

Abstract
Genome-wide association studies (GWAS) have uncovered >65 common variants associated with type 2 diabetes (T2D); however, their relevance for drug development is not yet clear. Of note, the first two T2D-associated loci (PPARG and KCNJ11/ABCC8) encode known targets of antidiabetes medications. We therefore tested whether other genes/pathways targeted by antidiabetes drugs are associated with T2D. We compiled a list of 102 genes in pathways targeted by marketed antidiabetic medications and applied Gene Set Enrichment Analysis (MAGENTA [Meta-Analysis Gene-set Enrichment of variaNT Associations]) to this gene set, using available GWAS meta-analyses for T2D and seven quantitative glycemic traits. We detected a strong enrichment of drug target genes associated with T2D (P = 2 × 10−5; 14 potential new associations), primarily driven by insulin and thiazolidinedione (TZD) targets, which was replicated in an independent meta-analysis (Metabochip). The glycemic traits yielded no enrichment. The T2D enrichment signal was largely due to multiple genes of modest effects (P = 4 × 10−4, after removing known loci), highlighting new associations for follow-up (ACSL1, NFKB1, SLC2A2, incretin targets). Furthermore, we found that TZD targets were enriched for LDL cholesterol associations, illustrating the utility of this approach in identifying potential side effects. These results highlight the potential biomedical relevance of genes revealed by GWAS and may provide new avenues for tailored therapy and T2D treatment design.
Introduction
Genome-wide association studies (GWAS) have uncovered >65 common DNA variants associated with type 2 diabetes (T2D) that collectively explain ∼10% of the genetic contribution to T2D susceptibility, mostly in populations of European ancestry (1,2). Hundreds of additional common variants (minor allele frequency >1%) are predicted to be associated with T2D, with modest odds ratios based on mixed-model and polygenic analyses of large GWAS meta-analyses (3–5). In addition, dozens of other single nucleotide polymorphisms (SNPs) have been found to be associated with various quantitative glucose and insulin-related traits (6–11), which are intermediate phenotypes of T2D and, hence, may help in better understanding the pathophysiology of diabetes. These GWAS have been extended by the use of large, custom-made genotyping arrays, which include the top SNP associations for a variety of metabolic traits that approached, but did not quite achieve, genome-wide significance (12); when deployed across large numbers of additional samples, genome-wide significance has been reached for 10 additional association signals with T2D (3) and 41 for a number of quantitative glycemic traits (13).
Several genes that lie near established SNPs associated with T2D or a related glycemic trait encode direct or indirect targets of antidiabetes medications (Table 1 and Supplementary Table 1). Specifically, the first two reproducible associations with T2D were reported for missense polymorphisms in two candidate gene regions (encoding the peroxisome proliferator–activated receptor γ [PPARG] and the islet ATP-sensitive potassium channel Kir6.2/sulfonylurea receptor SUR1 complex [KCNJ11/ABCC8]), which were selected because they happen to be targets of antidiabetic medications (thiazolidinediones [TZDs] and sulfonylureas, respectively) (14,15). This observation led us to ask whether additional genes that function in biological pathways or processes affected by various antidiabetes medications may also contain common variants associated with T2D or related glycemic traits, albeit less strongly (i.e., weaker effects, lower penetrance in the population), but when analyzed together, their combined effect may surpass statistical significance. In other words, we reasoned that proof of their successful modulation by pharmacological means might indicate their involvement in T2D pathophysiology.
Table 1.
Target genes and pathways of nine classes of FDA-approved antidiabetes medications
| Medication class | Mechanism of action | Physiological effect | Tissue | Number of target genes* | Target genes of antidiabetes medication class* | Target genes in LD to known T2D or glycemic trait–associated SNPs† | References |
|---|---|---|---|---|---|---|---|
| Insulin | Downstream signaling from insulin/IGF-I receptor | Signals glucose uptake | Liver, muscle, fat | 17 | ACACA, AKT1, FOXO1, GAB1, INSR, IRS1, IRS2, IRS4, PIK3CA, PIK3R1, SLC2A1, SLC2A2, SLC2A3, SLC2A4, SLC2A5, SHC1, TRIB3 | IRS1: T2D, fasting insulin, insulin resistance; SLC2A2 (GLUT2): fasting glucose | 11,19,42 |
| Metformin (biguanide) | AMP-activated protein kinase pathway; complex I inhibition | Improves insulin sensitivity; reduces gluconeogenesis | Liver, muscle | 6 | PRKAA2, SLC22A1, SLC22A2, SLC2A1, SLC2A4, STK11 | — | 20,30 |
| TZDs | PPARG receptor pathway | Fat, muscle, liver | 42 | ABCA1, ACSL1, ADIPOQ, ADIPOR1, ADIPOR2, APOA1, APOC3, CCL2, CD36, CPT1A, CPT1B, CPT2, CRP, EDN1, F3, FACL2, FGB, GK, HSD11B1, ICAM1, IL6, IRS1, IRS2, MMP9, NFKB1, NFKB2, NOS2A, PDK4, PIK3CA, PIK3R1, PPARA, PPARD, PPARG, PTGS1, RETN, RXRA, SCARB1, SLC2A4, SLC27A1, SORBS1, TNF, VCAM1 | PPARG: T2D; IRS1: T2D, fasting insulin | 11,21,22,42 | |
| Sulfonylureas | ATP-sensitive K channel inhibition | Increases insulin production and secretion from β-cells in pancreas and/or decreases glucagon secretion from α-cells in pancreas | Pancreas, liver, fat | 5 | ABCC8, ABCC9, KCNJ11, SLC2A4, TNF | KCNJ11: T2D ABCC8: T2D | 23–25 |
| GLP-1 receptor agonists | GLP-1 receptor pathway | Pancreas, GI tract | 18 | ATF4, BCL2, CASP12, CPA1, DDIT3, EIF2S1, GCG, GLP1R, HSPA5, JUNB, NGFB, NGFR, PDX1, PPP1R15A, PPYR1, PRKACA, XBP1, XIAP | PDX1: fasting glucose‡ | 16–18 | |
| DPP4 inhibitors | Inhibits DPP4 degradation (i.e., GLP-1, GIP) | Diffuse | 20 | ADCYAP1, CCL11, CCL5, CCL22, CXCL9, CXCL10, CXCL11, CXCL12, DPP4, DPP8, DPP9, FAP, GHRH, GIP, GIPR, GRP, GLP2R, NPY, PYY, TAC1 | GIPR: T2D‡, fasting glucose‡, 2-h glucose, 2-h insulin | 18,26 | |
| Amylin mimetics | Amylin receptor pathway | Pancreas, GI tract | 2 | IAPP, IDE | IDE: T2D | 27 | |
| Meglitinides | ATP-sensitive K channel inhibition | Pancreas, liver, fat | 2 | ABCC8, KCNJ11 | KCNJ11/ABCC8: T2D | 28 | |
| α-Glucosidase inhibitors | Inhibits α-glucosidase enzymes, α-amylase inhibition | Affects sugar absorption in gut by preventing digestion of carbohydrates | Small intestine, pancreas | 2 | AMY2A, GAA | — | 29 |
GI, gastrointestinal.
*Genes involved in pathways targeted by antidiabetes medications were compiled from the literature.
†Validated SNP associations are based on GWAS of European descent individuals; details of associations are in Supplementary Table 1.
‡These genes were added to our drug target gene list before their association with T2D or a related glycemic trait was reported (3). Genes in boldface refer to drug targets in LD to SNPs associated with T2D or glycemic traits.
Currently, multiple classes of antidiabetic medications are approved for clinical use, including insulin, biguanides, sulfonylureas, TZDs, meglitinides, α-glucosidase inhibitors, GLP-1 receptor agonists, dipeptidyl peptidase 4 (DPP4) inhibitors, and amylin mimetics (16–30). All medications help to decrease glucose blood levels through different mechanisms of action. These vary from increasing insulin secretion by pancreatic β cells to increasing insulin sensitization in target tissues (e.g., muscle, fat) and inhibiting glucose absorption in the gastrointestinal tract. Some of the more recently approved medication classes are the result of rational drug design (e.g., GLP-1 receptor agonists, DPP4 inhibitors), some from empirical experimentation (e.g., metformin), and others from a combination of the two (e.g., TZDs [ciglitazone discovered through in vivo compounds screening (31) and successive analogs designed against the subsequently identified drug target PPARG (32)]).
The targeted pathways and downstream effects of these drug classes have been investigated to a greater or lesser extent through human, animal, and cell culture studies (16–30). We attempted to leverage existing GWAS data sets to comprehensively test whether these pathways contain multiple genes, in addition to those already known, that harbor natural genetic perturbations that may influence risk of T2D. We further evaluated whether this approach could be used to predict unintended phenotypic effects of drug treatment by examining the genetic basis of a known nonglycemic effect of TZD drugs.
Research Design and Methods
Construction of the Antidiabetes Drug Target Gene Set
The source of genes to include in the drug target gene set were culled from PubMed searches (initially performed in 2007 [before the wave of GWAS publications] and repeated in 2010) of original reports and reviews on the mechanism of action of nine Food and Drug Administration (FDA)–approved antidiabetes medication classes at the time: insulin, biguanides (metformin), sulfonylureas, TZDs, meglitinides, α-glucosidase inhibitors, GLP-1 receptor agonists, DPP4 inhibitors, and amylin mimetics (16–30). More recent classes of antidiabetes drugs (cholesterol-binding resins, dopamine agonists, or sodium-glucose co-transporter-2 inhibitors) have not yet reached widespread use and, thus, were not studied here. Drug class targets and their downstream effectors were considered broadly for each drug class. Proteins were considered a potential target of the drug if they were directly affected by the drug or a direct downstream mediator of the known drug pathway, considering human, animal, and cell culture studies. There was significant overlap of target genes among certain drug classes because their mechanisms of action are largely the same (e.g., sulfonylureas, meglitinides). Proteins in the drug pathway whose genes did not have a validated human genome location were excluded from the analysis.
GWAS Meta-analyses Analyzed
The present analysis was based on the SNP association P values from the following GWAS meta-analyses: 1) DIAGRAMv3 (DIAbetes Genetics Replication And Meta-analysis version 3) T2D meta-analysis: 12,171 T2D cases and 56,862 controls across 12 GWAS of individuals of European descent and ∼2.5 × 106 genotyped and imputed SNPs (data can be downloaded at http://diagram-consortium.org/downloads.html) (3); 2) Metabochip T2D meta-analysis: 21,491 T2D cases and 55,647 controls across 25 studies of individuals of European descent and 1,178 T2D cases and 2,472 controls from one study of individuals of Pakistani descent (PROMIS [Pakistan Risk of Myocardial Infarction Study]), with a total of 22,669 cases and 58,119 controls (3) (the Metabochip custom array comprises 196,726 SNPs, 5,057 of which are T2D “replication” SNPs that capture the strongest independent autosomal association signals from the DIAGRAMv3 GWAS meta-analysis; individuals in the DIAGRAMv3 meta-analysis are independent from those in the Metabochip T2D meta-analysis); 3) seven MAGIC (Meta-Analyses of Glucose and Insulin-related traits Consortium) meta-analyses of glucose and insulin-related traits: 9–23 GWAS of 15,000–46,000 participants without diabetes and 2.3–2.7 × 106 genotyped and imputed SNPs, depending on the trait (data can be downloaded at www.magicinvestigators.org/downloads) (7–9); and 4) three GWAS meta-analyses of plasma LDL cholesterol (LDL-C), HDL cholesterol (HDL-C), and triglyceride levels: 46 GWAS in ∼95,000–100,000 individuals total (data can be downloaded at www.sph.umich.edu/csg/abecasis/public/lipids2010) (33). The global lipids blood measurements were taken after >8 h of fasting. The lower-bound minor allele frequency of SNPs in these meta-analyses is 1%.
Discovery Step 1: Gene Set Enrichment Analysis of GWAS SNP Data Using MAGENTA
To test whether a set of antidiabetes drug target genes contains multiple genes associated with T2D more than would be expected by chance, we applied a Gene Set Enrichment Analysis (GSEA) method (an approach originally developed for gene expression) that we previously adapted for GWAS data called Meta-Analysis Gene-set Enrichment of variaNT Associations (MAGENTA) (34). Intuitively, MAGENTA tests whether multiple genes associated with a disease or trait cluster in a given biological pathway or set of functionally related genes. It does so by testing whether the distribution of gene association P values of all genes in a gene set of interest is skewed toward low P values compared with the (close to uniform) P value distributions of randomly sampled gene sets of equivalent size (Fig. 1). A significant skewness below a given P value cutoff (enrichment cutoff) would suggest that the gene set is enriched for multiple genes associated with the tested complex disease or trait. To gain statistical and explanatory power, MAGENTA tests for enrichment not only of genes with strong SNP associations (e.g., ones that pass multiple hypothesis correction, P < 5 × 10−8) but also of genes with modest SNP associations (associations that have not yet reached genome-wide significance [e.g., P = 10−4–10−3] due to insufficient power afforded by finite GWAS sample sizes). This is supported by polygenic analyses of GWAS that suggest that various complex phenotypes, including T2D risk, are influenced by hundreds of modest associations that have not yet been detected due to insufficient statistical power (3–5).
Figure 1.

An overview of the study design, analytical steps, and questions addressed. The strategy addressed a number of key questions about the relationship between human genetic associations with T2D or related glycemic traits and antidiabetes drug targets. A similar strategy can be applied to other diseases and traits. 2-h glucose and 2-h insulin, glucose or insulin plasma levels measured 2 h after an oral glucose tolerance test; HbA1c, a measure for long-term glycemia; HOMA-B, a measure for β-cell function; HOMA-IR, a measure for insulin resistance.
We applied MAGENTA to all genotyped and imputed SNP associations from the aforementioned GWAS meta-analyses DIAGRAMv3, MAGIC, and global lipids GWAS meta-analyses, as described quantitatively and in detail by Segrè et al. (34). Briefly, first, MAGENTA scores all genes in the genome by assigning each gene the most significant local SNP association P value within −110 kilobases (kb) upstream and +40 kb downstream the transcript start and end sites, respectively (boundaries chosen to capture potential regulatory causal variants in addition to coding variants within the gene itself). The gene association scores are subsequently corrected for confounding effects, such as gene size, local SNP density, and linkage disequilibrium (LD)–related properties, using stepwise multivariate linear regression analysis (because larger genes are more likely to carry a SNP with a more significant P value than smaller genes by chance, as larger genes contain more SNPs) (34). Second, the adjusted gene association P values, used to rank genes in the genome with respect to their likelihood of association with the given trait, are used to estimate gene set enrichment P values for each gene set of interest. The gene set enrichment P value calculated by MAGENTA assesses the overrepresentation of highly ranked gene association P values above an enrichment cutoff, compared with multiple randomly sampled gene sets from the genome, with equal gene set size. The 75th percentile of the association P values of all genes in the genome (which corresponded, e.g., in DIAGRAMv3 to an adjusted gene P < 0.3) was used as the enrichment cutoff. Physical proximity along the chromosome between two or more genes in a given gene set was corrected for by collapsing them to one effective gene, retaining the gene with the most significant adjusted association P value. Only genes on autosomal chromosomes were analyzed, which led to the exclusion of three drug target genes on chromosome X. The HLA region was removed due to high LD and gene density in the region, making it difficult to disentangle the putative causal gene if an association signal exists in the region. One of the antidiabetes drug target genes, TNF lies in this region. The MAGENTA software package can be downloaded at www.broadinstitute.org/mpg/magenta.
The set of validated T2D SNPs used in this work (Supplementary Table 3) included 55 associated loci identified in GWAS meta-analyses of populations of European descent, including DIAGRAMv3 and Metabochip. Genes near the validated T2D SNPs were defined using the larger of two boundaries around each SNP: ±100 kb or an LD-based boundary defined by proceeding to r2 > 0.5 on either side of the SNP, then to the nearest recombination hotspot, and finally adding an additional 50 kb on either side. Genome build 36 (hg18) was used for chromosome positions.
Replication Step 2: Modified GSEA of Metabochip Meta-analysis
To test if the gene set enrichment results obtained by MAGENTA replicated in an independent study, we applied a modified GSEA method for genetic association data that we developed in Morris et al. (3) to a separate, larger meta-analysis of multiple association studies genotyped on the Metabochip array. A modified GSEA approach was needed to account for the bias in the Metabochip SNP design, which contains a subset of SNPs that unevenly cover the genome (196,725 replication and fine-mapping SNPs) compared with the less biased genome-wide SNP arrays (∼2 × 106 SNPs) used in DIAGRAMv3, MAGIC, and the lipid GWAS (analyzed in the discovery step). The Metabochip is a custom array designed to follow-up nominal associations for T2D and 22 other metabolic and cardiovascular traits in a more cost-effective manner than genome-wide SNP arrays and contains 5,057 T2D replication SNPs chosen based on the top independent association signals in DIAGRAMv3 (12). This has enabled the genotyping of top-ranked metabolic SNPs in an additional ∼21,500 T2D cases and ∼55,600 controls. Specifically, the modified GSEA method tests for enrichment of antidiabetes drug target genes among all genes near a set of top T2D-associated SNPs based on the Metabochip meta-analysis. First, for the enrichment cutoff, we used a set of high-confidence T2D SNPs/loci: 137 T2D loci, which included 53 established (P < 5 × 10−8); 6 highly probable (P < 5 × 10−7); and 78 probable (more modest) T2D loci with a posterior probability (confidence score) >75%, as chosen in Morris et al., based on a mixture model fitted to the Metabochip T2D meta-analysis z scores (for a list of the 137 T2D SNPs, see Supplementary Table 15 in Morris et al. [3], excluding the monogenic genes). Second, we tested two SNP-to-gene mapping definitions: 1) nearest gene and 2) an LD-based boundary defined by proceeding to r2 > 0.5 on either side of the SNP, then to the nearest recombination hotspot, and finally adding an additional 50 kb on either side. SNPs with no genes in LD were assigned the nearest gene. SNPs with the same nearest gene were collapsed to one locus. SNPs with more than one proximal gene in the drug target gene set were counted as one instance to reduce inflation of the gene set enrichment P value due to physical clustering of subsets of drug target genes along the genome (3). For each gene set gs and a set of T2D SNPs/loci l with m SNPs, we calculated the probability of observing at least k T2D loci of a total m loci with one or more proximal genes that belong to gene set gs, given that n of N Metabochip SNPs analyzed have one or more proximal genes in gene set gs (using a hypergeometric probability distribution) (Eq. 1):
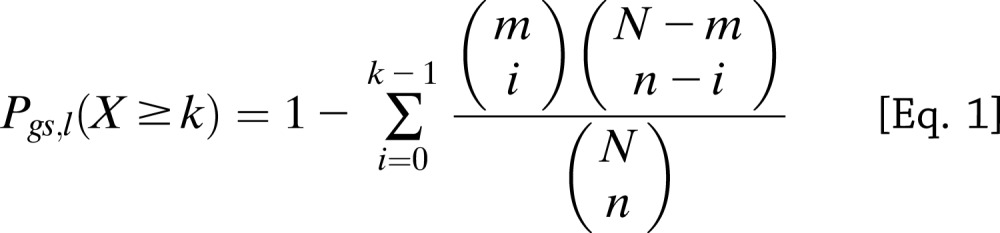 |
N refers to the number of Metabochip SNPs in the null set (defined next) plus the m top T2D SNPs/loci. To account for differences in coverage of the Metabochip replication SNPs across all genes in the genome, we generated an empirical null distribution of 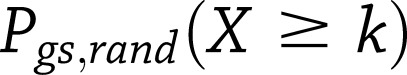 for
for 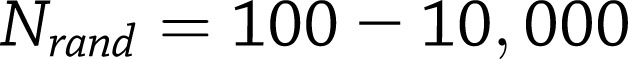 randomly sampled sets of SNPs, matched for SNP number and local gene density with the T2D SNPs/loci set l. The adjusted gene set enrichment P value
randomly sampled sets of SNPs, matched for SNP number and local gene density with the T2D SNPs/loci set l. The adjusted gene set enrichment P value 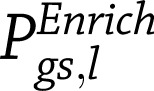 is the fraction of randomly sampled SNP sets of equal size to the T2D SNP set l with the same or more significant hypergeometric probability (Eq. 1) than that of the actual T2D SNP set (Eqs. 2–4):
is the fraction of randomly sampled SNP sets of equal size to the T2D SNP set l with the same or more significant hypergeometric probability (Eq. 1) than that of the actual T2D SNP set (Eqs. 2–4):
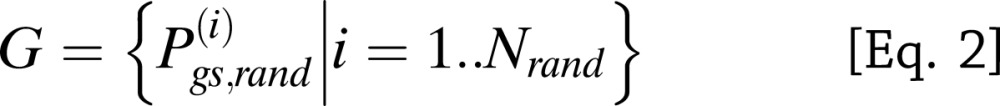 |
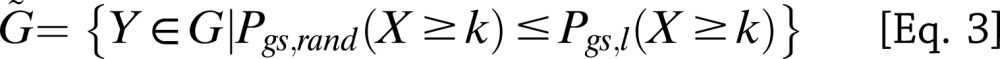 |
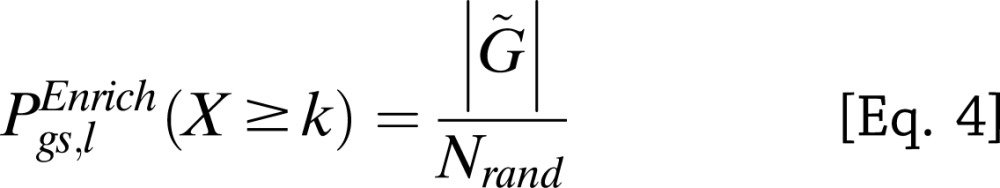 |
For the null SNP set, we used the full set of Metabochip replication SNPs after LD pruning (r2 < 0.05, using CEU [Utah residents with Northern and Western European ancestry] HapMap samples as the reference population), excluding the lead and proxy (CEU r2 ≥ 0.1) SNPs of previously established T2D SNPs, 5,057 T2D replication SNPs, SNPs near monogenic diabetes genes, and QT-interval replication SNPs (used as a negative control). This resulted in a set of 16,408 null SNPs. Similar GSEA results to those in Table 5 were obtained when using as the null SNP set 1,600 SNPs with the lowest posterior probability of being associated with T2D of 3,408 LD-pruned Metabochip T2D replication SNPs (posterior probability < 5% of belonging to an alternative distribution in the mixture model of Metabochip z scores) (data not shown).
Table 5.
Replication of T2D association enrichment signal in antidiabetes drug target set in an independent T2D meta-analysis (Metabochip)
| Genes mapped to established and high-confidence set of T2D SNPs* | Number of Metabochip replication SNPs (null set) near one or more drug target genes† | Number of top-ranked T2D SNPs near one or more drug target genes | Gene set enrichment P value | Genes near established or high-confidence T2D SNPs |
|---|---|---|---|---|
| Nearest gene | 48 | 5 | 0.003 | PPARG1, KCNJ111, IRS11, GIPR2, ACSL13 |
| Genes in LD‡ | 83 | 7 | 0.04 | PPARG1, KCNJ111, ABCC81, IDE1 IRS1 GIPR2, NFKB13, ACSL13 |
*T2D SNP set tested includes 137 loci: 59 established or highly probable SNPs and 78 high-confidence T2D SNPs based on Metabochip analysis (described in research design and methods).
†This set includes a null set of 16,408 LD-pruned Metabochip replication SNPs that does not contain SNPs in LD to previously established T2D SNPs, ∼5,000 T2D replication SNPs, monogenic diabetes genes, or QT-interval replication SNPs (see research design and methods ).
‡LD boundaries are defined in research design and methods.
1Genes near previously established T2D SNPs.
2Gene near T2D SNP found in the joint T2D meta-analysis of DIAGRAMv3 and Metabochip, which did not reach genome-wide significance in DIAGRAMv3 alone.
3Genes near T2D SNPs that have not yet reached genome-wide significance but have a high posterior probability of being associated with T2D based on Metabochip analysis (3).
Results
We tested the hypothesis that biological pathways targeted by antidiabetes medications may be enriched for multiple genes modestly associated with T2D, more than would be expected by chance, by applying the GSEA approach implemented in MAGENTA (34) to a compiled list of 102 direct or indirect target genes of one or more of the nine classes of antidiabetes medications (described in research design and methods). The antidiabetes drug target genes ranged from 1 to 41 per drug class (Table 1). The study design and analyses performed on the drug target gene set and its subclasses are described in Fig. 1.
Using the largest available T2D GWAS meta-analysis of ∼12,000 cases and ∼57,000 controls (DIAGRAMv3), we found that the full set of antidiabetes drug target genes was significantly enriched for multiple genes that carry SNPs modestly associated with T2D risk (P = 1.7 × 10−5, 1.8-fold enrichment [i.e., we predict that about one-half the target genes with association scores above the 75th percentile enrichment cutoff are modestly associated with T2D]) (Table 2 and Fig. 2). MAGENTA suggests that 18 of the 41 loci (44 genes) above the 75th percentile enrichment cutoff (expected number of genes above cutoff, 23) (see columns 3–5 in Table 2) are true associations with T2D risk, 4 of which map onto known T2D loci (see column 8 of Table 2) and 14 of which may contain new SNP associations with T2D. Additional follow-up analyses and experiments are needed to identify the 14 true-positive novel gene associations. The top-ranked 44 drug target genes and their most significant local T2D SNP P values are listed in Supplementary Table 2.
Table 2.
GSEA of T2D and glycemic trait associations in the antidiabetes drug target gene set
| GWAS meta-analysis | Nominal MAGENTA enrichment P value* | Number of OBS genes/loci above enrichment cutoff | Number of EXP genes/loci above enrichment cutoff | Excess number of genes/loci above enrichment cutoff (OBS − EXP)† | Enrichment fold (OBS/EXP) | Number of genes near validated GWAS SNPs‡ | Genes near validated GWAS SNPs‡ |
|---|---|---|---|---|---|---|---|
| T2D | 1.7 × 10−5 | 41** | 23 | 18 | 1.78 | 6*** | PPARG, IRS1§, KCNJ11/ABCC8‖, IDE, GIPR¶ |
| Fasting glucose | 0.078 | 31 | 24 | 7 | 1.29 | 1 | SLC2A2 |
| HOMA-IR | 0.11 | 29 | 24 | 5 | 1.21 | 0 | — |
| 2-h insulin | 0.24 | 27 | 24 | 3 | 1.13 | 1 | IRS1 |
| HOMA-B | 0.27 | 26 | 23 | 3 | 1.13 | 0 | — |
| Fasting insulin | 0.29 | 26 | 24 | 2 | 1.08 | 0 | — |
| 2-h glucose | 0.74 | 20 | 23 | 0 | 0.87 | 0 | — |
| HbA1c | 0.75 | 20 | 23 | 0 | 0.87 | 0 | — |
The 2-h glucose and 2-h insulin concentrations were measured after an oral glucose tolerance test. EXP, expected; OBS, observed.
*The gene set enrichment P value was calculated by MAGENTA using a 75th percentile enrichment cutoff.
**44 genes had scores above the enrichment cutoff, but 3 genes were removed from GSEA to correct for physical clustering along the genome (see Table 4).
***Only 4 loci contributed to enrichment signal. See next two footnotes for explanation.
†Estimated number of antidiabetes drug targets that may be true associations with T2D, 14 of which have not yet reached genome-wide significance.
‡Genes were mapped onto 55 established T2D SNPs using the larger of the two boundaries around each SNP: ±100 kb or LD r2 > 0.5 (see research design and methods and Supplementary Table 3).
§The gene association P value of IR1S did not surpass the enrichment cutoff because the established T2D GWAS SNP near IRS1 lies farther away than the gene boundaries used in MAGENTA (+110 kb/−40 kb).
‖KCNJ11/ABCC8 were collapsed to one effective gene in the GSEA due to their physical proximity.
¶GIPR was added to our drug target gene list before its association with T2D reached genome-wide significance in a joint meta-analysis of DIAGRAMv3 and Metabochip (3).
Figure 2.

Distribution of T2D gene association P values of antidiabetes drug targets. To visualize the enrichment of multiple modest associations with T2D among antidiabetes drug target genes, we plotted the noncumulative distribution of adjusted gene association P values (calculated with MAGENTA) for all the antidiabetes drug targets (99 autosomal genes), as shown in the first track (red line). The following two tracks display from top to bottom the individual gene P values (represented by vertical lines) for the insulin targets subset and the TZD targets subset. Common insulin and TZD targets are shown in blue. The dashed line marks the 75th percentile enrichment cutoff.
Given the positive results, we next asked whether the antidiabetes drug target gene set might also be enriched for multiple genes associated with glucose or insulin-related traits, which are intermediate phenotypes or risk factors of T2D. We repeated the aforementioned analysis by applying MAGENTA to seven GWAS meta-analyses of 15,000–46,000 nondiabetic individuals (MAGIC) for the following glycemic traits: fasting glucose levels, fasting insulin levels, 2-h glucose or 2-h insulin plasma levels following an oral glucose tolerance test, a measure for β-cell function (HOMA-B), a measure for insulin resistance (HOMA-IR), and a measure for long-term glycemia (glycated hemoglobin [HbA1c]). None of the glycemic traits showed a significant overrepresentation of multiple modest gene associations in the antidiabetes drug target gene set (Table 2).
Given the strong enrichment of drug target gene associations with T2D, we asked whether the observed enrichment signal was primarily driven by genes targeted by one or a subset of the nine classes of antidiabetes medication classes. To address this, we tested for enrichment of T2D associations in individual drug class target subsets for four of the nine classes of drugs that contained at least 10 target genes (chosen as the lower bound for statistical power considerations). We found that insulin targets and TZD targets were the primary drivers of the collective enrichment signal (P = 0.001, 2.5-fold enrichment, and P = 0.02, 1.6-fold enrichment, respectively) (Table 3 and Fig. 2). However, although not enriched, incretin targets were also among the top-ranked drug target genes based on their T2D association P values (e.g., the DPP4 inhibitors GIP, GLP2R, GRP, GIPR) (Table 4 and Supplementary Table 2).
Table 3.
GSEA of T2D associations in individual drug class target sets before and after removing genes in established T2D loci
| All genes analyzed |
Excluding genes in LD to validated T2D SNPs |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Antidiabetes drug target gene set | Number of genes analyzed* | Nominal MAGENTA enrichment P value | Enrichment fold (OBS/EXP) | Excess number of genes above enrichment cutoff (OBS − EXP) | Number of genes analyzed* | Nominal MAGENTA enrichment P value | Enrichment fold (OBS/EXP) | Excess number of genes above enrichment cutoff (OBS − EXP) | Genes in LD to validated T2D SNPs† |
| All nine classes of drugs | 92 | 1.7 × 10−5 | 1.8 | 18 | 87 | 4 × 10−4 | 1.7 | 15 | PPARG, IRS1‡, KCNJ11/ABCC8, IDE, GIPR |
| Insulin | 16 | 0.001 | 2.5 | 6 | 15 | 8 × 10−4 | 2.5 | 6 | IRS1 |
| TZDs | 38 | 0.02 | 1.6 | 6 | 36 | 0.01 | 1.7 | 6 | PPARG, IRS1 |
| DPP4 inhibitors | 18 | 0.15 | 1.4 | 2 | 17 | 0.22 | 1.5 | 2 | — |
| GLP-1 receptor agonists | 17 | 0.38 | 1.3 | 1 | 17 | 0.49 | 1.3 | 1 | — |
EXP, expected; OBS, observed.
*Number of genes analyzed after excluding genes in HLA region (one gene) and genes on sex chromosomes (three genes) and correcting for physical clustering of subsets of genes within a given gene set along the genome (six genes).
†Genes were mapped onto 55 established T2D SNPs using the larger of two boundaries around each SNP: ±100 kb or LD r2 > 0.5 (see research design and methods and Supplementary Table 3).
‡The gene P value of IRS1 did not pass the enrichment cutoff because the validated T2D GWAS SNP near IRS1 lies farther away than the gene boundaries used in the MAGENTA analysis.
Table 4.
Top-ranked antidiabetes target genes above enrichment cutoff based on their DIAGRAMv3 T2D gene association P values
| Gene | Description | T2D medication class | MAGENTA T2D gene association P value | Best local SNP rs number | Best local SNP DIAGRAMv3 P value | Gene in LD to established T2D SNPs |
|---|---|---|---|---|---|---|
| IDE | Insulin-degrading enzyme | Amylin mimetics | 1.55 × 10−15 | rs7911264 | 4.50 × 10−13 | + |
| PPARG | Peroxisome proliferator–activated receptor γ | TZDs | 6.32 × 10−9 | rs11709077 | 1.12 × 10−9 | + |
| KCNJ11* | Potassium inwardly rectifying channel, subfamily J, member 11 | Sulfonylureas, meglitinides | 6.88 × 10−4 | rs5215 | 4.36 × 10−6 | + |
| ABCC8* | ATP-binding cassette, subfamily C (CFTR/MRP), member 8 | Sulfonylureas | 1.22 × 10−3 | rs5215 | 4.36 × 10−6 | + |
| GIP | Gastric inhibitory polypeptide | DPP inhibitors | 7.62 × 10−3 | rs3809770 | 1.03 × 10−4 | − |
| ACSL1 | Acyl-CoA synthetase long-chain family member 1 | TZDs | 1.83 × 10−2 | rs735949 | 7.76 × 10−5 | − |
| IRS2 | Insulin receptor substrate 2 | Insulin, TZDs | 1.89 × 10−2 | rs1330545 | 1.02 × 10−4 | − |
| GLP1R | Glucagon-like peptide 1 receptor | GLP-1 receptor agonists | 2.39 × 10−2 | rs1929902 | 5.35 × 10−5 | − |
| GLP2R | Glucagon-like peptide 2 receptor | DPP inhibitors | 2.87 × 10−2 | rs17743194 | 4.28 × 10−5 | − |
| NFKB1 | Nuclear factor of κ light polypeptide gene enhancer in B-cells 1 | TZDs | 3.01 × 10−2 | rs4648055 | 2.04 × 10−4 | − |
| ADIPOQ | Adiponectin, C1Q, and collagen domain containing | TZDs | 3.22 × 10−2 | rs7649121 | 2.12 × 10−4 | − |
| CXCL9† | Chemokine (C-X-C motif) ligand 9 | DPP inhibitors | 3.83 × 10−2 | rs13131187 | 3.13 × 10−4 | − |
| CXCL11† | Chemokine (C-X-C motif) ligand 11 | DPP inhibitors | 3.86 × 10−2 | rs13131187 | 3.13 × 10−4 | − |
| CXCL10† | Chemokine (C-X-C motif) ligand 10 | DPP inhibitors | 3.89 × 10−2 | rs13131187 | 3.13 × 10−4 | − |
| SLC2A2 | Solute carrier family 2 (facilitated glucose transporter), member 2 | Insulin | 5.33 × 10−2 | rs8192675 | 6.99 × 10−4 | − |
| TRIB3 | Tribbles pseudokinase 3 | Insulin | 5.85 × 10−2 | rs1555318 | 4.42 × 10−4 | − |
| GAA | Glucosidase, α; acid | α-Glucosidase inhibitors | 6.14 × 10−2 | rs2361710 | 9.28 × 10−4 | − |
| GAB1 | GRB2-associated binding protein 1 | Insulin | 7.07 × 10−2 | rs300938 | 6.97 × 10−4 | − |
| VCAM1 | Vascular cell adhesion molecule 1 | TZDs | 8.35 × 10−2 | rs1932351 | 5.58 × 10−4 | − |
| NGF | Nerve growth factor (β-polypeptide) | GLP-1 receptor agonists | 8.94 × 10−2 | rs11466094 | 5.63 × 10−4 | − |
| CPT2 | Carnitine palmitoyltransferase 2 | TZDs | 9.48 × 10−2 | rs1288351 | 7.58 × 10−4 | − |
| NFKB2 | Nuclear factor of κ light polypeptide gene enhancer in B-cells 2 (pT9/p100) | TZDs | 9.64 × 10−2 | rs7897947 | 2.44 × 10−3 | − |
| DDIT3 | DNA damage–inducible transcript 3 | GLP-1 receptor agonists | 1.13 × 10−1 | rs813516 | 3.20 × 10−3 | − |
| GRP | Gastrin-releasing peptide | DPP inhibitors | 1.16 × 10−1 | rs9951619 | 5.96 × 10−4 | − |
| SLC2A4 | Solute carrier family 2 (facilitated glucose transporter), member T | Insulin, sulfonylureas, metformin, TZDs | 1.21 × 10−1 | rs4562 | 2.23 × 10−3 | − |
| TAC1 | Tachykinin, precursor 1 | DPP inhibitors | 1.28 × 10−1 | rs17168923 | 1.72 × 10−3 | − |
| SLC22A1 | Solute carrier family 22 (organic cation transporter), member 1 | Metformin | 1.34 × 10−1 | rs8191811 | 1.15 × 10−3 | − |
| ADIPOR1 | Adiponectin receptor 1 | TZDs | 1.45 × 10−1 | rs782810 | 1.62 × 10−3 | − |
| SLC2A3 | Solute carrier family 2 (facilitated glucose transporter), member 3 | Insulin | 1.45 × 10−1 | rs3782681 | 2.13 × 10−3 | − |
| GIPR | Gastric inhibitory polypeptide receptor | DPP inhibitors | 1.68 × 10−1 | rs8108269 | 3.12 × 10−3 | + |
| AMY2A | Amylase, α2A (pancreatic) | α-Glucosidase inhibitors | 1.70 × 10−1 | rs1058607 | 6.75 × 10−3 | − |
| CPT1A | Carnitine palmitoyltransferase 1A (liver) | TZDs | 1.82 × 10−1 | rs2507833 | 2.77 × 10−3 | − |
| FOXO1 | Forkhead box O1 | Insulin | 2.02 × 10−1 | rs12874490 | 3.00 × 10−3 | − |
| HSD11B1 | Hydroxysteroid (11-β) dehydrogenase 1 | TZDs | 2.03 × 10−1 | rs3737913 | 1.78 × 10−3 | − |
| ACACA | Acetyl-CoA carboxylase α | Insulin | 2.04 × 10−1 | rs3744589 | 2.17 × 10−3 | − |
| CCL22 | Chemokine (C-C motif) ligand 22 | DPP inhibitors | 2.05 × 10−1 | rs8102 | 3.70 × 10−3 | − |
| PTGS1 | Prostaglandin-endoperoxide synthase 1 (prostaglandin G/H synthase and cyclooxygenase) | TZDs | 2.06 × 10−1 | rs12005866 | 2.54 × 10−3 | − |
| PIK3CA | Phosphatidylinositol-T,5-bisphosphate 3-kinase, catalytic subunit α | Insulin, TZDs | 2.07 × 10−1 | rs6443629 | 3.79 × 10−3 | − |
| SLC2A5 | Solute carrier family 2 (facilitated glucose/fructose transporter), member 5 | Insulin | 2.13 × 10−1 | rs4908803 | 2.44 × 10−3 | − |
| PDX1 | Pancreatic and duodenal homeobox 1 | GLP-1 receptor agonists | 2.19 × 10−1 | rs9581940 | 2.37 × 10−3 | − |
| SCARB1 | Scavenger receptor class B, member 1 | TZDs | 2.46 × 10−1 | rs1031605 | 1.95 × 10−3 | − |
| PPARD | Peroxisome proliferator–activated receptor δ | TZDs | 2.55 × 10−1 | rs10484578 | 3.55 × 10−3 | − |
| CPA1 | Carboxypeptidase A1 (pancreatic) | GLP-1 receptor agonists | 2.61 × 10−1 | rs10263705 | 4.01 × 10−3 | − |
| IAPP | Islet amyloid polypeptide | Amylin mimetics | 2.82 × 10−1 | rs11045995 | 2.65 × 10−3 | − |
The listed genes are the top-ranked antidiabetes drug targets with an adjusted T2D gene association P value (based on DIAGRAMv3) above the 75th percentile enrichment cutoff. One-half of these genes are predicted by MAGENTA to be true associations with T2D that have not yet reached genome-wide significance.
*,†These are each a cluster of two or three genes physically adjacent to each other along the chromosome that share the same most significant local T2D SNP and hence were collapsed to one effective gene in the GSEA. TNF was excluded from the GSEA because it lies in the HLA region. IRS1 did not pass the enrichment cutoff because it lies >110 kb from the T2D GWAS SNP (beyond the SNP-to-gene mapping boundaries used), and hence its gene association score was weak.
Because 6 of the 102 drug target genes lie in five validated loci associated with T2D (Table 1 and Supplementary Table 1), we asked whether the T2D enrichment signal observed in the antidiabetes drug target set was mainly due to genes in known association regions or whether it was also driven by additional new associations of modest effect sizes that have not yet reached genome-wide significance (due to insufficient GWAS sample size). To test this, we excluded all genes near established T2D SNPs from the analysis (listed in Supplementary Table 3 for T2D SNP list and research design and methods for boundary definition) and reran MAGENTA on the full list of drug target genes and on the drug-specific target subsets. The enrichment signal still remained significant, although it decreased by an order of magnitude (P = 4 × 10−4, Bonferroni-corrected cutoff P < 0.003 accounting for 17 hypotheses tested in Tables 2 and 3), as may be expected when removing a portion of the signal. These results suggest that the enrichment is due not only to genes near known associations but also to ∼14 additional new genes of modest effects (odds ratio of modest associations above enrichment cutoff are in the range [0.85−1.24], with the exception of one value of 1.73; P values of best local SNPs range between 5.4 × 10−6 to 7 × 10−3).
To test the reproducibility of the T2D enrichment signal in the antidiabetes drug target set, we tested whether the results replicated in an independent association study of T2D. For this, we used the large-scale T2D association meta-analysis of ∼24,000 T2D cases and ∼58,000 controls genotyped on Metabochip and applied a modified GSEA method that accounts for the SNP bias of this custom array (see research design and methods for details). Of note, we observed a nominal enrichment of drug target genes among the nearest gene or the genes in LD to a set of 137 established or high-confidence T2D SNPs determined based on a mixture model of the Metabochip meta-analysis z scores (see research design and methods) (P = 0.003 and 0.04, respectively) (Table 5). Some of the new gene associations that replicated were ACSL1 and NFKB1 (TZD targets) and GIPR (which encodes the receptor for the incretin hormone GIP and reached genome-wide significance in the joint analysis of DIAGRAMv3 and Metabochip meta-analyses [3]).
Finally, we asked whether GSEA of human genetic association data could help to predict unintended, secondary phenotypic effects of drug treatment by testing for enrichment of associations in a drug target gene set, with a phenotype that is not directly targeted by the specific drug. We tested the hypothesis that targets of TZDs may be enriched for genetic associations with cardiovascular risk factors, such as circulating lipids. This is based on a potential effect of rosiglitazone on increased risk of myocardial infarction (35–37) (see FDA Advisory Committee Minutes at http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM369180.pdf) and on the observation that both rosiglitazone and pioglitazone affect lipid levels (22). Thus, we tested whether TZD target genes were enriched for multiple modest associations with circulating LDL-C, HDL-C, or triglyceride levels. We applied MAGENTA to three GWAS meta-analyses of LDL-C, HDL-C, and triglyceride blood levels across ∼100,000 individuals (33) and found that the 38 TZD targets were significantly enriched for genes associated with LDL-C levels with modest effect sizes (P = 0.0007, twofold enrichment) (Table 6). Of note, this result was specific to the TZD target set (see other antidiabetes drug class target sets in Table 6). We observed nominal enrichment of triglyceride levels among the TZD targets (P = 0.06) and the full set of antidiabetes drug targets (P = 0.03). No significant enrichment was found for HDL-C associations. In addition to the known LDL-C gene locus APOC3/APOA1, this analysis proposes that 8 of the 20 top-ranked genes based on their LDL-C association scores may be novel TZD targets associated with LDL-C plasma levels (listed in Supplementary Table 4).
Table 6.
Testing for potential nonglycemic effects of TZDs or other antidiabetes drug classes on global lipid plasma levels through GSEA of genetic associations
| LDL-C GWAS meta-analysis | Triglyceride GWAS meta-analysis | HDL-C GWAS meta-analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Antidiabetes drug target gene set | Number of genes analyzed* | Nominal MAGENTA enrichment P value | Excess number of genes above enrichment cutoff | Nominal MAGENTA enrichment P value | Excess number of genes above enrichment cutoff | Nominal MAGENTA enrichmentP value | Excess number of genes above enrichment cutoff | Antidiabetes drug target genes in LD to validated lipid SNPs‡ |
| TZDs | 38 | 0.0007† | 9 | 0.06 | 4 | 0.35 | 1 | APOA1 (TG, HDL-C, LDL-C), IRS1 (HDL-C, TG), SCARB1 (HDL-C) |
| All nine classes of drugs | 94–95 | 0.01 | 10 | 0.03 | 8 | 0.22 | 3 | APOA1 (TG, HDL-C, LDL-C), IRS1 (HDL-C, TG), SCARB1 (HDL-C) |
| Insulin | 16 | 0.19 | 2 | 0.35 | 1 | 0.35 | 1 | IRS1 (HDL-C, TG) |
| GLP-1 receptor agonists | 17 | 0.65 | 0 | 0.10 | 3 | 0.24 | 0 | — |
| DPP4 inhibitors | 18–20 | 0.91 | 0 | 0.67 | 0 | 0.88 | 0 | — |
TG, triglyceride.
*Number of genes analyzed after excluding genes in HLA region and genes on sex chromosomes and correcting for physical clustering along the genome of genes within a given gene set. The number varies a bit between GWAS meta-analyses due to slight differences in SNP coverage between traits. Because of the physical proximity of APOC3 and APOA1, these genes were collapsed into one effective gene in the enrichment analysis (choosing the more significant gene P value) to prevent inflation of the gene set enrichment P value.
†Passes Bonferroni correction, P < 0.003.
‡Based on 95 loci associated with global lipid traits in (33).
Discussion
Meta-analyses of GWAS have yielded dozens of genetic variants that are overrepresented in cases of T2D compared with nondiabetic controls (3,6,38–44). The robustness of the evidence for their association with T2D is based on a stringent threshold for genome-wide significance that accounts for the number of independent tests that are possible among the ∼106 common variants in the human genome (45). Because most common variants have modest effect sizes on common disease (e.g., disease risk odds ratio 1.05–1.10), although the adoption of this strict standard minimizes type I error, it leads to a high number of false-negative associations that remain undetected. The design of custom arrays that facilitate large-scale replication genotyping in many samples (12) can rescue some of these signals through increased power, but a large fraction (numbering in the hundreds to low thousands by some estimates) are yet to be discovered (3,4,39).
There is, therefore, a need to integrate additional tools to mine GWAS data sets in a hypothesis-driven but systematic manner, which can raise the prior probability of association while maintaining quantitative statistical standards. Other domains of biology can be brought to bear on GWAS data under the reasonable assumption that not every variant in the genome carries the same low prior probability of association with a given phenotype. In addition, queries that set each gene as the functional biological unit (gene-based tests) can increase statistical and explanatory power by considering association statistics of all variants that span a given gene and by collectively analyzing sets of genes that function in common pathways. The adaptation of GSEA to GWAS data sets as embedded in MAGENTA accomplishes all these goals, accounting for differences in genetic and physical properties between genes (34).
In this study, we applied MAGENTA to gene sets constructed under the reasonable assumption that genes whose protein products are targeted by drugs used to treat T2D are likely to influence glycemia when modified by naturally occurring variation. The present analysis was predicated on the initial observation that the first two reported and confirmed genetic associations with T2D implicated genes that encode T2D drug targets (14,15). Based on the empirical evidence of these existing T2D associations, we postulated that variants in other genes that encode drug targets have a higher likelihood of association with T2D than the genomic average, if they or variants in LD to them affect function or expression of the gene. Indeed, we found significant enrichment of modest common variant associations with T2D in pathways targeted by antidiabetes medications. The results were replicated in an independent study genotyped on the Metabochip, persisted after removing validated T2D loci, and suggest that 15–20 of the top-41 modest associations with T2D prioritized by the present GSEA are worthy of further investigation (about one-half of the 41 top-ranked loci listed in Table 4), mostly driven by genes involved in insulin and TZD signaling. These results are also consistent with the nominal enrichment observed in both the DIAGRAMv3 and the Metabochip meta-analyses for T2D associations in the PPAR signaling pathway, known to be targeted by TZDs (P < 0.04) (3). These findings highlight insulin sensitivity networks as a common nidus of potential T2D associations.
In addition, some of the top-ranked T2D associations lie near genes that encode hormones and their receptors, such as adiponectin and its receptor and incretins and their receptors (e.g., GIP, GIPR). Of note, some of the antidiabetes target genes are monogenic diabetes genes, including PDX1, INSR, KCNJ11, ABCC8, and PPARG (the latter three are also associated with the common form of T2D).
Although the present GSEA helped to hone in on a shortened list of candidate T2D-associated genes, additional functional analyses and experiments will be required to decipher which 15–20 of the ∼40 top-ranked drug target genes represent true T2D associations. Possible approaches include examining their expression levels in relevant T2D tissues or elucidating the phenotypic consequences of perturbing these genes in model systems. Further genetic studies with larger sample sizes may provide additional statistical support. Genes such as ACSL1, NFKB1, and GIPR that replicated in two independent genetic studies (DIAGRAMv3 and Metabochip) are top candidates for follow-up.
Because a large fraction of drugs that enter clinical trials today fail due to toxicity (46), we examined a test case to gauge the utility of this approach for detecting secondary, undesired phenotypic effects of drugs. Specifically, we provided human genetic support for a potential causal role of LDL-C blood level alterations in the potential increased incidence of myocardial infarction in people with T2D following treatment with TZDs (35). Although LDL-C blood levels have been shown to increase in response to TZDs (22), the clinical significance of this process is not yet clear. Further investigation of the top-ranked TZD target genes that are most likely to be associated with LDL-C based on MAGENTA analysis and that drove the observed enrichment signal may help to shed light on the LDL-dependent mechanism through which TZDs may affect risk of myocardial infarction in people with T2D. In the future, unbiased mining of genetic associations with a range of common diseases and traits may help to propose putative side effects of drugs for testing during drug development.
The finding that common DNA variants in genes that encode known drug targets are enriched for T2D associations supports the reciprocal notion that existing genetic associations from GWAS could guide us to novel relevant drug target genes or pathways. Furthermore, the present work may have useful applications for future genetic, pharmacogenetic, or drug development studies: 1) We expect that ongoing deep sequencing studies and/or larger GWAS focusing on functional variation might uncover novel genetic variation in our prioritized T2D-associated loci, 2) we highlight drugs and targets worthy of dedicated pharmacogenetic studies that might help to stratify the population into likely responders and nonresponders, 3) we suggest potential alternative drug targets for established drug classes, and 4) we provide additional evidence that might help to prioritize some of these genes in future GWAS for drug response if a suggestive signal of association with T2D is detected at one of these loci. One such example is SLC22A1, which encodes a liver-specific metformin transporter, because it is nominally associated with metformin response (47) and is among our top-predicted T2D-associated drug target genes.
The approach can be refined with more granular types of drug target definitions, such as genes whose expression varies in response to drug perturbations in relevant tissues or cell types, and can be applied to any complex disease or quantitative trait with available GWAS data and knowledge of drug targets. Of note, in concordance with the present findings, a recent study found that genes associated with rheumatoid arthritis or genes that interact with the disease genes through protein-protein interactions are enriched for targets of approved drugs for rheumatoid arthritis (48). An extension of this approach to examining low frequency or rare variation in drug target genes and pathways may be instrumental for personalized treatment design.
Supplementary Material
Article Information
Funding. For this project, A.V.S. and D.A. were supported by the American Diabetes Association Mentor-Based Postdoctoral Fellowship, N.W. by the National Institute of Diabetes and Digestive and Kidney Diseases training grant T32-DK-007028, and J.C.F. by a Doris Duke Clinical Scientist Development Award, a Massachusetts General Hospital Physician-Scientist Development Award, and the Massachusetts General Hospital Research Scholars Award.
Duality of Interest. D.A. received grant support from Pfizer. J.C.F. has received consulting honoraria from PanGenX and Pfizer. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. A.V.S. contributed to the design of the study and analyses, conduct of the analyses, and writing of the manuscript. N.W. contributed to the design of the study and analyses, literature search and compilation of the list of antidiabetes drug target genes, and writing of the manuscript. The DIAGRAM Consortium contributed the association summary statistics of the T2D GWAS and Metabochip meta-analyses. The MAGIC investigators contributed the association summary statistics for seven glycemic traits. D.A. contributed to the design of the study and analyses. J.C.F. contributed to the design of the study and analyses and writing of the manuscript. J.C.F. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract and oral form at the 71st Scientific Sessions of the American Diabetes Association, San Diego, CA, 24–28 June 2011.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-0703/-/DC1.
References
- 1.McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med 2010;363:2339–2350 [DOI] [PubMed] [Google Scholar]
- 2.Billings LK, Florez JC. The genetics of type 2 diabetes: what have we learned from GWAS? Ann N Y Acad Sci 2010;1212:59–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris AP, Voight BF, Teslovich TM, et al.; Wellcome Trust Case Control Consortium; Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators; Genetic Investigation of ANthropometric Traits (GIANT) Consortium; Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium . Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stahl EA, Wegmann D, Trynka G, et al.; Diabetes Genetics Replication and Meta-analysis Consortium; Myocardial Infarction Genetics Consortium . Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet 2012;44:483–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet 2011;88:294–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prokopenko I, Langenberg C, Florez JC, et al. Variants in MTNR1B influence fasting glucose levels. Nat Genet 2009;41:77–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dupuis J, Langenberg C, Prokopenko I, et al.; DIAGRAM Consortium; GIANT Consortium; Global BPgen Consortium; Anders Hamsten on behalf of Procardis Consortium; MAGIC Investigators . New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010;42:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saxena R, Hivert MF, Langenberg C, et al.; GIANT Consortium; MAGIC Investigators . Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 2010;42:142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soranzo N, Sanna S, Wheeler E, et al. Common variants at 10 genomic loci influence hemoglobin A₁(C) levels via glycemic and nonglycemic pathways [published correction appears in Diabetes 2011;60:1050–1051]. Diabetes 2010;59:3229–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strawbridge RJ, Dupuis J, Prokopenko I, et al.; DIAGRAM Consortium; GIANT Consortium; MuTHER Consortium; CARDIoGRAM Consortium; C4D Consortium . Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011;60:2624–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manning AK, Hivert M-F, Scott RA, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 2012;44:659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voight BF, Kang HM, Ding J, et al. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet 2012;8:e1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scott RA, Lagou V, Welch RP, et al.; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium . Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 2012;44:991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 2000;26:76–80 [DOI] [PubMed] [Google Scholar]
- 15.Gloyn AL, Weedon MN, Owen KR, et al. Large-scale association studies of variants in genes encoding the pancreatic β-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003;52:568–572 [DOI] [PubMed] [Google Scholar]
- 16.Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther 2007;113:546–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buteau J. GLP-1 receptor signaling: effects on pancreatic beta-cell proliferation and survival. Diabetes Metab 2008;34(Suppl. 2):S73–S77 [DOI] [PubMed] [Google Scholar]
- 18.Neumiller JJ. Differential chemistry (structure), mechanism of action, and pharmacology of GLP-1 receptor agonists and DPP-4 inhibitors. J Am Pharm Assoc (2003) 2009;49(Suppl. 1):S16–S29 [DOI] [PubMed] [Google Scholar]
- 19.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001;414:799–806 [DOI] [PubMed] [Google Scholar]
- 20.Bailey CJ, Turner RC. Metformin. N Engl J Med 1996;334:574–579 [DOI] [PubMed] [Google Scholar]
- 21.Rosen ED, Spiegelman BM. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem 2001;276:37731–37734 [DOI] [PubMed] [Google Scholar]
- 22.Yki-Järvinen H. Thiazolidinediones. N Engl J Med 2004;351:1106–1118 [DOI] [PubMed] [Google Scholar]
- 23.Müller G. The molecular mechanism of the insulin-mimetic/sensitizing activity of the antidiabetic sulfonylurea drug Amaryl. Mol Med 2000;6:907–933 [PMC free article] [PubMed] [Google Scholar]
- 24.Renström E, Barg S, Thévenod F, Rorsman P. Sulfonylurea-mediated stimulation of insulin exocytosis via an ATP-sensitive K+ channel-independent action. Diabetes 2002;51(Suppl. 1):S33–S36 [DOI] [PubMed] [Google Scholar]
- 25.Proks P, Reimann F, Green N, Gribble F, Ashcroft F. Sulfonylurea stimulation of insulin secretion. Diabetes 2002;51(Suppl. 3):S368–S376 [DOI] [PubMed] [Google Scholar]
- 26.Drucker DJ. Dipeptidyl peptidase-4 inhibition and the treatment of type 2 diabetes: preclinical biology and mechanisms of action. Diabetes Care 2007;30:1335–1343 [DOI] [PubMed] [Google Scholar]
- 27.Ryan GJ, Jobe LJ, Martin R. Pramlintide in the treatment of type 1 and type 2 diabetes mellitus. Clin Ther 2005;27:1500–1512 [DOI] [PubMed] [Google Scholar]
- 28.Malaisse WJ. Mechanism of action of a new class of insulin secretagogues. Exp Clin Endocrinol Diabetes 1999;107(Suppl. 4):S140–S143 [DOI] [PubMed] [Google Scholar]
- 29.Martin AE, Montgomery PA. Acarbose: an alpha-glucosidase inhibitor. Am J Health Syst Pharm 1996;53:2277–2290; quiz 2336–2337 [DOI] [PubMed]
- 30.Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med 2002;137:25–33 [DOI] [PubMed] [Google Scholar]
- 31.Chang AY, Wyse BM, Gilchrist BJ. Ciglitazone, a new hypoglycemic agent. II. Effect on glucose and lipid metabolisms and insulin binding in the adipose tissue of C57BL/6J-ob/ob and − + / ? mice. Diabetes 1983;32:839–845 [DOI] [PubMed] [Google Scholar]
- 32.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 1995;270:12953–12956 [DOI] [PubMed] [Google Scholar]
- 33.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Segrè AV, Groop L, Mootha VK, Daly MJ, Altshuler D; DIAGRAM Consortium; MAGIC Investigators . Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 2010;6:e1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007;356:2457–2471 [DOI] [PubMed] [Google Scholar]
- 36.Mahaffey KW, Hafley G, Dickerson S, et al. Results of a reevaluation of cardiovascular outcomes in the RECORD trial. Am Heart J 2013;166:240–249 [DOI] [PubMed]
- 37.Tannen R, Xie D, Wang X, Yu M, Weiner MG. A new “comparative effectiveness” assessment strategy using the THIN database: comparison of the cardiac complications of pioglitazone and rosiglitazone. Pharmacoepidemiol Drug Saf 2013;22:86–97 [DOI] [PubMed] [Google Scholar]
- 38.Zeggini E, Scott LJ, Saxena R, et al.; Wellcome Trust Case Control Consortium . Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 2008;40:638–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voight BF, Scott LJ, Steinthorsdottir V, et al.; MAGIC Investigators; GIANT Consortium . Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 2010;42:579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yasuda K, Miyake K, Horikawa Y, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 2008;40:1092–1097 [DOI] [PubMed] [Google Scholar]
- 41.Unoki H, Takahashi A, Kawaguchi T, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 2008;40:1098–1102 [DOI] [PubMed] [Google Scholar]
- 42.Rung J, Cauchi S, Albrechtsen A, et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet 2009;41:1110–1115 [DOI] [PubMed] [Google Scholar]
- 43.Kooner JS, Saleheen D, Sim X, et al.; DIAGRAM; MuTHER . Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet 2011;43:984–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saxena R, Elbers CC, Guo Y, et al.; Look AHEAD Research Group; DIAGRAM Consortium . Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci [published correction appears in Am J Hum Genet 2012;90:753]. Am J Hum Genet 2012;90:410–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol 2008;32:381–385 [DOI] [PubMed] [Google Scholar]
- 46.Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov 2013;12:581–594 [DOI] [PubMed] [Google Scholar]
- 47.Jablonski KA, McAteer JB, de Bakker PI, et al.; Diabetes Prevention Program Research Group. Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle interventions in the Diabetes Prevention Program. Diabetes 2010;59:2672–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okada Y, Wu D, Trynka G, et al.; RACI Consortium; GARNET Consortium . Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.