (Newsholme et al. 1985; Souba 1993; Colombo et al. 2010; Wang et al. 2011), we tested the effects of treatment with DON on the development of neurological sequelae. This study shows that SINV induces long-term neurological sequelae in mice that persist beyond active virus infection and that administration of DON partially mitigates development of these deficits.
Materials and Methods
Sindbis Virus Infection of Mice
Five week old male C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) were intranasally inoculated with 105 pfu of the TE strain of SINV (Lustig et al. 1988) in 20 μL PBS under light isoflurane anesthesia. Mock-infected control animals similarly received 20 μL PBS vehicle. Body weights were measured daily, and mice were observed for signs of clinical disease characterized by hunched posture, paresis, and abnormal gait. Mice were sacrificed on 5, 28, or 90 DPI (correlating with each phase of infection) following behavioral testing for the initial time course experiment or on 7 DPI for the treatment study. All studies were approved by the Johns Hopkins University Institutional Animal Care and Use Committee and carried out with strict adherence to the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health and the Policy on Humane Care and Use of Laboratory Animals by the U.S. Public Health Service.
Open Field
Animals were placed in the center of the open field arena (San Diego Instruments, San Diego, CA) at the beginning of the test and left undisturbed for 30 minutes. A 16 x 16 photobeam configuration was used to track the subject’s path within the arena. The total number of beams broken was used as a measure of locomotor activity, and the ratio of center to periphery breaks was used as a measure of anxiety (Potter et al. 2010; Marlatt et al. 2012). The investigator was blinded to the experimental conditions throughout testing.
Y-maze
The Y-maze consisted of three arms of equal length interconnected at 120°. This test measured working memory by scoring the number of alternations the mouse completed (animal visits all three arms without going into the same arm twice in a row) over five minutes (Melnikova et al. 2006). The investigator was blinded to the experimental conditions throughout testing.
Contextual and Cued Fear Conditioning
On day 1, mice were placed in the testing chamber for a total of 300 seconds; baseline was recorded from 0–120 seconds, and then three tone-shock pairings were applied. The first tone was given for 30 seconds between 120–150 seconds, which was paired with a two second foot shock (0.5 mA) during the last two seconds of the tone (148–150 sec). The second tone was given for 30 seconds between 180–210 seconds with the shock administered during the last two seconds (208–210 seconds). Finally, the third tone was given for 30 seconds between 240 to 270 seconds, with the shock between 268–270 seconds. On day 2, mice were placed into the same testing chamber and scored for percentage of freezing for five minutes during which time no tone or shock was given (contextual fear conditioning). Three hours after contextual testing, the mice were introduced to the same testing chamber with altered context. After a 300 second baseline (pretone), five tones were given for 30 seconds each at one-minute intervals without the shock pairing (tone). The percent time the mouse spent freezing was recorded during the tone phase just described and used as a measure of cued fear conditioning (Okun et al. 2010). The investigator was blinded to the experimental conditions throughout testing.
Quantification of Infectious Virus
Tissue homogenates were made using the left-brain hemispheres from three to six mice per group at each time point. Brains were placed in Lysing Matrix A tubes (MP Biomedicals, Santa Ana, CA), and ice-cold PBS was added to make 20% weight/volume concentrations. Tissues were dissociated at 6.0 M/s for 40 seconds using a FastPrep-24 homogenizer (MP Biomedicals) and centrifuged for 15 minutes at 13,200 rpm at 4°C. The clarified supernatant was used to quantify infectious virus via plaque assay. Ten-fold serial dilutions of homogenates were incubated on BHK cells for one hour followed by an agar overlay and evaluation of plaque formation after 48 hours.
Sindbis Viral RNA Quantification
Right brain hemispheres from three to four mice per group were dissociated in Lysing Matrix E tubes (MP Biomedicals) at 6.0 M/s for 40 seconds using a FastPrep-24 homogenizer (MP Biomedicals). RNA was isolated using the Qiagen RNeasy Lipid Tissue Mini kit (Valencia, CA), and cDNA was synthesized using random primers from a High Capacity cDNA Reverse Transcription Kit (Life Technologies, Grand Island, NY). Quantitative real-time PCR (qRT-PCR) was performed using TaqMan Universal PCR Master Mix and TaqMan probe nt 8760-5′–6-carboxyfluorescein (FAM)-CGCATACAGACTTCCGCCCAGT–6-carboxytetramethylrhodamine (TAMRA)-3′-8781 (Applied Biosystems, Carlsbad, CA) on 2.5 μL cDNA. Primers (forward, nt 8732-5′-TGGGACGAAGCGGACGATAA-3′-nt 8752; reverse, nt 8805-5′-CTGCTCCGCTTTGGTCGTAT-3′-nt 8786) were used to amplify the E2 structural region gene of SINV. Fifty reaction cycles were performed on a 7500 Fast Real-Time PCR System, and data were analyzed by relative quantitation using Sequence Detector software, version 1.4 (Applied Biosystems). Values were quantified via standard curve using ten-fold dilutions of a pGEM-3Z plasmid containing the E2 region and normalized to endogenous mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA.
Histology and Immunohistochemistry
Following euthanasia, three to four mice per group were perfused with ice cold PBS followed by 4% paraformaldehyde. Brains were collected and divided into three coronal sections using an Adult Mouse Brain Slicer (Zivic Instruments, Pittsburgh, PA) and fixed overnight in 4% paraformaldehyde at 4°C. Sectioned brains were then washed in PBS and embedded in paraffin. Three to four 10μm brain sections per mouse were stained with hematoxylin and eosin (H&E), coded, and scored blindly as previously described (Rowell and Griffin 1999) using a 0–3 scale. A score of 0 was given for slides with no detectable inflammation, a score of 1 for one to two small inflammatory foci per section, a score of 2 for moderate inflammatory foci in up to 50% of 10X fields, and a score of 3 for moderate to large inflammatory foci in greater than 50% of 10X fields. If excessive parenchymal cellularity was present, an additional point was added, allowing for a maximal score of 4.
For SINV antigen staining, 10 μm sections of brain were rehydrated and treated with 1mg/mL proteinase K (1:200 in distilled water) for 20 minutes, and endogenous peroxidase was quenched in 3% H2O2 for ten minutes. Tissues were blocked with 10% normal goat serum (NGS) in PBS for 20 minutes and stained with a rabbit polyclonal antibody to SINV ((Jackson et al. 1987) (1:200 in PBS + 5% NGS + 0.04% Triton-X) for 60 minutes, biotinylated anti-rabbit IgG secondary (5 mg/mL in PBS + 5% + 0.04% Triton-X) for 30 minutes, and avidin-biotin-horseradish peroxidase (HRP) complex (VECTASTAIN Elite ABC kit, Vector Labs, Burlingame, CA) for 40 minutes developed in 3,3′-diaminobenzidine (Vector Labs) for eight minutes. Slides were counterstained with hematoxylin for 60 seconds, dehydrated, and mounted with Permount (Fisher Scientific, Waltham, MA).
For TUNEL staining, 10 μm brain sections were rehydrated and treated with 1mg/mL proteinase K (1:200 in distilled water) for 30 minutes, and endogenous peroxidase was quenched in 3% H2O2 for five minutes. Sections were immersed in TdT Labeling Buffer and stained with TdT Labeling Reaction mix for 60 minutes at 37°C in a humidity chamber, immersed in TdT Stop Buffer, and stained with streptavidin-HRP solution for ten minutes (TACS 2 TdT kit, Trevigen Inc, Gaithersburg, MD). Tissues were developed in 3,3′-diaminobenzidine (Vector Labs) for seven minutes, counterstained with hematoxylin for 60 seconds, dehydrated, and mounted with Permount (Fisher Scientific).
For CD3+ cell staining, 10 μm brain sections were rehydrated and boiled in 0.01M Sodium Citrate (pH 6.0) for ten minutes for antigen retrieval. Endogenous peroxidase was quenched in 3% H2O2 for ten minutes, and sections were blocked with 10% NGS in PBS for 30 minutes. Sections were stained with polyclonal anti-human CD3 (1:400 in PBS + 5% NGS + 0.04% Triton-X, Dako, Carpenteria, CA) for 60 minutes, biotinylated anti-rabbit IgG secondary (5 mg/mL in PBS + 5% + 0.04% Triton-X) for 30 minutes, and avidin-biotin-HRP complex (VECTASTAIN Elite ABC kit, Vector Labs) for 40 minutes. Slides were developed in 3,3′-diaminobenzidine (Vector Labs) for ten minutes, counterstained with hematoxylin for 60 seconds, dehydrated, and mounted with Permount (Fisher Scientific).
Stained TUNEL and CD3 sections were coded, and the whole visible hippocampus on one brain section per mouse was outlined to determine the hippocampus area using a Nikon Eclipse E600 microscope and StereoInvestigator software (MBF Bioscience, Williston, VT). All TUNEL-positive or CD3-positive cells, as indicated by brown staining, were counted blindly within the outlined area, and results were analyzed as TUNEL-positive cells or CD3-positive cells per mm2 hippocampus. TUNEL-positive neuronal cell bodies in the granule cell layer of the dentate gyrus and in the pyramidal cell layer of the CA regions were also counted, and results were analyzed as TUNEL-positive neurons per mm2 hippocampus.
Drug Administration
Daily intraperitoneal injections of 6-diazo-5-oxo-l-norleucine (DON, Sigma-Aldrich, St. Louis, MO) were administered at doses of 0.3 or 0.6 mg/kg beginning the day of SINV inoculation (0 DPI) and continuing through 7 DPI. DON working solution was diluted in PBS each day from aliquots of a 100mM stock solution in PBS stored at −80°C. Control group animals received injections of PBS. On days of behavioral testing (5 and 6 DPI), the drug or vehicle was administered one hour prior to testing.
DON Measurement in Tissues
DON was spiked into mouse sera or brains from untreated animals to generate standards at concentrations from 10 nM to 100 μM. DON was derivatized into a more stable and lipophilic analyte by adding butanol with 3N HCl (250 μL) to standards and samples (50 μL). Brains were homogenized, and all samples were centrifuged at 16,000xg for five minutes to precipitate proteins. To carry out the derivatization reaction, an aliquot of supernatant (200 μL) was incubated at 60°C for 30 minutes and dried at 45°C under a nitrogen stream. Samples were resuspended in 50 μL 70%/30% water/acetonitrile, vortexed, and centrifuged again at 16,000xg for five minutes. Samples (2 μL) were subsequently injected and separated on an Agilent 1290 HPLC equipped with a C18 column over a 5.5 minute gradient from 30–70% acetonitrile + 0.1% formic acid and quantified on an Agilent 6520 QTOF mass spectrometer (Santa Clara, CA). Standards within the quantifiable range (30 nM–100 μM) were used to generate a standard curve. Samples below the limit of quantification were assigned a value of 0.
SINV-Specific Antibody Assay
SINV-specific IgM and IgG levels were evaluated in mouse sera by enzyme-linked immunosorbent assay (ELISA). Ninety-six well Maxisorp plates (Thermo Scientific Nunc, Marietta, OH) were coated overnight with a SINV TE-infected BHK cell lysate diluted in 50 mM NaHCO3 (pH 9.6). Wells were blocked for two hours at 37°C with 10% FBS in PBS-0.05% Tween 20, and diluted serum samples were added and incubated overnight at 4°C. Sera were diluted 1:100 for IgM detection and 1:10 for IgG detection in 10% FBS in PBS-0.05% Tween 20. Antibody was detected with HRP-conjugated goat anti-mouse IgM or IgG (Southern Biotech, Birmingham, AL) diluted 1:1000 in 10% FBS in PBS-0.05% Tween 20, for one hour at 37°C and developed using BD OptEIA TMB Substrate Reagent Set (San Jose, CA) as substrate and 2N H2SO4 as stop solution. Absorbance was read at 450nm, and the resulting values were plotted as the optical density (OD) values of SINV-infected samples minus the OD values for uninfected control sera.
Statistics
Statistical analyses were performed using Graphpad Prism 6 software (La Jolla, CA) or Statview (SAS institute inc.). For behavioral tests and body weights, an unpaired Student’s t-test was used for two-group comparisons and a one-way ANOVA with Bonferroni’s multiple comparisons post-test or two-way repeated measures ANOVA with Fisher’s PLSD post-hoc analysis was used to compare three or more groups. For clinical disease evaluation, a Mantel-Cox log-rank test was used to compare Kaplan-Meyer curves. Because small sample size precluded normal data distribution, nonparametric statistical analyses were employed for viral titer, RNA level, and histopathologic data. Mann-Whitney U tests were employed for two-group comparisons, and Kruskal-Wallis tests with Dunn’s multiple comparisons post-tests were used to compare three or more groups. A p value of ≤ 0.05 was considered significant in all analyses.
Results
Behavioral characterization of mice at different phases of Sindbis virus infection
SINV-infected mice were tested in the open field at 5, 28, and 90 DPI and compared to mock-infected control mice (Fig. 1a and 1b). Infected mice were hyperactive (Fig. 1a) and less anxious (Fig. 1b) compared to control mice 5 days after infection (p ≤ 0.01, Student’s t-test) but not at 28 or 90 DPI.
Fig. 1.

Assessment of locomotor activity, anxiety, and memory in SINV-infected mice. (a) In the open field test, SINV-infected mice were hyperactive 5 days after infection compared to mock-infected mice, but this characteristic was not observed at 28 or 90 DPI. (b) Infected mice also exhibited less anxiety in the open field at 5 DPI but not at 28 or 90 DPI as assessed by decreased ratio of center to periphery beam breaks. (c) Assessment of working memory in the Y-maze revealed that SINV had no effect on spontaneous alternation at 5, 28, or 90 DPI. (d,e) Assessment of hippocampal-dependent memory at 5 DPI showed that SINV-infected mice were severely impaired on both contextual (d) and cued (e) fear conditioning. Only contextual fear conditioning remained impaired at the 28 day and 90 day time points. (N=12–15/group; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001; Repeated measures ANOVA for cued fear conditioning and unpaired t-tests for all other tests; Data presented as mean ± SEM)
Assessment of spatial working memory on the Y-maze indicated that SINV infection had no effect on spontaneous alternation at 5, 28, or 90 DPI (Fig. 1c). At 5 DPI, the SINV-infected mice were severely impaired on both contextual (Fig. 1d; p ≤ 0.0001, Student’s t-test) and cued fear conditioning (Fig. 1e; p ≤ 0.0001, repeated measures ANOVA followed by Fisher’s PLSD). At the 28 and 90 DPI time points, contextual fear conditioning continued to be impaired (Fig. 1d; p ≤ 0.05, Student’s t-test) while an effect on cued conditioning was no longer detectable (Fig. 1e).
Cellular and molecular characterization of Sindbis virus infection over time
To interpret the findings from the behavioral testing, brains were collected and examined for the presence of virus, signs of inflammation, and evidence of cell death. Infectious virus was readily measureable at 5 DPI but undetectable at 28 and 90 DPI in brains of SINV-infected mice (Fig. 2a). In contrast, while viral RNA levels in the brain were highest at 5 DPI, SINV RNA was still measurable at 28 and 90 DPI (Fig. 2b).
Fig. 2.

SINV, inflammation, and cell death in the brain over time. (a) Infectious SINV was detectable by plaque assay at high levels in the brain at 5 DPI but undetectable at 28 and 90 DPI (dotted line represents limit of detection; ND = not detectable). (b) SINV RNA levels were highest at 5 DPI, lower at 28 DPI, and just above the level of detection at 90 DPI in the brain. (c) Representative photomicrographs of H&E-stained coronal brain sections from SINV-infected mice demonstrating the levels of parenchymal inflammation and perivascular cuffing in the hippocampus (white arrowheads denote perivascular cuffing; 200X magnification; scale bar = 100μm). (d) Representative photomicrographs of immunohistochemical staining for SINV antigen in infected mice at each time point. SINV protein was readily detectable in the hippocampus at 5 DPI, but not at 28 or 90 DPI (brown staining = SINV protein; 40X magnification; scale bar = 500μm). (e) Representative photomicrographs from TUNEL staining of the hippocampal dentate gyrus of SINV-infected mice at each time point. Multiple apoptotic cells were detected at 5 DPI but not at 28 and 90 DPI (brown nuclear staining = TUNEL-positive [denoted by black arrowheads]; 200X magnification; scale bar = 100μm). (f) Quantification of brain inflammation. Inflammation was greater at 5 and 28 DPI in SINV-infected than mock-infected control mice. (g) Quantification of TUNEL-positive cells in the hippocampus. Higher numbers of apoptotic/necrotic cells were detected in SINV-infected mice compared to mock-infected control mice at 5 DPI, but not at 28 or 90 DPI (For all bar graphs, N=3–4 mice per group per time point; data presented as mean ± SEM)
Moderate to marked levels of mononuclear cell infiltration and perivascular inflammation, as detected by H&E staining, were present throughout SINV-infected brains at 5 DPI; severity of inflammation decreased at 28 DPI, and by 90 DPI was at the level of uninfected controls (Fig.s 2c and 2f). SINV protein was widespread throughout the brain at 5 DPI and was especially abundant in the hippocampus, particularly in the granule cell layer of the dentate gyrus. At 28 and 90 DPI, SINV antigen was not detectable by immunohistochemical staining in the hippocampus (Fig. 2d) or other brain regions. The numbers of apoptotic cells in the hippocampus, as detected by TUNEL staining, were increased at 5 DPI in SINV-infected mice, but not at 28 or 90 DPI compared to mock-infected controls (Figs. 2e and 2g).
Concentrations of DON in serum and brain of Sindbis virus-infected mice
Due to the deficits in hippocampal-dependent memory seen in SINV-infected mice at 5 and 6 DPI, we next examined whether development of these signs could be prevented through administration of a glutamine antagonist, DON, that affects two different mechanisms known to contribute to the pathology seen during SINV infection: inflammatory damage induced by the immune response and glutamate excitotoxicity (Newsholme et al. 1985; Greene et al. 2008; Wang et al. 2011). After seven days of daily intraperitoneal drug administration at either a low dose (0.3 mg/kg) or high dose (0.6 mg/kg), DON was measured in the sera and brains of SINV-infected and mock-infected mice. DON was quantifiable in all serum samples from treated mice (Fig. 3a). The drug was also measurable in all six brain samples from mice dosed at 0.6 mg/kg, but in only two of the six brain samples from mice dosed at 0.3 mg/kg (Fig. 3b). Similar concentrations of DON were present in SINV- and mock-infected mice.
Fig. 3.
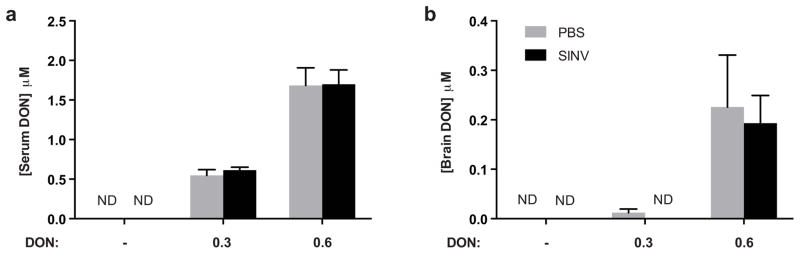
DON levels in tissues of SINV-infected and mock-infected mice. (a) Serum levels of DON were detectable at 7 DPI after high (0.6 mg/kg) and low-dose (0.3 mg/kg)-treatment (N=3 mice per group; ND = not detectable). (b) Brain levels of DON were detectable in the high dose (0.6 mg/kg) treatment group, but not consistently in the low dose (0.3 mg/kg) group (N=3 mice per group; ND = not detectable; data presented as mean ± SEM)
Effect of DON treatment on Sindbis virus-induced clinical disease
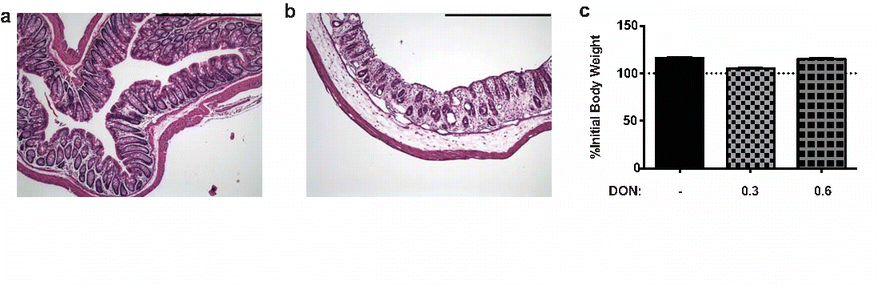
DON treatment was given for the first seven days following SINV infection. Clinical disease status was evaluated through daily observation for a characteristic combination of neurological signs (kyphosis, limb paresis, and abnormal gait) and by measurement of body weight. While almost all SINV-infected, untreated mice developed clinical signs of encephalomyelitis by 7 DPI, less than half of the DON-treated mice developed neurological signs by that time (Fig. 4a; p ≤ 0.0001; Mantel-Cox log rank test). Body weight differed between treatment groups at 4, 5, 6, and 7 DPI (Fig. 4b; p ≤ 0.0001; one-way ANOVA). SINV-infected mice in the low-dose (0.3 mg/kg) DON treatment group lost less weight than the untreated SINV-infected mice (p ≤ 0.01; Bonferonni’s multiple comparisons test). However, the weight loss was greater than for the mock-infected untreated and mock-infected low-dose (0.3 mg/kg) DON groups (p ≤ 0.0001 and p ≤ 0.01, respectively; Bonferonni’s multiple comparisons test). Mice in the high-dose (0.6 mg/kg) DON-treatment groups, regardless of infection status, lost the most weight over the seven-day time course, and marked loss of gastrointestinal (GI) tract architecture indicative of GI toxicity was seen histologically in those mice (Figs. S1a and S1b). By 28 DPI, SINV-infected mice treated with high-dose (0.6mg/kg) DON had regained the lost weight, as had mice in the untreated and low-dose (0.3mg/kg) DON-treated, SINV-infected groups (Fig. S1c).
Fig. 4.
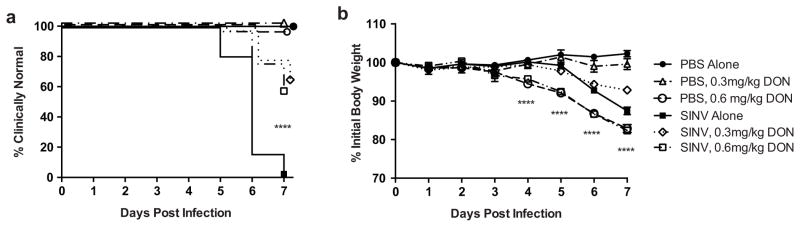
Clinical disease in DON-treated, SINV-infected mice. (a) Kaplan-Meyer plot of time to development of signs of neurologic disease. All untreated SINV-infected mice developed clinical signs by 7 DPI, while over half of the low and high dose DON-treated, SINV-infected mice remained clinically normal (****p ≤ 0.0001 by log-rank [Mantel Cox] test; N=9–32 mice over 3 separate independent experiments). (b) Changes in body weight. SINV-infected mice receiving low-dose (0.3 mg/kg) DON treatment lost less weight by 7 DPI compared to untreated SINV-infected mice, but not as much as mice receiving high-dose (0.6 mg/kg) DON treatment (****p ≤ 0.0001, one-way ANOVA with p ≤ 0.01 by Bonferroni’s multiple comparison tests; N=9–32 mice over three separate independent experiments; data presented as mean ± SEM)
Impact of DON treatment on behavioral effects induced by Sindbis virus
Because SINV infection most severely affected behavior during the first phase of infection, mice treated with DON were tested in the open field and in both contextual and cued fear conditioning experimental paradigms at 5 DPI.
In the open field test, there was a significant difference between groups in the number of beam breaks (Fig. 5a; p ≤ 0.0001, one-way ANOVA). Specifically, the untreated SINV-infected group had a greater number of beam breaks compared to all other groups (p ≤ 0.01, Fisher’s PLSD). The mock-infected untreated group and the low (0.3 mg/kg) and high (0.6 mg/kg) dose SINV-treated groups were not significantly different from each other, indicating that DON protected from SINV-induced hyperlocomotion. Interestingly, the mock-infected high dose (0.6 mg/kg) DON-treated group had fewer beam breaks compared to all other groups (p ≤ 0.01, Fisher’s PLSD), indicating that chronic DON treatment of control mice can also affect motor abilities. Anxiety levels, as measured by the ratio of beam breaks in the center of the arena to those in the periphery, were also significantly different between groups (Fig. 5b; p ≤ 0.001, one-way ANOVA). Specifically, the untreated SINV-infected group had an increased ratio compared to all other groups (p ≤ 0.05, Fisher’s PLSD). The other groups were not significantly different from each other, indicating that DON protected from SINV-induced alterations in anxiety levels and did not affect anxiety levels in control mice.
Fig. 5.

Effect of DON on locomotor activity, anxiety and hippocampal-dependent memory. (a) At 5 DPI in the open field test, the untreated SINV-infected mice had a significantly greater number of beam breaks compared to all other groups. The mock-infected untreated and the low (0.3 mg/kg) and high (0.6 mg/kg) dose SINV-treated groups were not significantly different from each other. The mock-infected high dose (0.6 mg/kg) DON treated group had significantly fewer beam breaks compared to all other groups. (b) In measurement of anxiety levels, the untreated SINV-infected group had a significantly increased ratio of beam breaks in the center of the arena to those in the periphery compared to all other groups. These other groups were not significantly different to each other. (c,d) High dose (0.6 mg/kg) DON partially reversed the deficits observed in contextual but not in cued fear conditioning. (c) For contextual fear conditioning, mock-infected groups were significantly different from all three SINV-infected groups. The DON-treated SINV-infected groups were not different from each other. The SINV-infected mice in the high dose (0.6 mg/kg) DON-treated group, but not the low dose (0.3 mg/kg), differed significantly from the SINV-infected untreated mice. (d) For cued fear conditioning, both mock-infected groups were significantly different from all three SINV-infected groups but were not significantly different from each other. There was no difference between the three SINV-infected groups. (* p ≤ 0.05; ** p ≤ 0.01; **** p ≤ 0.0001, ANOVA followed by Fisher’s PLSD; N=11–27/group; Data presented as mean ± SEM)
Overall, there was a significant difference between groups in contextual fear conditioning (Fig. 5c; p ≤ 0.0001, one-way ANOVA). Specifically, the mock-infected groups had a greater percentage of time spent freezing than the three SINV-infected groups (p ≤ 0.0001, Fisher’s PLSD). The two DON-treated SINV-infected groups were not significantly different from each other, but the SINV-infected mice treated with high dose (0.6 mg/kg) DON, but not the low dose (0.3 mg/kg), showed a significant improvement in contextual fear conditioning compared to the SINV-infected untreated group (p ≤ 0.01, Fisher’s PLSD), indicating partial protection by DON from the deficit induced by SINV infection. Treatment of mock-infected mice with DON (0.6 mg/kg) had no effect on performance.
Testing of cued fear conditioning revealed an overall significant difference between groups (Fig. 5d; p ≤ 0.0001, repeated measures ANOVA). There was a decrease in percent freezing in the three SINV-infected groups compared to both mock-infected groups (p ≤ 0.0001, Fisher’s PLSD). However, neither dose of DON had a detectable effect on protecting against this deficit. Treatment of mock-infected mice with DON (0.6 mg/kg) had no effect on performance.
Effect of DON treatment on viral load, inflammation, and cell death
Following behavior testing, mice were sacrificed at 7 DPI to examine the effects of DON treatment on SINV levels, inflammation, and cell death. While infectious virus was present in the brains of both untreated and DON-treated mice, virus titers differed significantly (Fig. 6a; p ≤ 0.01, Kruskal-Wallis test). Mice in the high-dose (0.6 mg/kg) DON group had almost ten-fold higher titers than untreated mice (p ≤ 0.05, Dunn’s multiple comparisons test). However, no difference was detected in levels of SINV RNA between groups at this time (Fig. 6b).
Fig. 6.

Effect of DON treatment on SINV, inflammation and cell death at 7 DPI. (a) Amounts of infectious SINV as determined by plaque formation were significantly higher in the brains of high dose (0.6 mg/kg) DON-treated mice than untreated mice (p ≤ 0.01 by Kruskal-Wallis test with *p ≤ 0.05 by Dunn’s multiple comparisons post test; N=4–6 mice per group; dotted line indicates the limit of detection). (b) SINV RNA levels in the brain as determined by qRT-PCR were not affected by DON treatment (N=3 mice per group). (c) Representative photomicrographs of hematoxylin & eosin-stained coronal brain sections demonstrating the effect of DON treatment on inflammatory cell infiltration into the hippocampus of SINV-infected mice (200X magnification; scale bar = 100μm). (d) Representative photomicrographs of immunohistochemical staining for SINV antigen in DON-treated or untreated, SINV-infected mice showing the comparable levels of SINV protein in the brain (SINV antigen = brown staining; 40X magnification; scale bar = 500μm). (e) Representative photomicrographs TUNEL staining of the dentate gyrus in the hippocampus of SINV-infected mice showing higher numbers of apoptotic/necrotic cells in untreated mice compared to DON-treated mice (DNA fragmentation = brown nuclear staining; 200X magnification; scale bar = 100μm). (f) Representative photomicrographs CD3 staining of the hippocampus of SINV-infected mice showing higher numbers of CD3+ cells in untreated mice compared to DON-treated mice (CD3+ cells = brown staining; 200X magnification; scale bar = 100μm). (g) Quantification of inflammation showed greater levels in the brains of untreated, SINV-infected mice compared to mock-infected control mice and high dose (0.6 mg/kg) DON-treated, SINV-infected mice (p ≤ 0.01 by Kruskal-Wallis test with * p ≤ 0.05 by Dunn’s multiple comparisons post test; N=3–6 mice per group). (h) Quantification of apoptotic/necrotic cells showing greater numbers in the hippocampus of untreated SINV-infected mice than mock-infected or DON-treated mice (p ≤ 0.05 by Kruskal-Wallis test with * p ≤ 0.05 by Dunn’s multiple comparisons post test; N=3 mice per group). (i) Quantification of apoptotic/necrotic neuronal cell bodies in the granule layer of the dentate gyrus and pyramidal layer of the CA regions, showing greater numbers in untreated SINV-infected mice than mock-infected or DON-treated mice (p ≤ 0.05 by Kruskal-Wallis test; N=3 mice per group). (j) Inflammation score and number of TUNEL+ cells are positively correlated (open symbols = SINV infection, closed symbols = PBS mock infection, circles = no treatment, squares = DON treatment; N=17 mice). (k) Quantification of CD3+ cells showing greater numbers in the hippocampus of untreated SINV-infected mice than mock-infected or DON-treated mice (p ≤ 0.05 by Kruskal-Wallis test; for all bar graphs, N=3 mice per group per time point). OD measurements at 450nm for SINV-specific IgM (l) and IgG (m) in serum show decreased to undetectable levels in DON-treated mice (p ≤ 0.05 by Kruskal-Wallis test; N=2–4 mice per group; for all bar graphs, data presented as mean ± SEM)
H&E-stained brain sections from SINV-infected mice were scored for inflammation and significantly differed between untreated and DON-treated groups (Figs. 6c and 6g; p ≤ 0.01, Kruskal-Wallis test). Specifically, untreated SINV-infected mice had higher scores than mock-infected mice or high dose (0.6 mg/kg) DON-treated, SINV-infected mice (p ≤ 0.05, Dunn’s multiple comparisons test). Immunohistochemical staining showed that SINV protein was present throughout the brain in untreated mice and in both groups of DON-treated mice (Fig. 6d). SINV was readily detected in the granule cell layer as well as the polymorphic layer of the dentate gyrus and in the pyramidal layer of both CA1 and CA3 of the hippocampus. The numbers of total apoptotic cells in the hippocampus (Figs. 6e and 6h) and apoptotic neurons in the granule cell layer of the dentate gyrus and pyramidal cell layers of the CA regions (Fig. 6i) also differed between infection and treatment groups (p ≤ 0.05, Kruskal-Wallis test), with the highest number of positive cells seen in the untreated SINV-infected group. Inflammation and cell death were positively correlated in mouse brains, regardless of infection or treatment status (Fig. 6j; Spearman ρ = 0.77, p ≤ 0.001). Infiltration of CD3+ T cells differed among groups (Figs. 6f and 6k; p ≤ 0.05, Kruskal-Wallis test), with higher numbers of positive cells in untreated SINV-infected brains. Treatment of mock-infected animals with DON did not affect either inflammation or cell death compared to untreated mock-infected mice (Figs. 6g, 6h, 6i, and 6k).
SINV-specific antibody levels were measured in sera of SINV-infected mice at 7 DPI. IgM and IgG were readily detectable in untreated, SINV-infected mice (Figs. 6l and 6m). However, of the SINV-infected, DON-treated groups, only low levels of IgM were found in the sera of low-dose (0.3mg/kg) DON-treated mice. IgM antibody in the high-dose (0.6mg/kg) DON-treated group and IgG antibody in either DON-treated group were undetectable.
Discussion
In the present study, we show that at the peak of active virus infection, SINV-infected mice demonstrated increased activity, reduced anxiety, and impaired hippocampal-dependent contextual and cued fear conditioning memory. Eleven weeks after the clearance of infectious virus when only viral RNA was present in the brain, SINV-infected mice continued to show significant memory impairment as indicated in contextual fear conditioning tests. Treatment for seven days after infection with the glutamine antagonist DON improved outcome without inhibiting virus replication or improving virus clearance. Rather, DON treatment decreased inflammatory cell infiltration and prevented cell death in the brain compared to untreated SINV-infected mice. These studies document the presence of neurologic deficits in a mouse model long after apparent recovery from viral encephalitis and indicate that neuroprotection with agents that decrease inflammation and excitotoxic damage is an encouraging approach to preventing sequelae.
During the early stages of infection, SINV mice were hyperactive and less anxious in the open field test compared to uninfected mice. Epidemic encephalitis can lead to behavioral abnormalities similar to ADHD, with damage to the brainstem suggested as a cause (Millichap 2008). Other areas that are important for locomotor activity are the basal ganglia and the cerebellum. The amygdala is a central area of processing fear and anxiety (Adhikari 2014). Indeed, in cases of viral encephalitis such as from human herpesvirus 6 (HHV6), there was significant damage to the medial temporal lobe including the amygdala (Provenzale et al 2008). Neurological sequelae in patients surviving alphavirus encephalomyelitis have been reported to include hyperactivity, emotional disinhibition, and temperament disturbances (Bruyn and Lennette 1953; Finley et al. 1955; Earnest et al. 1971; Villari et al. 1995), and symptoms similar to attention-deficit/hyperactivity disorder have been described in patients affected by encephalitis caused by other types of viruses, such as HIV (Nozyce 2006; Millichap 2008).
The most profound and prolonged abnormalities were identified using fear conditioning. Contextual and cued fear conditioning involve different brain circuitry, with contextual fear conditioning being most associated with the hippocampus (Phillips and LeDoux 1992; Sanders et al. 2003; Kim and Jung 2006; Wiltgen 2006; Crawley 2007). While SINV infects neurons in the cortex, brainstem, and spinal cord (Jackson et al. 1987), previous studies of mice infected with a lethal neuroadapted strain of SINV (NSV) have noted that hippocampal neurons are particularly vulnerable to progressive damage (Kimura and Griffin 2003). Hippocampal neurons are also particularly susceptible to glutamate-induced excitotoxic damage, which plays a role in SINV-induced cell death (Nadler et al. 1978; Olney et al. 1979; Nargi-Aizenman et al. 2004). In addition, the hippocampus plays a very important role in cognition, and factors affecting learning and memory processes can be probed using tasks such as fear conditioning (Crawley 2007; Maren et al. 2013). In our study, deficits in fear conditioning at 5 DPI correlated with demonstration of active virus replication, inflammation, and cell death in the hippocampus during the acute phase of infection. Impairments in contextual, but not cued, fear conditioning persisted for at least 90 days after infection, a time when the mice appeared to have fully recovered.
Glutamatergic drugs such as AMPA and NMDA receptor antagonists have been previously shown to enhance survival of hippocampal neurons and increase lifespan in NSV-infected mice by prevention of immune-mediated damage and inhibition of excitotoxicity (Nargi-Aizenman et al. 2004; Greene et al. 2008). In the current study, we tested the effects of a glutamine antagonist, DON, for its potential to mitigate the consequences of nonfatal SINV infection seen at the peak of infection. DON blocks the conversion of glutamine to glutamate, thus decreasing the potential for glutamate excitotoxicity in the brain, but also suppresses proliferation of lymphocytes, which require glutamine to support rapid cell division induced by antigen stimulation (Newsholme et al. 1985; Colombo et al. 2010; Wang et al. 2011). DON was examined as a potential chemotherapeutic agent in the 1970’s and 1980’s, but it was not well tolerated in clinical trials due primarily to GI toxicity (Sklaroff et al. 1980; Kovach et al. 1981; Earhart et al. 1982; Earhart et al. 1990; Souba 1993). In our study, mice in the high dose (0.6 mg/kg) DON treatment group developed severe intestinal pathology, likely explaining the marked weight loss seen regardless of infection status. These signs may also help explain why we observed a reduced motor ability in DON-treated mice. We do not believe that this influenced the cognitive tests since there was no difference in freezing levels between mock-infected mice with or without DON during fear conditioning testing. Previous studies using DON (Shijie et al. 2009; Shelton et al. 2010) have also reported issues with drug toxicity and dosing regimens were modified to counteract these negative side effects. In its present form, DON does not represent a viable treatment for alphavirus encephalomyelitis. However, by serving as a prototype, different glutamine antagonists or different formulations or derivatives of DON can be further evaluated as potential therapeutic options.
The glutamine antagonist DON partially prevented development of the deficit in contextual, but not cued, fear conditioning. This result suggests that cued and contextual conditioning are differentially susceptible to the effects of DON, which may be related to differences between anatomical structures and circuitry that support both types of learning. While the amygdala is essential for both cued and contextual fear conditioning, the hippocampus is not essential for cued fear conditioning but does contribute to contextual fear conditioning (Phillips and LeDoux 1992; Rudy 1993; Phillips and LeDoux 1994; Logue et al. 1997; Holt and Maren 1999). Indeed, this disconnect between effects on cued and contextual conditioning has been shown with other glutamate antagonists such as MK-801 (Gould et al. 2002; Goeldner et al. 2009).
Long-term persistence of a viral RNA reservoir in neurons presents the possibility of viral reactivation, relapse of disease, and progressive pathology (Levine and Griffin 1992). Both infectious virus and SINV RNA typically peak in the brains of infected mice at 3–5 DPI, and while infectious virus quickly decreases to undetectable levels by 8–10 DPI, RNA levels slowly decline but remain measureable for at least a year (Metcalf and Griffin 2011). At 7 DPI, a time when virus clearance has typically been initiated, mice receiving high-dose (0.6 mg/kg) DON had significantly higher titers than untreated mice, due to a delay in virus clearance likely associated with a decrease in immune cell infiltration into the CNS. While infectious virus clearance occurs quickly, resulting in a precipitous drop in titers over the course of a few days following clearance initiation, decline in viral RNA levels proceeds at a much more gradual rate over several weeks (Metcalf and Griffin 2011). Therefore, a significant difference in viral RNA levels among treatment groups may not be evident yet at this time in the clearance process.
Previous studies using the more neurovirulent NSV strain of SINV have shown that the immune response to infection plays a significant role in the neuronal damage and death seen during fatal alphavirus encephalomyelitis (Kimura and Griffin 2003), as well as in virus clearance. While neuronal cell death has been found to occur independent of DNA fragmentation in the motor neurons of the spinal cord during NSV infection, neuronal damage appears to follow the classical apoptotic pathway in the hippocampus and can be detected through TUNEL staining (Havert et al. 2000). In the present study, treatment with DON resulted in decreased inflammatory cell infiltration, including CD3+ T cells, and lower levels of cell death in the brain, despite increased levels of infectious virus and comparable levels of viral antigen and RNA. Moreover, fewer DON-treated mice developed neurological signs by 7 DPI compared to untreated SINV-infected mice. The numbers of apoptotic neurons in the granule cell layer of the dentate gyrus and pyramidal cell layers of the CA regions differed between infection and treatment groups, with the highest number of positive cells seen in the untreated SINV-infected group. This loss of neurons across the hippocampal subfields, areas key to intact cognitive abilities, helps to explain the significantly impaired performance this group displayed in behavior tests.
Despite obtaining high serum levels, DON did not penetrate the brain well, suggesting that the drug’s effect on alphavirus pathology primarily occurs through peripheral suppression of the antiviral immune response. DON treatment resulted in fewer CD3+ T cells infiltrating the brain and mostly-undetectable levels of circulating SINV-specific IgM and IgG compared to untreated SINV-infected mice, supporting this hypothesis. Decreased inflammatory cell infiltration into the brain reduces neuronal damage and thus explains the lack of clinical signs and weight loss in the low-dose (0.3 mg/kg) DON group. Previous studies have shown that alphavirus clearance from the CNS is primarily facilitated by antibody against the SINV E2 glycoprotein (Levine et al 1991) and the cytokine interferon gamma produced by NK cells and T cells (Binder and Griffin 2001). In contrast to untreated, SINV-infected mice, SINV-specific antibody levels were considerably lower or undetectable in the sera of DON-treated mice, and infectious virus titers were significantly higher in the brains of high-dose (0.6mg/kg) DON-treated mice at 7 DPI. Therefore, while compromised immune cell proliferation in the periphery and infiltration into the brain results in decreased CNS pathology, it also causes impaired virus clearance from neurons. This supports the importance of the immune response in the pathogenesis of the less virulent SINV TE strain, as well as NSV, and suggests that immunosuppression in the periphery plays an important role in the mitigation of nonfatal alphavirus encephalomyelitis by glutamine antagonism.
Although virus-induced behavioral alterations in mice have been previously reported (Amaral et al. 2011; Jurgens et al. 2012; Umpierre et al. 2014), to our knowledge, this is the first study of cognitive deficits and other behavioral abnormalities in a mouse model of alphavirus infection. The continued emergence of viral encephalomyelitis outbreaks around the world emphasizes the need to identify novel therapeutics to improve outcome using an animal model. These findings may also be applicable to treatment of other virus infections of the nervous system that share similar cognitive and pathological features including immune activation, inflammation and excitotoxicity (McArthur et al. 2010; Fitting et al. 2013; Potter et al. 2013). Additionally, the immunosuppressive nature of DON makes it a useful tool to further elucidate the mechanisms and pathogenesis of alphavirus-induced disease.
Supplementary Material
Gastrointestinal toxicity associated with high dose (0.6mg/kg) DON treatment. Representative photomicrographs of hematoxylin & eosin-stained large intestine from (a) untreated, mock-infected and (b) high dose (0.6mg/kg) DON, mock-infected mice at 7 DPI. High dose DON-treatment resulted in intestinal dilatation with loss of columnar epithelium and decreased cellularity (100X magnification; scale bar = 500μm). (c) SINV-infected mice receiving high dose (0.6mg/kg) DON regained body weight lost by 28 DPI (N=3–4 mice per group)
{kind=link}
Acknowledgments
The following grants were used to fund this research: NIH grants: R01 NS038932 (DEG), R01 NS087539 (DEG), T32 8T32OD011089 (VKB), P30 MH075673 (BSS), and R03 DA032470 (BSS), as well as a pilot grant from the Brain Science Institute of Johns Hopkins University School of Medicine. The funding sources played no role in the conduct of the research, preparation of the paper, or decision to submit the article for publication. The authors would like to thank Joseph Mankowski, Kelly Metcalf Pate, Lisa Mangus, and Claire Lyons along with the Retrovirus group at Johns Hopkins University for the use of and assistance with their microscope and imaging software. We would additionally like to express our appreciation to Sivabalan Manivannan for his assistance in preparing the DON stock solution.
Footnotes
The authors declare no competing financial interests.
References
- Adhikari A. Distributed circuits underlying anxiety. Front Behav Neurosci. 2014;8:112. doi: 10.3389/fnbeh.2014.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DC, Rachid MA, Vilela MC, Campos RD, Ferreira GP, Rodrigues DH, Lacerda-Queiroz N, Miranda AS, Costa VV, Campos MA, Kroon EG, Teixeira MM, Teixeira AL. Intracerebral infection with dengue-3 virus induces meningoencephalitis and behavioral changes that precede lethality in mice. J Neuroinflammation. 2011;8:23. doi: 10.1186/1742-2094-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder GK, Griffin DE. Interferon-γ-mediated site-specific clearance of alphavirus from CNS neurons. Science. 2001;293:303–306. doi: 10.1126/science.1059742. [DOI] [PubMed] [Google Scholar]
- Bruyn HB, Lennette EH. Western equine encephalitis in infants; a report on three cases with sequelae. Calif Med. 1953;79:362–366. [PMC free article] [PubMed] [Google Scholar]
- Colombo SL, Palacios-Callender M, Frakich N, De Leon J, Schmitt CA, Boorn L, Davis N, Moncada S. Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc Natl Acad Sci USA. 2010;107:18868–18873. doi: 10.1073/pnas.1012362107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. What’s wrong with my mouse?: behavioral phenotyping of transgenic and knockout mice. 2. Wiley-Interscience; Hoboken, NJ: 2007. [Google Scholar]
- Earhart RH, Amato DJ, Chang AY, Borden EC, Shiraki M, Dowd ME, Comis RL, Davis TE, Smith TJ. Phase II trial of 6-diazo-5-oxo-L-norleucine versus aclacinomycin-A in advanced sarcomas and mesotheliomas. Invest New Drugs. 1990;8:113–119. doi: 10.1007/BF00216936. [DOI] [PubMed] [Google Scholar]
- Earhart RH, Koeller JM, Davis HL. Phase I trial of 6-diazo-5-oxo-L-norleucine (DON) administered by 5-day courses. Cancer Treat Rep. 1982;66:1215–1217. [PubMed] [Google Scholar]
- Earnest MP, Goolishian HA, Calverley JR, Hayes RO, Hill HR. Neurologic, intellectual, and psychologic sequelae following western encephalitis. A follow-up study of 35 cases. Neurology. 1971;21:969–974. doi: 10.1212/wnl.21.9.969. [DOI] [PubMed] [Google Scholar]
- Finley KH, Longshore WA, Jr, Palmer RJ, Cook RE, Riggs N. Western equine and St. Louis encephalitis; preliminary report of a clinical follow-up study in California. Neurology. 1955;5:223–235. doi: 10.1212/wnl.5.4.233. [DOI] [PubMed] [Google Scholar]
- Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF. Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry. 2013;73:443–453. doi: 10.1016/j.biopsych.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeldner C, Reiss D, Wichmann J, Kieffer BL, Ouagazzal A-M. Activation of nociceptin opioid peptide (NOP) receptor impairs contextual fear learning in mice through glutamatergic mechanisms. Neurobiol Learn Mem. 2009;91:393–401. doi: 10.1016/j.nlm.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TJ, McCarthy MM, Keith RA. MK-801 disrupts acquisition of contextual fear conditioning but enhances memory consolidation of cued fear conditioning. Behav Pharmacol. 2002;13:287–294. doi: 10.1097/00008877-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Greene IP, Lee E-Y, Prow N, Ngwang B, Griffin D. Protection from fatal viral encephalomyelitis: AMPA receptor antagonists have a direct effect on the inflammatory response to infection. Proc Natl Acad Sci USA. 2008;105:3575–3580. doi: 10.1073/pnas.0712390105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin DE. Emergence and re-emergence of viral diseases of the central nervous system. Prog Neurobiol. 2010;91:95–101. doi: 10.1016/j.pneurobio.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin DE. Alphaviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6. Lippincott Williams & Wilkins; Philadelphia: 2013. pp. 652–686. [Google Scholar]
- Gubler DJ. The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res. 2002;33:330–342. doi: 10.1016/s0188-4409(02)00378-8. [DOI] [PubMed] [Google Scholar]
- Havert MB, Schofield B, Griffin DE, Irani DN. Activation of divergent neuronal cell death pathways in different target cell populations during neuroadapted sindbis virus infection of mice. J Virol. 2000;74:5352–5356. doi: 10.1128/JVI.74.11.5352-5356.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt W, Maren S. Muscimol inactivation of the dorsal hippocampus impairs contextual retrieval of fear memory. J Neurosci. 1999;19:9054–9062. doi: 10.1523/JNEUROSCI.19-20-09054.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hurk AF, Ritchie SA, Mackenzie JS. Ecology and geographical expansion of Japanese encephalitis virus. Annu Rev Entomol. 2009;54:17–35. doi: 10.1146/annurev.ento.54.110807.090510. [DOI] [PubMed] [Google Scholar]
- Jackson AC, Moench TR, Griffin DE, Johnson RT. The pathogenesis of spinal cord involvement in the encephalomyelitis of mice caused by neuroadapted Sindbis virus infection. Lab Invest. 1987;56:418–423. [PubMed] [Google Scholar]
- Jurgens HA, Amancherla K, Johnson RW. Influenza infection induces neuroinflammation, alters hippocampal neuron morphology, and impairs cognition in adult mice. J Neurosci. 2012;32:3958–3968. doi: 10.1523/JNEUROSCI.6389-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Jung MW. Neural circuits and mechanisms involved in Pavlovian fear conditioning: A critical review. Neurosci Biobehav Rev. 2006;30:188–202. doi: 10.1016/j.neubiorev.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Griffin DE. Extensive immune-mediated hippocampal damage in mice surviving infection with neuroadapted Sindbis virus. Virology. 2003;311:28–39. doi: 10.1016/S0042-6822(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Kovach JS, Eagan RT, Powis G, Rubin J, Creagan ET, Moertel CG. Phase I and pharmacokinetic studies of DON. Cancer Treat Rep. 1981;65:1031–1036. [PubMed] [Google Scholar]
- Lambrechts L, Scott TW, Gubler DJ. Consequences of the expanding global distribution of Aedes albopictus for dengue virus transmission. PLoS Negl Trop Dis. 2010;4:e646. doi: 10.1371/journal.pntd.0000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Griffin DE. Persistence of viral RNA in mouse brains after recovery from acute alphavirus encephalitis. J Virol. 1992;66:6429–6435. doi: 10.1128/jvi.66.11.6429-6435.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Hardwick JM, Trapp BD, Crawford TO, Bollinger RC, Griffin DE. Antibody-mediated clearance of alphavirus infection from neurons. Science. 1991;254:856–860. doi: 10.1126/science.1658936. [DOI] [PubMed] [Google Scholar]
- Logue SF, Paylor R, Wehner JM. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci. 1997;111:104–113. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- Lustig S, Jackson AC, Hahn CS, Griffin DE, Strauss EG, Strauss JH. Molecular basis of Sindbis virus neurovirulence in mice. J Virol. 1988;62:2329–2336. doi: 10.1128/jvi.62.7.2329-2336.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Phan KL, Liberzon I. The contextual brain: implications for fear conditioning, extinction and psychopathology. 2013:1–12. doi: 10.1038/nrn3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlatt MW, Potter MC, Lucassen PJ, van Praag H. Running throughout middle-age improves memory function, hippocampal neurogenesis, and BDNF levels in female C57BL/6J mice. Devel Neurobio. 2012;72:943–952. doi: 10.1002/dneu.22009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur JC, Steiner J, Sacktor N, Nath A. HIV-associated neurocognitive disorders: “mind the gap”. Ann Neurol. 2010 doi: 10.1002/ana.22053. NA–NA. [DOI] [PubMed] [Google Scholar]
- Melnikova T, Savonenko A, Wang Q, Liang X, Hand T, Wu L, Kaufmann WE, Vehmas A, Andreasson KI. Cycloxygenase-2 activity promotes cognitive deficits but not increased amyloid burden in a model of Alzheimer’s disease in a sex-dimorphic pattern. Neuroscience. 2006;141:1149–1162. doi: 10.1016/j.neuroscience.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Metcalf TU, Griffin DE. Alphavirus-induced encephalomyelitis: antibody-secreting cells and viral clearance from the nervous system. J Virol. 2011;85:11490–11501. doi: 10.1128/JVI.05379-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millichap JG. Etiologic Classification of attention-deficit/hyperactivity disorder. Pediatrics. 2008;121:e358–e365. doi: 10.1542/peds.2007-1332. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271:676–677. doi: 10.1038/271676a0. [DOI] [PubMed] [Google Scholar]
- Nargi-Aizenman JL, Havert MB, Zhang M, Irani DN, Rothstein JD, Griffin DE. Glutamate receptor antagonists protect from virus-induced neural degeneration. Ann Neurol. 2004;55:541–549. doi: 10.1002/ana.20033. [DOI] [PubMed] [Google Scholar]
- Newsholme EA, Crabtree B, Ardawi MS. Glutamine metabolism in lymphocytes: its biochemical, physiological and clinical importance. Q J Exp Physiol. 1985;70:473–489. doi: 10.1113/expphysiol.1985.sp002935. [DOI] [PubMed] [Google Scholar]
- Nozyce ML. A behavioral and cognitive profile of clinically stable HIV-infected children. Pediatrics. 2006;117:763–770. doi: 10.1542/peds.2005-0451. [DOI] [PubMed] [Google Scholar]
- Okun E, Griffioen K, Barak B, Roberts NJ, Castro K, Pita MA, Cheng A, Mughal MR, Wan R, Ashery U, Mattson MP. Toll-like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proc Natl Acad Sci USA. 2010;107:15625–15630. doi: 10.1073/pnas.1005807107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Fuller T, de Gubareff T. Acute dendrotoxic changes in the hippocampus of kainate treated rats. Brain Res. 1979;176:91–100. doi: 10.1016/0006-8993(79)90872-2. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Lesions of the dorsal hippocampal formation interfere with background but not foreground contextual fear conditioning. Learn Mem. 1994;1:34–44. doi: 10.1101/lm.1.1.34. [DOI] [PubMed] [Google Scholar]
- Potter MC, Figuera-Losada M, Rojas C, Slusher BS. Targeting the glutamatergic system for the treatment of HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol. 2013;8:594–607. doi: 10.1007/s11481-013-9442-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter MC, Yuan C, Ottenritter C, Mughal M, van Praag H. Exercise is not beneficial and may accelerate symptom onset in a mouse model of Huntington’s disease. PLoS Curr. 2010;2:RRN1201. doi: 10.1371/currents.RRN1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowell JFJ, Griffin DED. The inflammatory response to nonfatal Sindbis virus infection of the nervous system is more severe in SJL than in BALB/c mice and is associated with low levels of IL-4 mRNA and high levels of IL-10-producing CD4+ T cells. J Immunol. 1999;162:1624–1632. [PubMed] [Google Scholar]
- Rudy JW. Contextual conditioning and auditory cue conditioning dissociate during development. Behav Neurosci. 1993;107:887–891. doi: 10.1037//0735-7044.107.5.887. [DOI] [PubMed] [Google Scholar]
- Sanders MJ, Wiltgen BJ, Fanselow MS. The place of the hippocampus in fear conditioning. Eur J Pharmacol. 2003;463:217–223. doi: 10.1016/S0014-2999(03)01283-4. [DOI] [PubMed] [Google Scholar]
- Shelton LM, Huysentruyt LC, Seyfried TN. Glutamine targeting inhibits systemic metastasis in the VM-M3 murine tumor model. Int J Cancer. 2010;127:2478–2485. doi: 10.1002/ijc.25431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shijie J, Takeuchi H, Yawata I, Harada Y, Sonobe Y, Doi Y, Liang J, Hua L, Yasuoka S, Zhou Y, Noda M, Kawanokuchi J, Mizuno T, Suzumura A. Blockade of glutamate release from microglia attenuates experimental autoimmune encephalomyelitis in mice. Tohoku J Exp Med. 2009;217:87–92. doi: 10.1620/tjem.217.87. [DOI] [PubMed] [Google Scholar]
- Silverman MA, Misasi J, Smole S, Feldman HA, Cohen AB, Santagata S, McManus M, Ahmed AA. Eastern equine encephalitis in children, Massachusetts and New Hampshire, USA, 1970–2010. Emerg Infect Dis. 2013;19:194–201. doi: 10.3201/eid1902.120039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklaroff RB, Casper ES, Magill GB, Young CW. Phase I study of 6-diazo-5-oxo-L-norleucine (DON) Cancer Treat Rep. 1980;64:1247–1251. [PubMed] [Google Scholar]
- Souba WW. Glutamine and cancer. Ann Surg. 1993;218:715–728. doi: 10.1097/00000658-199312000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele KE, Reed DS, Glass PJ, Hart MK, Ludwig GV, Pratt WD, Parker MD, Smith JF. Alphavirus encephalitides. In: Dembek ZF, editor. Medical aspects of biological warfare. Office of the Surgeon General, US Army Medical Department Center and School, Borden Institute; Washington, DC: 2007. pp. 1–30. [Google Scholar]
- Umpierre AD, Remigio GJ, Dahle EJ, Bradford K, Alex AB, Smith MD, West PJ, White HS, Wilcox KS. Impaired cognitive ability and anxiety-like behavior following acute seizures in the Theiler’s virus model of temporal lobe epilepsy. Neurobiol Dis. 2014;64:98–106. doi: 10.1016/j.nbd.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villari P, Spielman A, Komar N, McDowell M, Timperi RJ. The economic burden imposed by a residual case of eastern encephalitis. Am J Trop Med Hyg. 1995;52:8–13. doi: 10.4269/ajtmh.1995.52.8. [DOI] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Reisen WK. Present and future arboviral threats. Antiviral Res. 2010;85:328–345. doi: 10.1016/j.antiviral.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver SC, Salas R, Rico-Hesse R, Ludwig GV, Oberste MS, Boshell J, Tesh RB. Re-emergence of epidemic Venezuelan equine encephalomyelitis in South America. VEE Study Group. Lancet. 1996;348:436–440. doi: 10.1016/s0140-6736(96)02275-1. [DOI] [PubMed] [Google Scholar]
- Wiltgen BJ. Context fear learning in the absence of the hippocampus. J Neurosci. 2006;26:5484–5491. doi: 10.1523/JNEUROSCI.2685-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gastrointestinal toxicity associated with high dose (0.6mg/kg) DON treatment. Representative photomicrographs of hematoxylin & eosin-stained large intestine from (a) untreated, mock-infected and (b) high dose (0.6mg/kg) DON, mock-infected mice at 7 DPI. High dose DON-treatment resulted in intestinal dilatation with loss of columnar epithelium and decreased cellularity (100X magnification; scale bar = 500μm). (c) SINV-infected mice receiving high dose (0.6mg/kg) DON regained body weight lost by 28 DPI (N=3–4 mice per group)