

Abstract
The herpes simplex virus type 1 (HSV-1) latency associated transcript (LAT) encodes several microRNAs. One of these, miR-H2, overlaps and is antisense to the ICP0 gene, and appears to decrease expression of the ICP0 protein. To determine if miR-H2 plays a role in the HSV-1 latency-reactivation cycle, we constructed a mutant, McK-ΔH2, in which this microRNA has been disrupted without altering the predicted amino acid sequence of ICP0. McK-ΔH2 produced increased amounts of ICP0. Although replication of McK-ΔH2 was similar to that of its wt McKrae parental virus in RS cells and mouse eyes, McK-ΔH2 was more neurovirulent in Swiss Webster mice than McKrae based on the percent of mice that died from herpes encephalitis following ocular infection. In addition, using a mouse TG explant model of induced reactivation, we show here for the first time that miR-H2 appears to play a role in modulating HSV-1 reactivation. Although the percent of TG from which virus reactivated by day 10 after explant was similar for McK-ΔH2, wt McKrae, and the marker rescued virus McK-ΔH2Res, at earlier times significantly more reactivation was seen with McK-ΔH2. Our results suggest that in the context of the virus, miR-H2 downregulates ICP0 and this moderates both HSV-1 neurovirulence and reactivation.
INTRODUCTION
Herpes simplex virus type 1 (HSV-1) is an important human pathogen causing much disease worldwide. In the U.S., Herpes simplex encephalitis (HSE) is the leading cause of sporadic lethal encephalitis in immune competent individuals, with an untreated death rate of ~70%(1,2). Even with treatment the death rate is ~19%. Additionally, over 50% of survivors have significant neurological deficits. Herpes simplex stromal keratitis (HSK) is the most frequent serious viral eye infection in developed countries and the leading cause of corneal blindness due to an infectious agent(3,4). Like HSV-2, HSV-1 also causes genital herpes. HSE, HSK, and genital herpes most commonly occur after reactivation of HSV from latency, rather than from primary infection. Following primary ocular infection, HSV-1 ascends through axons and persists throughout life as a latent infection in sensory neurons of the trigeminal ganglia (TG). There is no effective HSV-1 vaccine and long term oral acyclovir is only partially protective(5). Thus, a better understanding of the molecular biology of HSV-1 neurovirulence, latency, and reactivation, is highly desirable for development of more efficacious therapies to reduce HSV-1 related encephalitis and blindness.
During neuronal latency, the only HSV-1 gene that is abundantly and consistently detected is the latency associated transcript (LAT)(6,7). LAT plays an important role in the HSV-1 latency-reactivation cycle, since mutants not expressing LAT have reduced spontaneous and in vivo induced reactivation in rabbits and reduced ex vivo reactivation in the mouse TG explant induced reactivation model(8-11). LAT has anti-apoptosis activity(12-19) that appears to be a major factor in how it enhances the reactivation phenotype, since the wild type (i.e., LAT(+)-like) high reactivation phenotype can be restored to a LAT(−) mutant by substitution of various anti-apoptosis genes in place of LAT(14,16-18). LAT also has immune evasion properties including decreasing and/or delaying interferon production, exhaustion of CD8+ T-cells in TG, blocking granzyme B CD8+ T-cell killing, increasing HVEM expression, and inhibiting maturation of dendritic cells(20-25), all of which may contribute to how LAT enhances latency and reactivation.
Recently, 8 “LAT” microRNAs (miRs H1 to H8) mapping in or near the LAT locus, were reported(26,27). miR-H2 is expressed in the LAT direction and overlaps part of the major exon of the HSV-1 ICP0 gene in an antisense orientation. ICP0 is an immediate early (IE) gene that is critical for transactivation of HSV-1 early and late genes. It has long been hypothesized that downregulation of ICP0 by LAT via an antisense mechanism might be important in how LAT regulates latency/reactivation(6,7), but evidence for such antisense downregulation of ICP0 has not been reported. Interestingly, miR-H2 was reported to downregulate ICP0 translation, but not transcription, in a transient transfection assay(27).
In this report we have constructed an HSV-1 mutant in which we used codon redundancy to knock out (KO) miR-H2 without altering the predicted sequence of the overlapping ICP0 open reading frame (ORF). This mutant was made on a wild type (wt) HSV-1 strain McKrae background and is designated McK-ΔH2. We show here that compared to its wt McKrae parental virus and its marker rescued McK-ΔH2Res virus, McK-ΔH2 expresses more ICP0 protein, confirming that miR-H2 down regulates ICP0 expression in the context of the virus. We also found that McK-ΔH2 has increased neurovirulence, as judged by mouse survival following ocular infection. Importantly, we show here for the first time that an miR-H2 KO mutant (i.e., McK-ΔH2 mutant) reactivates in the mouse explant TG reactivation model earlier than wt virus or McK-ΔH2Res. This suggests that miR-H2 normally functions to decrease HSV-1 pathogenesis and to help maintain HSV-1 latency.
MATERIALS AND METHODS
Cell lines
Rabbit skin (RS) cells were maintained in Eagle minimal essential medium (MEM) with 2 mM L-glutamine, 0.1 mM nonessential amino acids, 1mM sodium pyruvate, 10% fetal bovine serum (Promega Scientific), penicillin (100 U/ml), and streptomycin (100 μg/ml) (Sigma, St. Louis, MO).
Viruses
All parental, mutant, and marker rescued viruses were triply plaque purified and passaged only one or two times in rabbit skin (RS) cells prior to use. Wild-type McKrae (wt) has been previously described(9). McKrae is the parental virus of McK-ΔH2.
Construction of McK-ΔH2
McK-ΔH2 was constructed by homologous recombination following co-transfection of infectious purified HSV-1 McKrae genomic DNA with a plasmid containing the 21 nt alterations of interest along with sufficient flanking wt HSV-1 sequences for efficient recombination using our standard procedures(9,14,16,18,28-34). Briefly, 21 of the 75 H2 miRNA precursor nts were changed in a way that does not change the predicted amino acid sequence of the overlap ICP0 ORF. A XhoI restrict enzyme site was also introduced into the altered H2 miRNA precursor sequence (Fig. 2). A 469 nt sequence corresponding to the LAT sequence containing the modified H2 miRNA precursor was synthesized commercially and cloned into pUC57 between appropriate flanking sequences (Genescrip Corp.). This synthesized LAT DNA was further cloned into plasmid pBSK-LAT6.5K containing a Not I fragment of 6506 nts corresponding to nt 118442 to nt 124948 of the McKrae genome. This generated the ΔH2 targeting construct, pH2mut, containing the LAT core promoter and the altered miR-H2 precursor region. pH2mut was co-transfected with infectious purified HSV-1 McKrae genomic DNA into RS cells. The virus McK-ΔH2 was generated by homologous recombination. Viruses from the co-transfection were plated, and isolated plaques were picked and screened for the altered H2 sequence by restriction digestion and Southern analysis. Selected plaques were plaque purified for 6 rounds and reanalyzed by restriction digestion and Southern analysis to ensure that both copies of LAT contained the altered H2 sequence. A final plaque was purified and designated McK-ΔH2. Using the same methods, the McK-ΔH2Res rescued virus, in which the altered miR-H2 sequence was restored to wt in both copies of LAT, was generated by co-transfection and homologous recombination of infectious McK-ΔH2 virus DNA with the original intact Not I-containing plasmid. As above, plaques were isolated from the co-transfection mix and screened by restriction digestion and Southern analysis. One plaque was triple plaque purified and reanalyzed by restriction digestion and Southern analysis to ensure that both copies of miR-H2 had been restored.
Fig. 2.

Altered codon usage of the ICP0 ORF region corresponding to the miR-H2 precursor RNA. A. Sequences are for the ICP0 region complementary to the H2 precursor miRNA. Changed nts are blue. Nts complementary to the H2 miRNA are red. H2 miRNA precursor: 21 of 75 nts (28%) are changed. H2 miRNA: 7 of 24 nts (29%) are changed. Seed sequence: 2 of 7 nts are changed. The predicted ICP0 amino acid sequence shown immediately under the “altered” nucleotide sequence is identical for the “original” (wt) and the “altered” (mutated) nucleotide sequences. B. The altered sequence changes the predicted structure of the RNA made from the complementary LAT DNA strand. Virgo software version 2.0 no longer predicts a miRNA precursor in this location. Thus, no H2 miRNA will be made. The amino acid sequence
Mice
Eight- to 10-week-old Swiss-Webster or C57BL/6 female mice (Jackson Labs) were used. Viral infections were done without corneal scarification as we previously described(18,31,32).
Titration of virus in tears of infected mice
Tear films were collected from both eyes of 10 mice per group on days 3, 5, and 7 post infection (p.i.) using a Dacron-tipped swab. Each swab was placed in 0.5 ml of tissue culture medium and squeezed, and the amount of virus was determined by a standard plaque assay on RS cells.
In vitro TG explant reactivation assay
Mice were sacrificed at 30 days p.i., and individual trigeminal ganglia (TG) were removed and cultured in tissue culture medium as we described previously ((32)). Aliquots of medium were removed from each culture daily for up to 10 days and plated on indicator cells (RS cells) to assay for the appearance of reactivated virus. As the medium from the explanted TG cultures was plated daily, the time at which reactivated virus first appeared in the explanted TG cultures could be determined.
DNA extraction and PCR analysis for HSV-1 gB DNA
DNA was isolated from homogenized individual TG using a DNeasy Blood and Tissue Kit (Qiagen, Cat. No. 69504) according to the manufacturer's instructions. PCR analysis was done using gB specific primers (forward, 5′-AACGCGACGCACATCAAG-3′; reverse, 5′-CTGGTACGCGATCAGAAAGC-3′; and probe, 5′-FAM-CAGCCGCAGTACTACC-3′, where FAM is 6-carboxyfluorescein). All the primers and probe were synthesized by Sigma-Aldrich Corp. The amplicon length for this primer set is 72 bp. Relative copy numbers for the gB DNA were calculated using standard curves.
Stem-loop RT-PCR analysis for miR-H2
The expression of miR-H2 in RS cells infected with either McK-ΔmiR-H2 or wt McKrae were assayed by quantitative stem-loop RT-PCR as previously described(27). The RT-loop primer 5’-TCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGTCGC-3’, forward primer 5’-TCATAACCTGAGCCAGGGACGA -3’, and probe (6-FAM) TACGACAGTCGCACT (MGB) were synthesized by Sigma. Total RNA was extracted using Direct-zol™ RNA MiniPrep Kit (Zymo Research). RNA was reverse transcribed with the TaqMan® MicroRNA Reverse Transcription Kit (Life Technologies) and miRNA-specific RT primers. Aliquots of cDNA were assayed on an ABI 7900HT fast real-time PCR system (Applied Biosystems).
Western blots
Total cell extracts were separated by Novex 4-20% tris glycine sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, Life Technologies) and transferred to a polyvinylidene difluoride membrane. The membrane was incubated with mouse monoclonal anti-ICP0 (EastCoast Bio.) at a 1:8,000 dilution, or polyclonal rabbit anti-GAPDH antibody at a 1:10,000 dilution (GeneTex, Irvine, CA) and then washed, and the antibody bound to the blots was visualized by chemiluminescence (Thermo Scientific) with goat anti-mouse immunoglobulin G (IgG) or goat anti-rabbit IgG conjugated to horseradish peroxidase according to the instructions of the manufacturer (Thermo Scientific).
Southern blots
Briefly, viral DNA was double-digested with BamHI and XhoI. The restriction fragments were separated in a 0.8% agarose gel, transferred to Nylon membrane, rinsed in 2×SSC (1×SSC is 0.15M NaCl plus 0.015M sodium citrate) for 5 min, and cross-linked to the membrane by UV light. DNA-DNA hybridization was performed with 32P-labeled probes.
RESULTS
Codon redundancy to KO miR-H2 without altering ICP0
miR-H2 overlaps the very important HSV-1 ICP0 open reading frame (ORF) in an anti-sense direction (Fig. 1). This complicates construction of a miR-H2 KO mutant, since simply deleting the DNA region encoding miR-H2 would disrupt ICP0 and likely produce a mutant phenotype unrelated to the miR-H2 KO. We therefore used an innovative approach of employing codon redundancy to alter the sequence of the ICP0 ORF region that overlaps miR-H2 (i.e., we altered the ICP0 ORF codon usage of this region) as shown in Fig. 2A). The desired sequence, in which 21 of the 75 miR-H2 precursor nts (28%) were changed without altering the predicted amino acid sequence of ICP0, was synthesized commercially. The changes made resulted in this region of ICP0 having a codon usage similar to that of a typical human gene. This dramatically altered the predicted RNA structure (Fig. 2B). Computer analysis suggests that this sequence will not code for a miRNA. In the extremely unlikely event that a miRNA is made, it would not have the correct function (see next paragraph). The mutant synthesized DNA was cloned into a plasmid (pBSK-LAT6.5) containing the DNA sequence corresponding to McKrae LAT nts 118442-124948. This resulted in a plasmid containing LAT nts 118442-124948 with the region corresponding to the miR-H2 precursor replaced with the mutated DNA sequence. This plasmid was designated pH2mut.
Fig. 1.
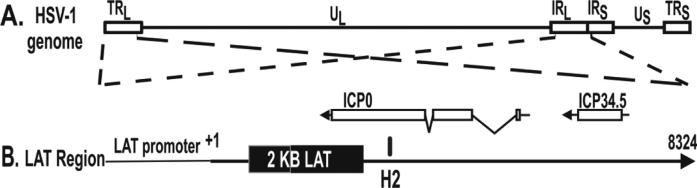
Location of LAT miRNA-H2. A. The HSV-1 genome has a Unique Long (UL) region flanked by inverted repeats (TRL=Terminal Repeat Long; IRL= Inverted Repeat Long), and a Unique Short (US) region flanked by short repeats. B: Expansion of the LAT region. The viral genome contains 2 identical copies of the LAT region, one in each long repeat. For simplicity, only one copy is shown. Their opposite orientation of the two LAT regions is indicated by the crossing dashed lines indicating the blow up of the LAT regions. The relative location of miR-H2 is shown as a thick vertical line. The primary transcript is ~8.3 kb. The important viral genes ICP0 and ICP34.5 overlap portions of LAT in an antisense direction. miR-H2 overlaps the major ICP0 exon in an antisense direction.
To confirm that the mutated DNA no longer encoded a miR-H2 with the wild type miR-H2 sequence's ability to down regulate ICP0, RS cells were co-transfected with a plasmid expressing ICP0 and a plasmid expressing either no microRNA, wt miR-H2, or mutant miR-H2 (from pH2mut). A Western blot showing the resulting relative amount of ICP0 protein is shown in Fig. 3. Compared to the amount of ICP0 protein in the absence of miR-H2, the expression of miR-H2 efficiently downregulated ICP0 protein level. Alteration of the sequence of the ICP0 ORF region that overlaps miR-H2 as described above, completely abolished this downregulation. GAPDH is a loading control. These results strongly support a previous report(27) that plasmids expressing wt miR-H2 downregulate ICP0 protein levels in transient expression assays. More importantly, these results indicate, that as expected, the mutant miR-H2 sequence has lost the ability to downregulate ICP0 protein expression. After confirming that the mutated miRNA sequence KO'd miR-H2 function, rabbit skin (RS) cells were co-transfected with infectious wt McKrae genomic DNA and pH2mut. Mutant virus was isolated and stocks prepared as described in Materials and Methods in a manner similar to what we have previously described for construction of other HSV-1 mutant viruses(9,18,30).
Fig. 3.

A plasmid expressing the mutated H2 microRNA no longer down regulates expression of ICP0. RS cells were co-transfected with and ICP0 expressing plasmid and a plasmid expressing wt H2 of the mutated H2. Lanes: 1=no ICP0; 2=empty plasmid; 3=wt miR-H2; 4=mutated miR-H2.
Confirmation of the McK-ΔH2 genomic structure by Southern blot analysis
The mutant miR-H2 sequence in pH2mut was chosen to both KO miR-H2 without altering ICP0 and to introduce a XhoI restriction site for use in screening and confirming the genomic structure of the resulting mutant, designated McK-ΔH2. Following 6 rounds of plaque purification McK-ΔH2 genomic DNA was purified, digested with BamHI and XhoI and compared to identical digestion of wt McKrae. The resulting DNA fragments were separated on a 0.8% agarose gel, denatured, and transferred to a nylon membrane for Southern blot analysis (Fig. 4A). A NotI-XhoI DNA restriction fragment corresponding to LAT nts 118442 to 123031 was radiolabeled with 32P-dCTP by nick translation and used as a probe. In wild-type McKrae, BamHI and XhoI digestion cuts both copies of the LAT region (one in each long repeat). In Fig. 4A, the wild-type LAT fragments are 9708 and 6098 bp. Since we introduced an additional XhoI site in McK-ΔH2 within the miR-H2 region, the corresponding DNA fragments from McK-ΔH2 are predicted to be 8517 and 4900 bp respectively. Comparison of the left and center lanes in the Southern blot (Fig. 4A) confirms that the mobility of the McK-ΔH2 fragments are consistent with the predicted sizes. Thus, both copies of miR-H2 contain the desired mutant sequence. McK-ΔH2 was then marker rescued by co-transfection of its genomic DNA with plasmid pBSK-LAT6.5K containing the wt sequence version of LAT nts 118442-124948 as we previously described for marker rescue of other HSV-1 mutant viruses(9) and as described in Materials and Methods. Comparison of the rightmost lane (McK-ΔH2Res) with the left and middle lanes in Fig. 4A shows that the marker rescued McK-ΔH2Res virus no longer contains the XhoI restriction site. The original wt miR-H2 sequence has thus been restored (rescued) in both copies. To further confirm that McK-ΔH2 does not express miR-H2, RS cells were infected with either McK-ΔH2 or wt McKrae and stem-loop RT-PCR analysis was performed as described in Materials and Methods. No miR-H2 product was detected in cells infected with the miR-H2 KO mutant McK-ΔH2 (Fig. 4B).
Fig. 4.

Southern blot analysis of McK-ΔH2 and McK-ΔH2Res. RS cells were infected and viral DNA extracted, digested with BamHI and XhoI and Southern blots performed as described in Materials and Methods. The LAT (ICP0/miR-H2) bands produced by the BamHI and XhoI digestion from the two repeats regions are of different sizes. Since altering the ICP0 sequence to KO miR-H2 introduced a XhoI restriction site at this location, both bands from McK-ΔH2 are smaller than the corresponding bands from wt virus and McK-ΔH2Res.
Increased expression of ICP0 in McK-ΔH2
RS cells were infected at an MOI of 5 with McK-ΔH2, McK-ΔH2Res, or wt McKrae. Cultures were harvested at 2, 4, 6, and 10 hrs post infection (p.i.). Cell extracts were analyzed on Western blots using mAb specific for ICP0 (Fig. 5). GAPDH is included as a loading control. More ICP0 appears to be present in McK-ΔH2 infected cells compared to both McKrae and McK-ΔH2Res infected cells. This is most easily seen at 4 and 6 hrs p.i. At 4 hrs p.i. only trace amounts of ICP0 are seen in McKrae and McK-ΔH2Res infected cells, while in McK-ΔH2 infected cells an obvious ICP0 band is seen at this time. At 6 hrs p.i., the ICP0 band from McK-ΔH2 infected cells is clearly more abundant than it is with McKrae and McK-ΔH2Res. At 10 hrs p.i. the McK-ΔH2 ICP0 band is broader and again it appears that there is more ICP0 with miR-H2 than with wt McKrae or marker rescued McK-ΔH2Res. Thus, the miR-H2 KO mutant, McK-ΔH2, appeared to express more ICP0 protein than either wt McKrae or marker rescued McK-ΔH2Res.
Fig. 5.

Increased ICP0 protein levels expressed by McK-ΔH2. RS cells were infected, harvested at the times indicated post infection (Hrs pi), and Western blots were done using an anti-ICP0 mAb to detect ICP0 as described in Materials and Methods. GAPDH is a loading control.
Although unlikely, the increased ICP0 levels with McK-ΔH2 could have been due to increased stability of the altered ICP0 mRNA or to increased translation efficiency of the altered ICP0 mRNA. To test this, RS cells were transfected with a plasmid expressing wt ICP0 mRNA or a plasmid expressing the altered ICP0 mRNA (Fig. 6A). The plasmid expressing the complete ICP0 mRNA containing the mutated sequence present in McK-ΔH2, did not result in increased ICP0 protein levels compared to the corresponding wt sequence plasmid. A plasmid expressing GFP was co-transfected with the ICP0 plasmids as a transfection efficiency control. GAPDH is a loading control. Neither plasmid expressed significant miR-H2 (not shown) indicating that the miR-H2 promoter is not present in the plasmid. As an additional control, the amount of ICP0 plasmid in the cells was determined by qPCR (Fig. 6B). No differences were detected, supporting similar transfection efficiencies. These results strongly suggest that the altered ICP0 codon usage did not increase ICP0 levels independent of the miR-H2 KO.
Fig. 6.

Similar expression of ICP0 protein by a wt ICP0 plasmid and the mutated (ΔmiRH2) ICP0 plasmid. A. RS cells were transfected with a wt ICP0 or a ΔmiR-H2 ICP0 expressing plasmid. No significant miR-H2 was detected (not shown). GAPDH is a loading control. A GFP plasmid was co-transfected as a transfection efficiency control. Results are representative of 3 experiments. B. qPCR for plasmid levels was an additional transfection efficiency control.
Replication of McK-ΔH2 in tissue culture and mouse eyes
The ICP0 protein made by McK-ΔH2 should theoretically be unaltered, since the substituted mutant sequence still encodes for the same amino acid sequence. Nonetheless, it was important to confirm normal ICP0 function to ensure that any phenotype exhibited by the McK-ΔH2 mutant was not due to alteration of ICP0. ICP0 is not considered to be an essential virus protein, because ICP0 deletion mutants can replicate, albeit poorly, when cells are infected at a high multiplicity of infection (MOI) such as 50 or 100. However, at a low MOI (i.e., 0.01 pfu/cell), ICP0 is a critical (if not technically an essential gene) since ICP0 deletion mutants do not replicate under these conditions in most cells, including RS cells. Therefore, to confirm that ICP0 function was normal, RS cells were infected at an MOI of 0.01 with McK-ΔH2, wt McKrae, or McK-ΔH2Res (Fig. 7A). McK-ΔH2 replicated at least as well as the other viruses. This confirmed that ICP0 is fully functional, since at this MOI replication of McK-ΔH2 would be obviously defective if ICP0 function was abnormal. In fact, in multiple experiments McK-ΔH2 appeared to replicate slightly better than either wt McKrae or McK-ΔH2Res, as might be expected of a mutant that overexpressed ICP0, but the results were not significantly different.
Fig. 7.

Replication of McK-ΔH2 in tissue culture and in rabbit eyes. Panel A: RS cell monolayers were infected with the indicated virus at an MOI of 0.01. At the times indicated the monolayer and tissue culture media were freeze thawed 2X and the amount of virus determined by standard plaque assays on RS cells. Each time point was done in triplicate. Panel B: Rabbits were ocularly infected with 2 × 105 pfu/eye of the indicated virus. Tears swabs were collected on the indicated days and the amount of virus determined by plaque assays on RS cells. Each time point represents the average of 10 eyes.
Swiss Webster mice were infected with 2×105 pfu/eye. Eye swabs were collected on days 3, 5, and 7 p.i. and the amount of virus determined by standard plaque assays on RS cells (Fig. 7B). Again, no significant differences were detected for McK-ΔH2 vs wt virus or marker rescued McK-ΔH2Res. Thus, McK-ΔH2 appeared to replicate at least as well as wt and marker rescued virus in tissue culture and in mouse eyes.
Neurovirulence (based on mouse survival)
Swiss Webster mice (female, ~8 weeks of age) were infected with 2×105 pfu/eye using eye drops without corneal scarification as we previously described(14). Survival was monitored for 10 days. Mice that develop significant neurological symptoms that would lead to death were immediately euthanized and these mice were considered to have died from the infection. Thus, death is thus not really an endpoint. In these studies, we did not distinguish between neuroinvasion, the ability of the virus to get to the CNS, and the formal definition of neurovirulence, the ability of the virus to kill mice after it gets to the CNS. Rather we simply define neurovirulence here as the ability of the virus to kill mice by encephalitis following ocular infection. The results were analyzed using a Kaplan-Meier survival curve analysis (Fig. 8). By these criteria, McK-ΔH2 was more neurovirulent than its parental wt virus (P=0.023). This suggests that miR-H2 expression acts to decrease HSV-1 neurovirulence.
Fig. 8.

Neurovirulence of McK-ΔH2 determined by survival of Swiss Webster mice. Mice were ocularly infected with 2 × 105 pfu/eye of the indicated virus. The percent of mice surviving during the first 10 days pi was determined and plotted as a Kaplan-Meier survival curve. Wt-McKrae: 20 mice; McK-ΔH2: 18 mice.
Reactivation of McK-ΔH2 from latency
Swiss Webster mice were ocularly infected with 2×105 pfu/eye of McK-ΔH2 or wt McKrae. Thirty days p.i. when latency was well established, surviving mice were euthanized. Individual trigeminal ganglia (TG) were explanted into tissue culture media. An aliquot of media was removed daily and plated on RS indicator cells to look for the time of first appearance of reactivated virus. Reactivation of McK-ΔH2 was detected from 21 of 22 (95%) latently infected TG on day 4 after TG explant. In contrast, wt McKrae was detected in only 18 of 30 (60%) of latently infected TG during this time and this difference was significantly different (Fig. 9A; P=0.004, Fisher exact test). On day 5, 20/30 TG from the McKrae group had reactivated and this was still significantly different from the 21/22 in the McK-ΔH2 group (P=0.02). By day 6, 22/30 McKrae TG had produced reactivated virus. This did not reach significance compared to the 21/22 in the McK-ΔH2 group (P=0.06). By day 9, reactivation was detected in an additional TG (23/30) while no additional reactivation was detected in the McK-ΔH2 group (21/22) (P=0.11). Thus, in this experiment, compared to TG latently infected with wt virus, more TG latently infected with McK-ΔH2 appeared to reactivate earlier following TG explant. At later times, although there was a trend towards more reactivation with McK-ΔH2 compared to wt McKrae, the difference did not reach significance.
Fig. 9.

Reactivation of McK-ΔH2. Panel A: Swiss Webster mice were ocularly infected with 2×105 pfu/eye of the indicated virus. 30 days pi surviving mice were euthanized and TG removed for induction of reactivation by explantation into tissue culture media. Aliquots of the tissue culture media were collected daily and plated on RS indicator cells to determine the time of first appearance of reactivated virus. The cumulative percent of TG from which virus had reactivated are plotted. Wt McKrae: 30 TG; McK-ΔH2: 22 TG. Panel B: C57BL/6 mice were infected with the indicated virus except that 1×106 pfu/eye were used, and reactivation determined as in panel A. 50 TG per group.
A second experiment was performed to compare McK-ΔH2 to its marker rescued virus McK-ΔH2Res (Fig. 9B). We also used a different strain of mice, C57BL/6 instead of Swiss Webster, to see if the apparent more rapid reactivation by McK-ΔH2 would occur in more than one mouse strain. C57BL/6 mice, which are more resistant to ocular HSV-1 infection than are outbred Swiss Webster mice, were infected with 1×106 pfu/eye of miR-H2 or McK-ΔH2Res and reactivation from latency was determined as above. Reactivation of McK-ΔH2 was detected from 20 of 50 (40%) of latently infected TG within 3 days of TG explant. In contrast, reactivation of McK-ΔH2Res was detected in only 9 of 50 (18%; P=0.027) latently infected TG during this time. The results of these experiments strongly suggest that McK-ΔH2 reactivated from latency more rapidly than either its wt parent or its marker rescued virus.
Establishment of latency
To determine if the faster reactivation of McK-ΔH2 might be due to increased establishment of latency, the amount of latency was examined in C57BL/6 mice infected as above, on day 30 pi. Using probes for gB DNA, latency levels were analyzed by qPCR in individual TG as described in Materials and Methods. No significant differences were seen between any of the groups, suggesting that all three viruses produced similar amounts of latency.
DISCUSSION
To our knowledge, this is the first report showing that an HSV-1 mutant that does not express the LAT miR-H2 reactivates more rapidly in the mouse TG explant model than its wt parent or its marker rescued virus (Fig. 9). This strongly suggests that miR-H2 decreases reactivation and/or helps maintain latency. The results reported here also show that in the absence of miR-H2, expression of ICP0 is increased in the context of the virus (Fig. 5). This indicates that miR-H2 normally functions to repress ICP0 expression. This is consistent with previous reports showing that miR-H2 can down regulate ICP0 expression in transient transfection assays with plasmids(26,27). Interestingly, despite increased ICP0 expression, McK-ΔH2 did not appear to replicate more efficiently in RS cells in tissue culture (Fig. 7A). This was consistent with a recent report in which a different miR-H2 mutant did not have altered replication in NIH 3T3 cells as judged by viral DNA levels(35). McK-ΔH2 also appeared to have unaltered replication in mouse eyes (Fig. 7B). In contrast, McK-ΔH2 had increased neurovirulence in Swiss Webster mice as judged by decreased survival due to viral encephalitis following ocular infection (Fig. 8). This is consistent with a report that was published while this paper was being prepared. A neuron-specific mouse microRNA, miR-138 was found to repress expression of ICP0. M138, a mutant HSV-1 virus with the miR-138 target sites disrupted, had enhanced expression of ICP0 (similar to McK-ΔH2) and increased encephalitis and death in mice(36). Thus a simple explanation for the increased neurovirulence of our McK-ΔH2 mutant is that it was the result of increased ICP0 expression in the brain in the absence of down regulation by miR-H2.
Previous studies(26,27) that showed downregulation of ICP0 by miR-H2 in transient assays did not report any direct effect of miR-H2 on other immediate early (IE) HSV-1 genes. The results reported here are consistent with this. However, since ICP0 is a transactivator of other IE genes, in the context of the virus, KO of miR-H2 might indirectly result in some increased expression of other IE genes. However, such indirect effects would be expected to be minor, as we were unable to detect any significant difference in virus replication in tissue culture or mouse eyes infected with McK-ΔH2 compared to its parental McKrae wt virus.
The H2 microRNA is located anti-sense to ICP0 on a region of LAT that we previously showed did not appear to play a major role in latency-reactivation. This was based on a mutant (LAT3.3A that was originally designated LAT15a)(29). LAT3.3A expresses just the first ~1.5 kb of LAT. Thus, the region of LAT from which miR-H2 is derived is not expressed by this mutant. LAT3.3A was reported to be indistinguishable from its parental wt virus as regards to replication, neurovirulence, and reactivation(29). We therefore might expect that if LAT3.3A were directly compared to McK-ΔH2, using the same analysis used in this report, it is possible that LAT3.3A might appear to have a subtle reactivation deficit at early times. It should be pointed out here that the increased reactivation with McK-ΔH2 compared to wt and marker rescued viruses was rather subtle. A significant difference was only detected at one or two early time points after TG explant (Fig. 9). When the cumulative amount of virus reactivation was analyzed at later times, McK-ΔH2 was no longer significantly different from wt or McK-ΔH2Res. Thus, more TG latently infected with McK-ΔH2 appeared to reactivate very early but, at later times, reactivation of wt and McK-ΔH2Res caught up to McK-ΔH2. The subtle increase in the reactivation phenotype of McK-ΔH2 compared to its wt parent and its marker rescued virus may suggest a direct effect of miR-H2 at the time of reactivation, since we did not detect any significant difference in establishment of latency among these three viruses.
On first glance it may seem counterintuitive that a mechanism would evolve in HSV-1 that decreases neurovirulence and reactivation from latency. However, both functions are likely to be beneficial for the virus and hence subject to selective pressure. Decreasing neurovirulence increases host survival. This is a typical evolutionary adaptation for a virus that has co-evolved with its host for a long period of time, as have herpes simplex and humans. Viruses that have recently jumped to a new host often are very virulent and kill the host rapidly (e.g., Ebola). This decreases the ability of the virus to spread throughout the host population and outbreaks are usually self-limiting. Evolutionary forces would also be expected to select for viruses that can establish latency (host survival, virus survival, sequestration from host immune responses) with sporadic reactivations (spread to other hosts, maintain low host immune responses). Thus, natural selection should favor a viral means of maintaining latency and modulating reactivation, as appears to be the case for miR-H2. In short, miR-H2's ability to modulate ICP0 likely helps maintain HSV-1 in a latent state. Moreover, release of ICP0 downregulation from miR-H2 would be a likely mechanism to trigger HSV-1 reactivation. In this light, several reports have presented data suggesting that down regulation of HSV-1 LAT (and hence presumably down regulation of miR-H2) or the bovine herpes virus homologue, LR, is a trigger for reactivation(37-41). We hypothesize that down regulation or shut off of miR-H2 may play a key role in triggering the virus to reactivate from latency.
It was important to ensure that the increased ICP0 levels seen with McK-ΔH2 were due to knock out of miR-H2 and not a result of increased stability or increased translation efficiency of the altered ICP0 mRNA sequence. This was done by comparing the amount of ICP0 made following transfection with a plasmid expressing ICP0 from the wt sequence versus a plasmid expressing ICP0 from the mutated sequence (Fig. 6). The levels of ICP0 protein were similar with both plasmids, strongly suggesting that transfection and translation efficiencies of both sequences were similar. This supports the hypothesis that miR-H2 can down regulate expression of ICP0. It is important to point out that neither of the plasmids nor the inserted ICP0 sequences contains a promoter capable of driving expression of the miR-H2. Thus, the results are not altered by potential expression of this microRNA. These results strongly suggest that the altered ICP0 codon usage did not increase ICP0 levels independent of the miR-H2 KO.
Interestingly, in a 1987 paper that was probably the first to examine the effects of ICP0 mutants on HSV-1 replication, no difference in virus replication or plaques was seen when wt HSV-1 strain KOS was grown in normal Vero cells compared to Vero cells engineered to express ICP0 (cell line 0-28)(42). This is consistent with the results reported here that our McK-ΔH2 mutant that expresses increased amounts of ICP0 replicated similarly to its parental wt virus in both tissue culture and mouse eyes. This suggests that neurovirulence and reactivation assays in mice, both of which revealed differences between McK-ΔH2 and wt McKrae, may be more sensitive to the effect of elevated ICP0 levels than are assays for virus replication.
The role of the other LAT miRNAs in neurovirulence and latency-reactivation remain to be determined. Two additional miRNAs (H7 and H8)(26,27) overlap the large ICP0 intron and may also modify ICP0 expression. Two miRNAs (H3 and H4)(26,27) overlap the ICP34.5 gene, with one overlapping the ICP34.5 ORF and the other overlapping the non-coding ICP34.5 leader sequence. ICP34.5 is an important HSV-1 neurovirulence gene that also plays a role in HSV-1 latency(43,44). If the ICP34.5 miRNAs alter expression of ICP34.5 they would also be expected to effect neurovirulence and latency. One miRNA near the end of LAT does not overlap any known gene (other than LAT), while two additional miRNAs are located in the LAT promoter region(26,27). miRNAs can influence expression of genes that they do not overlap. Thus, it is possible that one of more of the LAT miRNAs plays a role in modifying expression of one or more host genes and/or non-overlapping viral genes.
In summary, the findings reported here support the hypothesis that some viral microRNAs may play significant roles in the HSV-1 latency-reactivation cycle. Specifically, miR-H2 appeared to act to decrease HSV-1 neurovirulence and decrease reactivation from latency (or help maintain latency).
Fig. 10.

Virus genome copies in latently infected TG. C57BL/6 mice were infected as above. 30 days pi, TG were harvested and the amount of HSV-1 genome copies in individual TG was determined by qPCR using probes specific for the HSV-1 gB gene. 6 TG per group.
ACKNOWLEDGMENTS
This study was supported by Public Health Service NIH grants R01EY013191, 1R56AI098985, 1R56AI093133, RO1EY019896, and RO1EY14900, and The Discovery Center for Eye Research. We thank Dr. Nigel Fraser for reading this manuscript and providing helpful comments.
Footnotes
CONFLICT OF INTEREST
All of the authors declare that they have no conflict of interest.
LITERATURE CITED
- 1.Whitley RJ. Herpes simplex virus. In: Scheld W, Whitley RJ, Durack D, editors. Infections of the central nervous system. 2nd ed. Lippincott-Raven; Philadelphia: 1997. pp. 73–89. [Google Scholar]
- 2.Hjalmarsson A, Blomqvist P, Skoldenberg B. Herpes simplex encephalitis in Sweden, 1990-2001: incidence, morbidity, and mortality. Clin Infect Dis. 2007;45:875–880. doi: 10.1086/521262. [DOI] [PubMed] [Google Scholar]
- 3.Nesburn AB. Report of the corneal disease panel: Vision research: A national plan 1983-1987. Vol II, Part III., vol. II, part III. C.V. Mosby Co.; St. Louis: 1983. [Google Scholar]
- 4.Smith RE, McDonald HR, Nesburn AB, Minckler DS. Penetrating keratoplasty: changing indications, 1947 to 1978. Arch Ophthalmol. 1980;98:1226–1229. doi: 10.1001/archopht.1980.01020040078009. [DOI] [PubMed] [Google Scholar]
- 5.Herpetic, Eye, Disease, Study, and Group Acyclovir for the prevention of recurrent herpes simplex virus eye disease. Herpetic Eye Disease Study Group [see comments]. N Engl J Med. 1998;339:300–306. doi: 10.1056/NEJM199807303390503. [DOI] [PubMed] [Google Scholar]
- 6.Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol. 1987;61:3820–3826. doi: 10.1128/jvi.61.12.3820-3826.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science. 1987;235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- 8.Hill JM, Sedarati F, Javier RT, Wagner EK, Stevens JG. Herpes simplex virus latent phase transcription facilitates in vivo reactivation. Virology. 1990;174:117–125. doi: 10.1016/0042-6822(90)90060-5. [DOI] [PubMed] [Google Scholar]
- 9.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol. 1994;68:8045–8055. doi: 10.1128/jvi.68.12.8045-8055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Block TM, Deshmane S, Masonis J, Maggioncalda J, Valyi-Nagi T, Fraser NW. An HSV LAT null mutant reactivates slowly from latent infection and makes small plaques on CV-1 monolayers. Virology. 1993;192:618–630. doi: 10.1006/viro.1993.1078. [DOI] [PubMed] [Google Scholar]
- 11.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol. 1989;63:2893–2900. doi: 10.1128/jvi.63.7.2893-2900.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perng G, Jones C, Ciacci-Zanella H, Henderson G, Yukht A, Slanina S, Hofman F, Ghiasi H, Nesburn A, Wechsler S. Virus induced neuronal apoptosis blocked by the herpes simplex virus latency associated transcript (LAT). Science. 2000;287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- 13.Inman M, Perng GC, Henderson G, Ghiasi H, Nesburn AB, Wechsler SL, Jones C. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J Virol. 2001;75:3636–3646. doi: 10.1128/JVI.75.8.3636-3646.2001. PMC:114855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perng GC, Maguen B, Jin L, Mott KR, Osorio N, Slanina SM, Yukht A, Ghiasi H, Nesburn AB, Inman M, Henderson G, Jones C, Wechsler SL. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J Virol. 2002;76:1224–1235. doi: 10.1128/JVI.76.3.1224-1235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng W, Jin L, Henderson G, Perng GC, Brick DJ, Nesburn AB, Wechsler SL, Jones C. Mapping herpes simplex virus type 1 latency-associated transcript sequences that protect from apoptosis mediated by a plasmid expressing caspase-8. J Neurovirol. 2004;10:260–265. doi: 10.1080/13550280490468690. [DOI] [PubMed] [Google Scholar]
- 16.Jin L, Perng GC, Mott KR, Osorio N, Naito J, Brick DJ, Carpenter D, Jones C, Wechsler SL. A herpes simplex virus type 1 mutant expressing a baculovirus inhibitor of apoptosis gene in place of latency-associated transcript has a wild-type reactivation phenotype in the mouse. J Virol. 2005;79:12286–12295. doi: 10.1128/JVI.79.19.12286-12295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin L, Perng GC, Carpenter D, Mott KR, Osorio N, Naito J, Brick DJ, Jones C, Wechsler SL. Reactivation phenotype in rabbits of a herpes simplex virus type 1 mutant containing an unrelated antiapoptosis gene in place of latency-associated transcript. J Neurovirol. 2007;13:78–84. doi: 10.1080/13550280601164333. [DOI] [PubMed] [Google Scholar]
- 18.Jin L, Carpenter D, Moerdyk-Schauwecker M, Vanarsdall AL, Osorio N, Hsiang C, Jones C, Wechsler SL. Cellular FLIP can substitute for the herpes simplex virus type 1 latency-associated transcript gene to support a wild-type virus reactivation phenotype in mice. J Neurovirol. 2008;14:389–400. doi: 10.1080/13550280802216510. PMC:2980827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmed M, Lock M, Miller CG, Fraser NW. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J Virol. 2002;76:717–729. doi: 10.1128/JVI.76.2.717-729.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng W, Henderson G, Inman M, BenMohamed L, Perng GC, Wechsler SL, Jones C. The locus encompassing the latency-associated transcript of herpes simplex virus type 1 interferes with and delays interferon expression in productively infected neuroblastoma cells and trigeminal Ganglia of acutely infected mice. J Virol. 2005;79:6162–6171. doi: 10.1128/JVI.79.10.6162-6171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen SJ, Hamrah P, Gate D, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, BenMohamed L, Ahmed R, Wechsler SL, Ghiasi H. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus 1. J Virol. 2011;85:4184–4197. doi: 10.1128/JVI.02290-10. PMC:3126262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chentoufi AA, Kritzer E, Tran MV, Dasgupta G, Lim CH, Yu DC, Afifi RE, Jiang X, Carpenter D, Osorio N, Hsiang C, Nesburn AB, Wechsler SL, BenMohamed L. The herpes simplex virus 1 latency-associated transcript promotes functional exhaustion of virus-specific CD8+ T cells in latently infected trigeminal ganglia: a novel immune evasion mechanism. J Virol. 2011;85:9127–9138. doi: 10.1128/JVI.00587-11. PMC:3165846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang X, Chentoufi AA, Hsiang C, Carpenter D, Osorio N, BenMohamed L, Fraser NW, Jones C, Wechsler SL. The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J Virol. 2011;85:2325–2332. doi: 10.1128/JVI.01791-10. PMC:3067767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chentoufi AA, Dervillez X, Dasgupta G, Nguyen C, Kabbara KW, Jiang X, Nesburn AB, Wechsler SL, Benmohamed L. The Herpes Simplex Virus Type 1 Latency-Associated Transcript Inhibits Phenotypic and Functional Maturation of Dendritic Cells. Viral Immunol. 2012 doi: 10.1089/vim.2011.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allen SJ, Rhode-Kurnow A, Mott KR, Jiang X, Carpenter D, Rodriguez-Barbosa JI, Jones C, Wechsler SL, Ware CF, Ghiasi H. Interactions between Herpesvirus Entry Mediator (TNFRSF14) and Latency-Associated Transcript during Herpes Simplex Virus 1 Latency. J Virol. 2014;88:1961–1971. doi: 10.1128/JVI.02467-13. PMC:3911542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Umbach JL, Nagel MA, Cohrs RJ, Gilden DH, Cullen BR. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J Virol. 2009;83:10677–10683. doi: 10.1128/JVI.01185-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perng GC, Chokephaibulkit K, Thompson RL, Sawtell NM, Slanina SM, Ghiasi H, Nesburn AB, Wechsler SL. The region of the herpes simplex virus type 1 LAT gene that is colinear with the ICP34.5 gene is not involved in spontaneous reactivation. J Virol. 1996;70:282–291. doi: 10.1128/jvi.70.1.282-291.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perng GC, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3- kilobase primary transcript. J Virol. 1996;70:976–984. doi: 10.1128/jvi.70.2.976-984.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perng GC, Slanina SM, Ghiasi H, Nesburn AB, Wechsler SL. A 371-nucleotide region between the herpes simplex virus type 1 (HSV-1) LAT promoter and the 2-kilobase LAT is not essential for efficient spontaneous reactivation of latent HSV-1. J Virol. 1996;70:2014–2018. doi: 10.1128/jvi.70.3.2014-2018.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perng GC, Esmaili D, Slanina SM, Yukht A, Ghiasi H, Osorio N, Mott KR, Maguen B, Jin L, Nesburn AB, Wechsler SL. Three herpes simplex virus type 1 latency-associated transcript mutants with distinct and asymmetric effects on virulence in mice compared with rabbits. J Virol. 2001;75:9018–9028. doi: 10.1128/JVI.75.19.9018-9028.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perng GC, Slanina SM, Ghiasi H, Nesburn AB, Wechsler SL. The effect of latency-associated transcript on the herpes simplex virus type 1 latency-reactivation phenotype is mouse strain-dependent. J Gen Virol. 2001;82:1117–1122. doi: 10.1099/0022-1317-82-5-1117. [DOI] [PubMed] [Google Scholar]
- 33.Perng GC, Mott KR, Osorio N, Yukht A, Salina S, Nguyen QH, Nesburn AB, Wechsler SL. Herpes simplex virus type 1 mutants containing the KOS strain ICP34.5 gene in place of the McKrae ICP34.5 gene have McKrae-like spontaneous reactivation but non-McKrae-like virulence. J Gen Virol. 2002;83:2933–2942. doi: 10.1099/0022-1317-83-12-2933. [DOI] [PubMed] [Google Scholar]
- 34.Samoto K, Ehtesham M, Perng GC, Hashizume K, Wechsler SL, Nesburn AB, Black KL, Yu JS. A herpes simplex virus type 1 mutant with gamma 34.5 and LAT deletions effectively oncolyses human U87 glioblastomas in nude mice. Neurosurgery. 2002;50:599–605. doi: 10.1097/00006123-200203000-00031. discussion 605-596. [DOI] [PubMed] [Google Scholar]
- 35.Flores O, Nakayama S, Whisnant AW, Javanbakht H, Cullen BR, Bloom DC. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J Virol. 2013;87:6589–6603. doi: 10.1128/JVI.00504-13. PMC:3676078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan D, Flores O, Umbach JL, Pesola JM, Bentley P, Rosato PC, Leib DA, Cullen BR, Coen DM. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell host & microbe. 2014;15:446–456. doi: 10.1016/j.chom.2014.03.004. PMC:4142646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du T, Zhou G, Roizman B. Induction of apoptosis accelerates reactivation of latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc Natl Acad Sci U S A. 2012;109:14616–14621. doi: 10.1073/pnas.1212661109. PMC:3437834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du T, Zhou G, Roizman B. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc Natl Acad Sci U S A. 2011;108:18820–18824. doi: 10.1073/pnas.1117203108. PMC:3219146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rock D, Lokensgard J, Lewis T, Kutish G. Characterization of dexamethasone-induced reactivation of latent bovine herpesvirus 1. J Virol. 1992;66:2484–2490. doi: 10.1128/jvi.66.4.2484-2490.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaber T, Workman A, Jones C. Small noncoding RNAs encoded within the bovine herpesvirus 1 latency-related gene can reduce steady-state levels of infected cell protein 0 (bICP0). J Virol. 2010;84:6297–6307. doi: 10.1128/JVI.02639-09. PMC:2903259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinani D, Jones C. Localization of sequences in a protein (ORF2) encoded by the latency-related gene of bovine herpesvirus 1 that inhibits apoptosis and interferes with Notch1-mediated trans-activation of the bICP0 promoter. J Virol. 2011;85:12124–12133. doi: 10.1128/JVI.05478-11. PMC:3209353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sacks WR, Schaffer PA. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J Virol. 1987;61:829–839. doi: 10.1128/jvi.61.3.829-839.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perng GC, Thompson RL, Sawtell NM, Taylor WE, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. An avirulent ICP34.5 deletion mutant of herpes simplex virus type 1 is capable of in vivo spontaneous reactivation. J Virol. 1995;69:3033–3041. doi: 10.1128/jvi.69.5.3033-3041.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perng GC, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. High-dose ocular infection with a herpes simplex virus type 1 ICP34.5 deletion mutant produces no corneal disease or neurovirulence yet results in wild-type levels of spontaneous reactivation. J Virol. 1996;70:2883–2893. doi: 10.1128/jvi.70.5.2883-2893.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]