

Abstract
Recent preclinical studies showed the potential of nicotinamide adenine dinucleotide (NAD+) precursors to increase oxidative phosphorylation and improve metabolic health, but human data are lacking. We hypothesize that the nicotinic acid derivative acipimox, an NAD+ precursor, would directly affect mitochondrial function independent of reductions in nonesterified fatty acid (NEFA) concentrations. In a multicenter randomized crossover trial, 21 patients with type 2 diabetes (age 57.7 ± 1.1 years, BMI 33.4 ± 0.8 kg/m2) received either placebo or acipimox 250 mg three times daily dosage for 2 weeks. Acipimox treatment increased plasma NEFA levels (759 ± 44 vs. 1,135 ± 97 μmol/L for placebo vs. acipimox, P < 0.01) owing to a previously described rebound effect. As a result, skeletal muscle lipid content increased and insulin sensitivity decreased. Despite the elevated plasma NEFA levels, ex vivo mitochondrial respiration in skeletal muscle increased. Subsequently, we showed that acipimox treatment resulted in a robust elevation in expression of nuclear-encoded mitochondrial gene sets and a mitonuclear protein imbalance, which may indicate activation of the mitochondrial unfolded protein response. Further studies in C2C12 myotubes confirmed a direct effect of acipimox on NAD+ levels, mitonuclear protein imbalance, and mitochondrial oxidative capacity. To the best of our knowledge, this study is the first to demonstrate that NAD+ boosters can also directly affect skeletal muscle mitochondrial function in humans.
Introduction
Acipimox is a nicotinic acid analog used to treat hyperlipidemia (1,2). Besides its effects on cholesterol and triglyceride levels, acipimox treatment acutely decreases plasma nonesterified fatty acid (NEFA) concentrations by 46–57%, which may lower ectopic fat accumulation in type 2 diabetes (T2D) (3,4) and thereby improve whole-body insulin sensitivity (5). Furthermore, elevated plasma NEFA concentrations have been shown to deteriorate mitochondrial function (6), and lowering plasma NEFA levels may, therefore, restore mitochondrial function. The latter is important because several features of muscle mitochondrial function can be impaired in patients with T2D and may be related to insulin sensitivity (7–9). Indeed, some studies showed sustained beneficial effects of acipimox treatment on plasma NEFA and insulin sensitivity (3,10). A major shortcoming of these studies, however, is that the last dose of acipimox was given on the morning of the test days, making it impossible to separate sustained effects from the acute effect of the last acipimox dose given. Indeed, even after treatment with acipimox four times daily for several weeks, a rebound increase in plasma NEFA levels can be observed (11). Next to this rebound effect, acipimox is known to induce flushing in a large number of patients (12), which will limit the clinical relevance of acipimox for the treatment of insulin resistance and mitochondrial dysfunction and an urge for the development of alternative treatments to improve mitochondrial oxidative capacity. In that respect, nicotinamide adenine dinucleotide (NAD+) precursors, such as nicotinamide riboside (NR) (13,14) or nicotinamide mononucleotide (NMN) (15), have been reported to improve oxidative metabolism in animal models. NR or NMN supplementation in mice resulted in elevated NAD+ levels, sirtuin 1 (SIRT1) activation, and beneficial adaptation in mitochondrial gene expression profiles (13,15). Longer-term NR or NMN supplementation in mice fed a high-fat diet additionally improved mitochondrial function and metabolic health (13,15). Furthermore, NR did not activate the GPR109A receptor, which is responsible for the flushing observed with acipimox treatment (16).
So far, NR and NMN have not been tested for efficacy or safety in human clinical trials or for effects on mitochondrial metabolism. However, besides the known effects of acipimox on lipolysis, acipimox is also an NAD+ precursor. Because both NR/NMN and acipimox are NAD+ precursors and acipimox is already available for clinical use in humans, we examined the potential for such NAD+ precursors in improving mitochondrial function in humans. To this end, in this proof-of-concept study, we treated T2D patients with acipimox for 2 weeks and examined the sustained effects on mitochondrial metabolism in detail. To rule out the acute lipid-lowering effects of the last dose of acipimox, patients underwent measurements after an overnight fast, omitting acipimox in the morning.
We found that despite an anticipated rebound effect on plasma NEFA levels, acipimox has pronounced effects in vivo, ex vivo, and in cell culture experiments on mitochondrial function comparable to the effects of other NAD+ precursors, such as NR and NMM, as found in animals. These results suggest a strong potential for novel, safe NAD+ precursors to boost mitochondrial function in humans.
Research Design and Methods
In the present multicenter, randomized, double-blind, placebo-controlled crossover trial, 21 patients with T2D were included (10 from the Maastricht University Medical Center and 11 from the German Diabetes Center in Düsseldorf) (Fig. 1). All T2D patients were treated with placebo (celluose mycrocrystal PH 102) and acipimox (250 mg three times daily) for 2 weeks each in random order. Between interventions, a 4-week washout period was maintained. During both 2-week intervention periods, patients were asked to stop their oral glucose-lowering medication. At the end of both 2-week periods, patients were provided with a standardized meal the day before the clamp test. Patients were also advised to refrain from physical exercise 3 days before the test days. After 2 weeks of acipimox and placebo treatment, measurements were performed to assess mitochondrial function, ectopic lipid accumulation, and insulin sensitivity.
Figure 1.

Study design flowchart. After screening and subject characterization, patients were randomly assigned to either 2 weeks of acipimox treatment or 2 weeks of placebo treatment. At the end of both treatments, mitochondrial function, ectopic lipid accumulation, and insulin sensitivity were assessed. After a washout period of 4 weeks, patients entered in the other intervention arm such that all patients served as their own control.
Subjects
Both male and postmenopausal female humans were included (Table 1). Before inclusion in the study, participants underwent physical examination and anthropometry measurements and completed a medical history questionnaire, including a history of cardiovascular, renal, and pulmonary disease; cancer; and duration of diabetes. Additionally, routine medical laboratory tests, including hematology, and a maximal aerobic capacity test with concurrent electrocardiogram were performed as previously described (17). Body composition was determined using hydrostatic weighing in Maastricht according to the method of Siri (18). In Düsseldorf, body composition was measured with DEXA scan. T2D patients had well-controlled diabetes (HbA1c 7.08 ± 0.16% [54 ± 1.51 mmol/mol]) and were either on monotherapy with metformin (n = 16), on metformin combined with sulfonylurea (n = 4), or on diet only (n = 1). Patients were allowed to take other medications, such as the statins rosuvastin (n = 2), atorvastatin (n = 1), and simvastatin (n = 1), and antihypertensive (n = 10) and uricostatic (n = 6) drugs. Other medications used included omeprazole, acetylsalicylic acid, antimigraine medication, and antidepressant drugs (Supplementary Table 1).
Table 1.
Subject characteristics
| Characteristic | Value |
|---|---|
| Male/female (n) | 18/3 |
| Age (years) | 57.7 ± 1.1 |
| Weight (kg) | 100.5 ± 2.9 |
| BMI (kg/m2) | 33.4 ± 0.8 |
| Fat mass (%) | 35.1 ± 1.2 |
| VO2max (mL/min/kg) | 24.0 ± 1.2 |
| Systolic BP (mmHg) | 149 ± 2.6 |
| Diastolic BP (mmHg) | 92.8 ± 1.9 |
| FPG (mmol/L) | 7.4 ± 0.2 |
| HbA1c (%) | 7.1 ± 0.2 |
| HbA1c (mmol/mol) | 54.0 ± 1.5 |
| AST (units/L) | 23.7 ± 1.7 |
| ALT (units/L) | 35.9 ± 3.2 |
| y-GT (units/L) | 41.6 ± 3.5 |
| Total cholesterol (mmol/L) | 8.4 ± 3.5 |
| HDL cholesterol (mmol/L) | 1.2 ± 0.1 |
| LDL cholesterol (mmol/L) | 2.6 ± 0.2 |
| Triglycerides (mmol/L) | 2.0 ± 0.2 |
| NEFA (μmol/L) | 672 ± 70 |
Data are mean ± SE unless otherwise indicated. ALT, alanine aminotransferase; AST, aspartate aminotransferase; BP, blood pressure; FPG, fasting plasma glucose; y-GT, γ-glutamyl transferase.
Patients with a diagnosis of T2D for at least 1 year were included. None of the participants were following a weight loss dietary program, and all patients had stable body weight for the last 6 months before the study. The Maastricht University Medical Ethics Committee (the Netherlands) and the Medical Association North Rhine in Düsseldorf (Germany) approved the study, and written informed consent was obtained from all participants before screening. The study was performed according to the principles expressed in the Declaration of Helsinki.
Hyperinsulinemic-Euglycemic Clamp
All participants underwent a two-step 6-h hyperinsulinemic-euglycemic clamp (10 and 40 mU/m2/min) (19). After an overnight fast, participants received a primed continuous infusion of [6,6-2H2] glucose (0.04 mg/kg/min) to determine rates of endogenous glucose production (EGP) and whole-body glucose disposal (WGD) rates as previously described (20). After 180 min, a low-insulin infusion was started (10 mU/m2/min) with the coinfusion of 0.1 μg/kg/min of somatostatin (21) for 3.5 h until a steady state was reached, after which time blood sampling and indirect calorimetry was performed for 30 min. Thereafter, a high-insulin infusion was started (40 mU/m2/min) with the coinfusion of 0.1 μg/kg/min somatostatin for 1.5 h, after which steady state was reached and blood sampling and indirect calorimetry repeated. During the clamp, oxygen consumption and carbon dioxide production were measured with an automated respiratory gas analyzer (calibrated daily) using a ventilated hood system (Omnical; IDEE, Maastricht, the Netherlands). Whole-body glucose and fat oxidation rates were calculated using stoichiometric equations based on measured oxygen consumption and carbon dioxide concentrations (22) with the assumption that protein oxidation was negligible.
Muscle Biopsies and Analysis
Muscle biopsy specimens were taken from the m. vastus lateralis before the clamp study under local anesthesia (2% lidocaine) using the Bergström technique (23). Any visible nonmuscle material was dissected from muscle tissue. One portion was immediately frozen in liquid nitrogen for biochemical analyses, one portion was frozen in nitrogen-cooled isopentane and embedded in Tissue-Tek for immunohistochemical analyses, and both samples were stored at −80°C until further use. The remaining muscle tissue was used for mitochondrial respiration analysis.
In the muscle tissue obtained, lipid accumulation was assessed histochemically in cross sections using a modified oil red O staining for fluorescence microscopy (24). In addition, ∼30 mg of the muscle tissue was used for high-resolution respirometry to determine ex vivo mitochondrial function. ATP content was measured in muscle tissue as described (14,25). Furthermore, immunoblotting was used to detect protein levels of mitochondrial-encoded cytochrome C oxidase I (MTCO1); succinate dehydrogenase complex, subunit A (SDHA); and heat shock protein 60 (HSP60) (14,25). MTCO1 and SDHA antibodies were from Abcam, and HSP60 antibody was from Santa Cruz Inc. Coomassie staining of proteins was used as a loading control and performed using a standard protocol.
Mitochondrial Respiration in Permeabilized Muscle Fibers
A small portion of the muscle biopsy sample (∼30 mg) was immediately placed in ice-cold biopsy preservation medium (BIOPS; OROBOROS Instruments, Innsbruck, Austria). Muscle fibers were permeabilized with saponin as previously described (7). After completion of the permeabilization protocol, muscle fibers were transferred into ice-cold mitochondrial respiration buffer (MiR05; OROBOROS Instruments). Subsequently, mitochondrial function ex vivo was determined by measuring oxygen consumption polarographically using a two-chamber Oxygraph (OROBOROS Instruments). Oxygen consumption, or oxygen flux, reflects the first derivative of the oxygen concentration (nmol/mL) in the respiration chambers (pmol/s/mg) corrected for muscle tissue wet weight (2–5 mg). To evaluate mitochondrial oxidative capacity, different substrate protocols were applied. In every protocol, 4.0 mmol/L malate was added to obtain state 2 respiration followed by the addition of 8.0 mmol/L glutamate as a substrate for complex I, which was combined with or without 40.0 μmol/L palmitoylcarnitine. In addition, an excess of 1.6 mmol/L ADP was added to evaluate state 3 respiration of complex I. (State 3 respiration reflects substrate oxidation coupled to energy production.) Then, 8.0 mmol/L succinate was added to obtain state 3 respiration of complex I and II. Finally, titrations (in steps of 0.5 μL of 1.0 mmol/L) of the chemical uncoupler fluoro carbonyl cyanide phenylhydrazone (FCCP) were added to evaluate maximal respiratory capacity state U.
Measures of Mitochondrial Density
Mitochondrial DNA (mtDNA) copy number was determined as a marker for mitochondrial density using quantitative real-time PCR based on the TaqMan probe method. mtDNA copy number was calculated from the ratio of NADH dehydrogenase subunit 1 to lipoprotein lipase (mtDNA/nuclear DNA [nDNA]) as previously described (7).
Plasma Assays
Blood collected in tubes containing EDTA was immediately centrifuged, and the plasma was stored at −80°C until assayed. Plasma NEFA and glucose concentrations were measured with enzymatic assays on Cobas Bio Fara and Mira analyzers (NEFA: Wako NEFA-C test kit [Wako Chemicals, Neuss, Germany]; glucose: hexokinase method [Roche, Basel, Switzerland]). Insulin concentration was determined by radioimmunoassay (Linco Research, St. Charles, MO). Cholesterol, LDL, and triglyceride levels were measured colorimetrically (Roche, Vienna, Austria).
RNA Analysis
Total RNA was isolated from human vastus lateralis muscle biopsy specimens using the acidic guanidinium thiocyanate method, purified with the RNeasy RNA isolation kit (QIAGEN), and prepared simultaneously for array analysis with all RNA integrity numbers ≥8.0. Microarrays were analyzed using the Human Gene 2.0 ST platform (Affymetrix). Gene Set Enrichment Analysis (GSEA) (Broad Institute) was performed using ontology sets designed by the Reactome consortium (26). These general gene sets were supplemented with specific custom NAD+ booster gene sets designed and detailed in previous studies (13,14,27,28).
In Vitro Cell Culture Study
C2C12 myoblasts were differentiated into myotubes as described (29) in 6-well plates. Four-day–differentiated myotubes were treated with acipimox 10 mol/L in normal glucose (4.5 g/L) DMEM containing 1% albumin-bound oleic acid (Sigma), 10% FCS, 2% HEPES, 1% nonessential amino acids, and 1% penstrep. NAD+ was extracted 3 h after the acipimox incubation and measured using high-performance liquid chromatography as described (30). Western blot analysis was performed 24 h after the acipimox incubation. Mitochondrial respiration was measured 24 h after the acipimox incubation using high-resolution respirometry as aforementioned (OROBOROS Instruments). Respirometry was corrected for cell count and viability of the cells loaded.
Calculations
Determination of atom percent enrichment of 2H was done as previously described (31) after deproteinization. Steele single-pool non–steady-state equations were used to calculate WGD and EGP (32). Volume of distribution was assumed to be 0.160 L/kg for glucose. Nonoxidative glucose disposal (NOGD) was calculated as WGD − carbohydrate oxidation.
Statistics
Data are reported as mean ± SE. Statistical analyses were performed using SPSS version 16.0.2 for MacOSX (IBM Corporation, Chicago, IL). Differences between the interventions were analyzed with a two-tailed paired Student t test. Statistical significance was set a priori at P < 0.05.
Results
Effect of Acipimox on Plasma Metabolite Concentrations
Patients were treated with acipimox or placebo for 2 weeks. To test compliance with and efficacy of the treatment, we determined plasma cholesterol and triglyceride levels. Despite the short duration of the intervention, cholesterol and triglyceride levels tended to decrease upon acipimox treatment compared with placebo, indicating that administration of acipimox was effective in the present patients (total cholesterol 5.6 ± 0.3 vs. 5.1 ± 0.3 mmol/L [P = 0.08], TG 2.6 ± 0.4 vs. 1.7 ± 0.3 mmol/L [P = 0.08], for placebo vs. acipimox). We next determined the effect of acipimox treatment on plasma NEFA levels, which we observed to increase rather than decrease upon acipimox treatment, consistent with the previously described rebound effect of acipimox (11,33). Fasting plasma NEFA concentrations increased from 0.77 ± 0.05 mmol/L with placebo to 1.13 ± 0.10 mmol/L with acipimox (P < 0.01) (Fig. 2A). Of note, plasma NEFA levels remained significantly elevated throughout the insulin-stimulated period of the hyperinsulinemic clamp (Fig. 2A).
Figure 2.
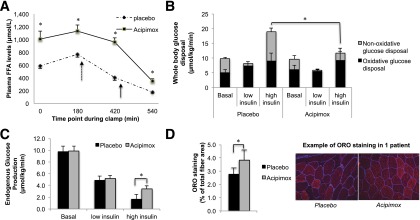
Metabolic effects of 2 weeks of acipimox treatment or placebo in T2D patients. A: Effect of acipimox treatment for 2 weeks on plasma NEFA concentrations in T2D patients, both in the fasted state as during a hyperinsulinemic-euglycemic clamp. Dashed and solid arrows indicate the start of low (10 mU/m2/min) and high (40 mU/m2/min) infusion of insulin, respectively. B: WGD rates divided into oxidative glucose disposal and NOGD. C and D: EGP and skeletal muscle lipid content as measured by ORO staining in the vastus lateralis muscle. *P < 0.05. FFA, free fatty acid; ORO, oil red O.
Insulin Sensitivity
Acipimox has been suggested to improve insulin sensitivity; however, elevations of plasma NEFA levels are generally associated with an induction of insulin resistance (34). Therefore, we examined whether sustained acipimox treatment influenced insulin sensitivity when the acute effect of acipimox was omitted. In noninsulin-stimulated condition and at low insulin concentrations, we observed no significant differences in insulin sensitivity and metabolic flexibility between the acipimox- and placebo-treated T2D patients. However, consistent with elevated plasma NEFA levels, at high insulin concentrations (40 mU/m2/min), WGD and NOGD were lower upon acipimox treatment (WGD 20.0 ± 2.6 vs. 13.5 ± 2.1 [P = 0.03] and NOGD 9.9 ± 2.6 vs. 2.5 ± 3.1 μmol/kg/min [P = 0.02] for placebo vs. acipimox) (Fig. 2B), whereas insulin-stimulated oxidative glucose disposal was not affected (Fig. 2B). Additionally, EGP was less suppressed by insulin upon acipimox treatment (1.7 ± 0.8 vs. 3.4 ± 0.5 μmol/kg/min for placebo vs. acipimox, P = 0.05) (Fig. 2C).
The effect of high NEFA levels on insulin sensitivity has been previously ascribed to fat accumulation in skeletal muscle (34). Therefore, we examined the effect of acipimox treatment on skeletal muscle lipid content and found that intramyocellular lipid content was increased from 2.8 ± 0.5 to 3.8 ± 0.8% of total fiber area upon acipimox treatment (P < 0.05) (Fig. 2D).
Mitochondrial Capacity
Although acipimox induced a rebound increase of plasma NEFA levels, which was associated with the development of insulin resistance, we hypothesized that acipimox may have a direct effect on mitochondrial oxidative capacity independent of its confounding effects on NEFA levels. Therefore, we first examined whether 2 weeks of acipimox treatment resulted in increases in mitochondrial content. mtDNA copy number was unaffected by acipimox treatment (2,305 ± 218 vs. 2,353 ± 202 arbitrary units for placebo vs. acipimox, P = 0.84). To investigate functional improvements in mitochondrial respiration, we next performed high-resolution respirometry in permeabilized muscle fibers. Ex vivo mitochondrial respiration increased upon acipimox treatment for both state 3 and maximal uncoupled respiration on malate/glutamate/succinate (Fig. 3A). Upon the addition of octanoylcarnitine, only maximal uncoupled respiration was significantly higher in the acipimox-treated condition (Fig. 3B). Because mtDNA content was not different between the interventions, a similar tendency with increased mitochondrial respiration upon acipimox treatment was observed after normalization for mitochondrial respiration to mtDNA content (data not shown).
Figure 3.
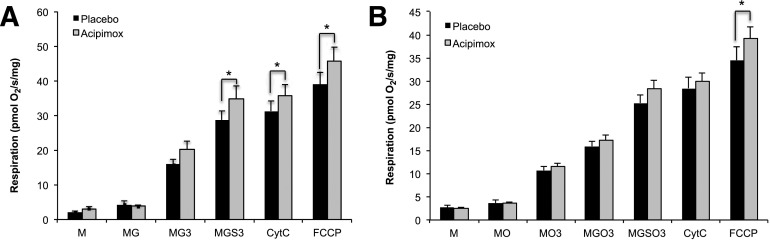
Two weeks of acipimox treatment improves mitochondrial respiration. Effect of acipimox treatment for 2 weeks on ex vivo mitochondrial function measured in permeabilized fibers upon addition of complex I and II substrates without (A) and with (B) addition of octanoylcarnitine as a substrate. Data are uncorrected for mtDNA. *P < 0.05. 3, state 3 after addition of ADP; CytC, cytochrome C; G, glutamate; M, malate; O, octanoylcarnitine; S, succinate.
To find further evidence for a direct effect of acipimox on mitochondrial metabolism, we performed microarray analysis on vastus lateralis muscle biopsy specimens of a small and randomly chosen subset of patients (n = 3). Unbiased GSEA showed a robust and significant enrichment for energy metabolism in acipimox-treated patients. In particular, acipimox treatment resulted in a highly significant enrichment of genes involved in the tricarboxylic acid (TCA) cycle and electron transport chain (ETC), suggesting a specific effect of acipimox on mitochondrial metabolism (Fig. 4A). In fact, of all predefined gene sets, only those directly related to mitochondrial metabolism were significantly enriched (familywise error rate, P < 0.05). However, no predefined gene sets specifically address NAD+ metabolism, so we examined a custom-designed gene set comprising genes known to be induced upon treatment with other compounds that boost cellular NAD+ content, such as NR (13,14) and Poly (ADP-ribose) polymerase (PARP) inhibitors (13,14,27,28). This custom gene set was highly enriched in the acipimox-treated patients, with particular similarity to gene sets targeted by PARP inhibitors or other NAD+ precursors (Fig. 4B). Furthermore, ATP content was markedly increased in the acipimox-treated patients (35.2 ± 21.3 vs. 70.0 ± 24.5 relative light units/mg protein for placebo vs. acipimox, P < 0.01) (Fig. 4C).
Figure 4.

NAD+ signature after 2 weeks of acipimox treatment. A: GSEA of muscle biopsy specimens from humans before and after 2 weeks of acipimox treatment. In the Reactome gene set list, only one set is significantly changed between cohorts: TCA cycle and ETC, which is upregulated in treated samples. B: We added a custom-designed gene set more directly related to NAD+ metabolism. The NAD+ booster gene set is significantly enriched in treated samples. C: ATP content measured by CellTiter-Glo chemiluminescence kit in muscle biopsy specimens, corrected for protein loading, of placebo- and acipimox-treated T2D patients. **P < 0.01. D: Protein levels of mtDNA-encoded MTCO1 and nDNA-encoded SDHA show a significant mitonuclear imbalance, leading to the induction of UPRmt, as reflected by the induction of HSP60 expression in muscle biopsy specimens of T2D patients after placebo or acipimox treatment. Coomassie blue staining served as a loading control. FWER, familywise error rate; ns, not significant; PARPi, PARP inhibitor; RLU, relative light unit.
Recently, it has been hypothesized that part of the beneficial effects of NAD+ boosting on mitochondrial metabolism may involve the presence of a stoichiometric imbalance between mitochondrial proteins encoded by the mtDNA and nDNA (i.e., a mitonuclear protein imbalance) (25). This imbalance will lead to the induction of the mitochondrial unfolded protein response (UPRmt), an adaptive and hermetic signaling pathway that ultimately repairs and improves mitochondrial function and metabolic health in animal models ranging from the nematode Caenorhabditis elegans to the mouse (14,25,35). In a subset of subjects, we examined the presence of mitonuclear protein imbalance and UPRmt. Acipimox treatment tended to increase the ratio of MTCO1 (an mtDNA-encoded ETC protein) and SDHA (an nDNA–encoded ETC component) protein content (MTCO1/SDHA 0.26 ± 0.19 vs. 0.75 ± 0.30 for placebo vs. acipimox treatment, P = 0.07) and increased protein content of the mitochondrial chaperone HSP60, which is induced during UPRmt (0.54 ± 0.36 vs. 1.14 ± 0.15 for placebo vs. acipimox, P < 0.05) (Fig. 4D). To further investigate whether acipimox has a direct effect on muscle mitonuclear protein imbalance, we used differentiated C2C12 cells incubated with acipimox or an empty vehicle. First, we confirmed that acipimox is able to increase NAD+ levels in muscle cells (Fig. 5A). Next, 24-h incubation of C2C12 cells with 10 mmol/L acipimox increased HSP60 (Fig. 5B) and the MTCO1/SDHA ratio (Fig. 5C). Finally, to evaluate functional effects of acipimox in C2C12 cells, we performed mitochondrial respiration assays in these cells 24 h after acipimox 10 mmol/L acipimox incubation. FCCP-driven maximal state U respiration was increased upon acipimox, mimicking the results observed in the muscle biopsy specimens obtained from the T2D participants (Fig. 5D). Together, the data obtained in C2C12 cells strengthen the concept that acipimox, as an NAD+ precursor, has direct effects on skeletal muscle mitochondrial respiration and metabolism independent of plasma NEFA levels.
Figure 5.

Effect of acipimox on mitochondrial function in C2C12 cells. A: NAD+ levels after 3 h of acipimox 10 mmol/L incubation in myotubes (n = 6). B: Evaluation of ATP5A, Uqcrc2, MTCO1, SDHA, and HSP60 protein levels in C2C12 cells 24 h after acipimox 10 mmol/L incubation. HSP90 and Ponceau staining served as a loading control. C: MTCO1/SDHA ratio after acipimox 10 mmol/L incubation. D: Evaluation of mitochondrial respiration at the basal condition or after oligomycin and FCCP administration in C2C12 cells 24 h after acipimox 10 mmol/L incubation (n = 6). *P < 0.05, **P < 0.01. Oligom, oligomycin; Veh, vehicle.
Discussion
We showed that treatment for 2 weeks with a standard clinical dosage of acipimox, an NAD+ precursor, caused a rebound effect in plasma NEFA concentrations when measured after omission of the morning dose of acipimox following overnight fasting, which was paralleled by reduced insulin sensitivity. Despite this rebound effect, acipimox still improved mitochondrial oxidative capacity and specifically activated a set of mitochondrial genes that overlaps with the gene sets induced by NAD+ boosters, such as NR, NMN, and PARP inhibitors (13–15,27,35), in animal studies. These changes were accompanied by a significant mitonuclear protein imbalance and the activation of the UPRmt in human muscle biopsy specimens and in cultured myotubes. To the best of our knowledge, this study is the first to demonstrate that as in animals treated with NAD+ boosters (14,35), UPRmt is present in humans. These data hence show that NAD+ precursors activate mitochondrial metabolism in humans using similar signaling pathways as elucidated in a variety of model organisms from worms to mice. Furthermore, the present data urge testing of new NAD+ precursors in human clinical trials that are devoid of the side effects observed after sustained acipimox treatment, such as flushing and a rebound rise in NEFA levels.
The observation of elevated NEFA levels and impaired insulin action contrasts some other studies that showed a long-term reduction of plasma NEFA concentrations by acipimox (3,10). Two major differences exist between these studies and the present study. First, we administered acipimox three times a day (i.e., after every meal) according to clinical and medicinal guidelines, whereas other studies used a dosage of four times a day. Second, in all the previous studies, the last dose of acipimox was given on the test day just before blood sampling and experimental testing, which may obscure the sustained effects of acipimox and result in acute effects of a morning dose. Of note, it has been shown that acipimox treatment can lead to a rebound effect, with plasma NEFA rising in the morning, even with a dosage of four times a day (11). Thus, it was shown that the administration of acipimox resulted in a sustained daytime rise in plasma NEFA, but did not change plasma NEFA concentrations over a 24-h period (11). This compensatory free fatty acid rise may be necessary to maintain energy production, which is interesting because mainly nocturnal concentrations of plasma NEFAs are elevated in T2D patients (36), with insulin resistance being more pronounced in the morning than in the afternoon (37,38). Hence, the increase in plasma NEFA concentrations upon acipimox is most likely a reflection of a nocturnal rebound effect, which was more pronounced in the present study because we did not administer acipimox in the morning of the experimental test days. Thus, it is likely that previous reports on the effect of acipimox on plasma NEFA concentrations and insulin sensitivity actually reflect the remnant effect of the last dose taken rather than the prolonged treatment. Of note, the reduction in insulin sensitivity upon acipimox treatment in the present study was completely accounted for by a reduction in insulin-stimulated NOGD, whereas insulin-stimulated glucose oxidation was not affected by the elevated plasma NEFA levels with acipimox treatment, suggesting that acipimox prevented the decline in oxidative capacity normally observed with high NEFA levels.
In that respect, the rebound rise in plasma NEFA provides a model to study the direct effects of acipimox on mitochondrial function, independent of reduced plasma NEFA levels. We and others have previously shown that acute elevation of plasma NEFA may actually decrease mitochondrial biogenesis and function (21,39,40). In concert with a direct effect of the NAD+ precursor acipimox on mitochondrial function, we observed that in humans, acipimox increased ex vivo mitochondrial oxidative capacity and ATP production despite the potential deleterious elevated plasma NEFA concentrations. A direct effect of acipimox on muscle mitochondrial oxidative capacity and ATP production was confirmed in C2C12 myotubes. Acipimox is a nicotinic acid analog and, thereby, can increase intracellular NAD+ concentrations in skeletal muscle. Reports have shown that stimulation of the NAD+ biosynthesis pathway through NR or NMN has beneficial effects on mitochondrial function (13,15,35). Indeed, in C2C12 myotubes, acipimox had a direct effect on enhancing NAD+ levels. Using an unbiased microarray approach, we also observed that acipimox had clear and significant effects on mitochondrial gene expression in skeletal muscle biopsy specimens from T2D participants. Most striking was the observation that acipimox induced a gene expression signature that was similar to that we observed previously when animals were treated long term with the NAD+ boosters, such as NR (13) and PARP inhibitors (27,35). Furthermore, we show that 2 weeks of acipimox treatment induced a mitonuclear protein imbalance both in cell culture and in ex vivo analysis, which is indicative of an activation of the UPRmt, a pathway that we recently showed to be involved in mediating the beneficial effects of NAD+ on metabolic health (14).
Taken together, the present results show for the first time in humans that an NAD+ precursor like acipimox activates mitochondrial metabolism in human skeletal muscle and induces a mitonuclear protein imbalance and UPRmt. This beneficial impact of acipimox on mitochondrial metabolism is all the more striking because it occurs despite the negative effects of acipimox on plasma NEFA levels and insulin sensitivity. These findings suggest that although acipimox itself may not be a suitable candidate, new NAD+ precursors devoid of such side effects, such as NR and NMN, may potentially act as mitochondrial boosters. Human clinical trials with such agents are urgently needed given that interventions targeting mitochondrial function, such as caloric restriction (41), exercise (42), and resveratrol (43), have been shown to be effective in improving metabolic health.
In conclusion, a 2-week intervention with clinical dosages of acipimox led to a rebound rise in plasma NEFA concentration, which negatively affected insulin sensitivity. Regardless of this negative effect, skeletal muscle mitochondrial oxidative capacity and ATP production improved, most likely through alterations in NAD+ levels and activation of mitochondrial metabolism and the UPRmt. Future research is needed to investigate the safety and efficacy of NAD+ precursors to boost mitochondrial function and improve metabolic health in human subjects, such as T2D patients.
Supplementary Material
Article Information
Acknowledgments. The authors thank the Molecular Resource Center of Excellence at The University of Tennessee Health Science Center for running the microarrays. The authors also thank Christina Wolff for excellent assistance with clamp experiments and patient recruitment in Düsseldorf.
Funding. This study was supported by the Center for Translational Molecular Medicine project PREDICCt (Biomarkers for the Prediction and Early Diagnosis of Diabetes and Diabetes-Related Cardiovascular Complications) (grant 01C-104) and the Netherlands Heart Foundation, Dutch Diabetes Research Foundation, and Dutch Kidney Foundation. E.Ph., V.B.S.-H., and P.S. are supported by Vici and Veni research grants for innovative research from the Netherlands Organisation for Scientific Research (grants 918.96.618 and 916.11.136). J.A. is the Nestlé Chair in Energy Metabolism. The research in the laboratory of J.A. is supported by the École Polytechnique Fédérale de Lausanne, the National Institutes of Health (R01-AG-043930), and the Swiss National Science Foundation (31003A-140780). E.Pi. was funded by the Academy of Finland.
Duality of Interest. No conflicts of interest relevant to this article were reported.
Author Contributions. T.v.d.W. contributed to the study design; data collection, processing, and analysis; and writing of the manuscript. E.Ph., L.B., E.R.R., P.N., L.M.S., S.P., B.H., N.M., E.Pi., and V.B.S.-H. contributed to the data collection, processing, and analysis and writing of the manuscript. E.G.W., A.B., R.L., and J.-H.H. contributed to the study content and review of the manuscript. J.S. contributed to the study design. M.K.C.H., M.R., and P.S. contributed to the study design and revision of the manuscript. J.A. contributed to the revision of the manuscript. M.R. and P.S. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Clinical trial reg. no. NCT00943059, clinicaltrials.gov.
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-0667/-/DC1.
References
- 1.Sirtori CR, Gianfranceschi G, Sirtori M, et al. Reduced triglyceridemia and increased high density lipoprotein cholesterol levels after treatment with acipimox, a new inhibitor of lipolysis. Atherosclerosis 1981;38:267–271 [DOI] [PubMed] [Google Scholar]
- 2.Taskinen MR, Nikkilä EA. Effects of acipimox on serum lipids, lipoproteins and lipolytic enzymes in hypertriglyceridemia. Atherosclerosis 1988;69:249–255 [DOI] [PubMed] [Google Scholar]
- 3.Bajaj M, Suraamornkul S, Romanelli A, et al. Effect of a sustained reduction in plasma free fatty acid concentration on intramuscular long-chain fatty Acyl-CoAs and insulin action in type 2 diabetic patients. Diabetes 2005;54:3148–3153 [DOI] [PubMed] [Google Scholar]
- 4.Bajaj M, Medina-Navarro R, Suraamornkul S, Meyer C, DeFronzo RA, Mandarino LJ. Paradoxical changes in muscle gene expression in insulin-resistant subjects after sustained reduction in plasma free fatty acid concentration. Diabetes 2007;56:743–752 [DOI] [PubMed] [Google Scholar]
- 5.Phielix E, Jelenik T, Nowotny P, Szendroedi J, Roden M. Reduction of non-esterified fatty acids improves insulin sensitivity and lowers oxidative stress, but fails to restore oxidative capacity in type 2 diabetes: a randomised clinical trial. Diabetologia 2014;57:572–581 [DOI] [PubMed] [Google Scholar]
- 6.Schrauwen P, Schrauwen-Hinderling V, Hoeks J, Hesselink MK. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta 2010;1801:266–271 [DOI] [PubMed]
- 7.Phielix E, Schrauwen-Hinderling VB, Mensink M, et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008;57:2943–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002;51:2944–2950 [DOI] [PubMed] [Google Scholar]
- 9.Szendroedi J, Schmid AI, Chmelik M, et al. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med 2007;4:e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Worm D, Henriksen JE, Vaag A, Thye-Rønn P, Melander A, Beck-Nielsen H. Pronounced blood glucose-lowering effect of the antilipolytic drug acipimox in noninsulin-dependent diabetes mellitus patients during a 3-day intensified treatment period. J Clin Endocrinol Metab 1994;78:717–721 [DOI] [PubMed] [Google Scholar]
- 11.Saloranta C, Taskinen MR, Widen E, Härkönen M, Melander A, Groop L. Metabolic consequences of sustained suppression of free fatty acids by acipimox in patients with NIDDM. Diabetes 1993;42:1559–1566 [DOI] [PubMed] [Google Scholar]
- 12.Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr 2008;28:115–130 [DOI] [PubMed] [Google Scholar]
- 13.Cantó C, Houtkooper RH, Pirinen E, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 2012;15:838–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mouchiroud L, Houtkooper RH, Moullan N, et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013;154:430–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab 2011;14:528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benyó Z, Gille A, Kero J, et al. GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J Clin Invest 2005;115:3634–3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuipers H, Verstappen FT, Keizer HA, Geurten P, van Kranenburg G. Variability of aerobic performance in the laboratory and its physiologic correlates. Int J Sports Med 1985;6:197–201 [DOI] [PubMed] [Google Scholar]
- 18.Siri WE. The gross composition of the body. Adv Biol Med Phys 1956;4:239–280 [DOI] [PubMed] [Google Scholar]
- 19.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979;237:E214–E223 [DOI] [PubMed] [Google Scholar]
- 20.Mensink M, Blaak EE, van Baak MA, Wagenmakers AJ, Saris WH. Plasma free fatty acid uptake and oxidation are already diminished in subjects at high risk for developing type 2 diabetes. Diabetes 2001;50:2548–2554 [DOI] [PubMed] [Google Scholar]
- 21.Brehm A, Krssak M, Schmid AI, Nowotny P, Waldhäusl W, Roden M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes 2006;55:136–140 [PubMed] [Google Scholar]
- 22.Frayn KN. Calculation of substrate oxidation rates in vivo from gaseous exchange. J Appl Physiol 1983;55:628–634 [DOI] [PubMed] [Google Scholar]
- 23.Bergström J, Hermansen L, Hultman E, Saltin B. Diet, muscle glycogen and physical performance. Acta Physiol Scand 1967;71:140–150 [DOI] [PubMed] [Google Scholar]
- 24.Koopman R, Schaart G, Hesselink MK. Optimisation of oil red O staining permits combination with immunofluorescence and automated quantification of lipids. Histochem Cell Biol 2001;116:63–68 [DOI] [PubMed] [Google Scholar]
- 25.Houtkooper RH, Mouchiroud L, Ryu D, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013;497:451–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bai P, Cantó C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab 2012;16:290–295 [DOI] [PubMed] [Google Scholar]
- 28.Bai P, Canto C, Brunyánszki A, et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab 2011;13:450–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantó C, Gerhart-Hines Z, Feige JN, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009;458:1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramsey KM, Yoshino J, Brace CS, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009;324:651–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stingl H, Krssák M, Krebs M, et al. Lipid-dependent control of hepatic glycogen stores in healthy humans. Diabetologia 2001;44:48–54 [DOI] [PubMed] [Google Scholar]
- 32.Steele R. Influences of glucose loading and of injected insulin on hepatic glucose output. Ann N Y Acad Sci 1959;82:420–430 [DOI] [PubMed] [Google Scholar]
- 33.Saloranta C, Groop L, Ekstrand A, Franssila-Kallunki A, Eriksson J, Taskinen MR. Different acute and chronic effects of acipimox treatment on glucose and lipid metabolism in patients with type 2 diabetes. Diabet Med 1993;10:950–957 [DOI] [PubMed] [Google Scholar]
- 34.Roden M, Price TB, Perseghin G, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 1996;97:2859–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pirinen E, Cantó C, Jo YS, et al. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab 2014;19:1034–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reaven GM, Hollenbeck C, Jeng CY, Wu MS, Chen YD. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes 1988;37:1020–1024 [DOI] [PubMed] [Google Scholar]
- 37.Bolli GB. The dawn phenomenon: its origin and contribution to early morning hyperglycemia in diabetes mellitus. Diabete Metab 1988;14:675–686 [PubMed] [Google Scholar]
- 38.Shapiro ET, Polonsky KS, Copinschi G, et al. Nocturnal elevation of glucose levels during fasting in noninsulin-dependent diabetes. J Clin Endocrinol Metab 1991;72:444–454 [DOI] [PubMed] [Google Scholar]
- 39.Hoeks J, Hesselink MK, Russell AP, et al. Peroxisome proliferator-activated receptor-gamma coactivator-1 and insulin resistance: acute effect of fatty acids. Diabetologia 2006;49:2419–2426 [DOI] [PubMed] [Google Scholar]
- 40.Hoeks J, van Herpen NA, Mensink M, et al. Prolonged fasting identifies skeletal muscle mitochondrial dysfunction as consequence rather than cause of human insulin resistance. Diabetes 2010;59:2117–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heilbronn LK, de Jonge L, Frisard MI, et al.; Pennington CALERIE Team . Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: a randomized controlled trial. JAMA 2006;295:1539–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meex RC, Schrauwen-Hinderling VB, Moonen-Kornips E, et al. Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes 2010;59:572–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Timmers S, Konings E, Bilet L, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 2011;14:612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.