

Abstract
Stimulation of endogenous β-cell expansion could facilitate regeneration in patients with diabetes. In mice, connective tissue growth factor (CTGF) is expressed in embryonic β-cells and in adult β-cells during periods of expansion. We discovered that in embryos CTGF is necessary for β-cell proliferation, and increased CTGF in β-cells promotes proliferation of immature (MafA−) insulin-positive cells. CTGF overexpression, under nonstimulatory conditions, does not increase adult β-cell proliferation. In this study, we tested the ability of CTGF to promote β-cell proliferation and regeneration after partial β-cell destruction. β-Cell mass reaches 50% recovery after 4 weeks of CTGF treatment, primarily via increased β-cell proliferation, which is enhanced as early as 2 days of treatment. CTGF treatment increases the number of immature β-cells but promotes proliferation of both mature and immature β-cells. A shortened β-cell replication refractory period is also observed. CTGF treatment upregulates positive cell-cycle regulators and factors involved in β-cell proliferation, including hepatocyte growth factor, serotonin synthesis, and integrin β1. Ex vivo treatment of whole islets with recombinant human CTGF induces β-cell replication and gene expression changes consistent with those observed in vivo, demonstrating that CTGF acts directly on islets to promote β-cell replication. Thus, CTGF can induce replication of adult mouse β-cells given a permissive microenvironment.
Introduction
Identification of novel factors that enhance β-cell proliferation and mass regeneration in vivo while retaining optimal function would serve as an ideal strategy for remediation of all forms of diabetes. Adult β-cell mass adapts to changing physiological demands, such as pregnancy and obesity (1). β-Cell mass expansion and regeneration occur primarily by replication of existing β-cells (2–4). The proportion of replicative β-cells declines dramatically with age (1). This age-dependent decline in basal proliferation and reduced ability of β-cells to re-enter the cell cycle limits the regenerative potential of adult β-cells (2). Processes that mediate the age-dependent decrease in proliferative and regenerative capacity remain poorly understood (3–5). Factors involved in β-cell replication in response to stimuli such as pregnancy, high-fat diet (HFD) feeding, and β-cell destruction have been identified (6). Understanding the underlying mechanisms or signaling pathways would move us closer to in vivo β-cell mass regeneration as a therapy.
The β-cell proliferative factor connective tissue growth factor (CTGF/CCN2) is a member of the CCN family of secreted extracellular matrix–associated proteins (7). TGF-β and integrin signaling are enhanced by CTGF; CTGF antagonizes BMP and Wnt (8–11). Depending on the growth factor milieu in the microenvironment, CTGF can regulate several cellular processes including proliferation, adhesion, extracellular matrix remodeling, and angiogenesis (12). In the pancreas, CTGF is expressed in ductal epithelium, vascular endothelium, and embryonic insulin-producing cells; expression in β-cells is silenced soon after birth (13). Our laboratory showed that CTGF is required for β-cell proliferation during embryogenesis and that transgenic overexpression of CTGF in embryonic insulin-producing cells increases β-cell proliferation and mass (14). In contrast, induction of CTGF in adult β-cells, under normal conditions, does not increase β-cell proliferation or mass (15). However, CTGF is re-expressed in adult β-cells during pregnancy and in response to HFD feeding (13) (R.E. Mosser and M. Gannon, unpublished observations), suggesting that it plays a role in β-cell compensation during known periods of β-cell mass expansion.
In this study, we examined the potential of CTGF to promote adult β-cell mass proliferation in vivo after partial β-cell destruction and ex vivo. We show that CTGF induction after 50% β-cell destruction increases β-cell proliferation, resulting in 50% β-cell mass recovery. CTGF increases the number of immature β-cells, promoting proliferation of both mature and immature β-cells. In conjunction, CTGF shortens the β-cell replicative refractory period, allowing single β-cells to undergo multiple rounds of cell division. Gene expression analyses revealed that CTGF elicits its effects via upregulation of cell-cycle regulators, TGF-β signaling components, and key growth factors known to enhance β-cell replication. These studies have implications on how the islet microenvironment allows for β-cell responsiveness to proproliferative factors.
Research Design and Methods
Animals
Generation of rat insulin promoter (RIP)-rtTA (16), TetO-CTGF (14), and RIP-diphtheria toxin receptor (DTR) (17) transgenic mice were described previously. Primers are available upon request. The Vanderbilt University Institutional Animal Care and Use Committee approved all mouse studies.
Intraperitoneal Glucose Tolerance Tests
Intraperitoneal glucose tolerance tests were performed as described (18).
Immunolabeling
Pancreata were dissected, fixed, and processed as in Golson et al. (19). Insulin/5-chloro-2’-deoxyuridine (CldU)/5-iodo-2’-deoxyuridine (IdU) was performed as in Teta et al. (20). See Table 1 for immunolabeling details. Imaging was with a ScanScope FL scanner (Aperio Technologies, Inc.) and quantified using Metamorph 6.1 (Molecular Devices). Unless otherwise noted, five sections >250 μm apart were selected and immunolabeled with ≥4,000 cells per animal quantified.
Table 1.
Antibody information
| Antibody | Source | Dilution/temperature/time | Antigen retrieval | Special note |
|---|---|---|---|---|
| Guinea pig anti-insulin | DakoCytomation | 1:500/4°C/ON | None | — |
| Rabbit antiglucagon | Sigma-Aldrich | 1:500/4°C/ON | None | — |
| Rabbit anti-Ki67 | Abcam | 1:500/RT/ON | 1× Na citrate (10 mmol/L; pH 6.0) | Boil 14 min on high |
| Rabbit anti-MafA | Bethyl Laboratories | 1:500/RT/ON | 1× TEG buffer (pH 9.0) | 1 min on high, 7.5 min on 10% |
| Rabbit anti-MafB | Bethyl Laboratories | 1:500/RT/ON | 1× TEG buffer (pH 9.0) | 1 min on high, 7.5 min on 10% |
| Rabbit anti-CD31 | Abcam | 1:100/4°C/ON | 1× Na citrate (10 mmol/L; pH 6.0) | Boil 14 min on high |
| Mouse anti–E-cadherin | BD Pharmingen | 1:500/RT/ON | 1× TEG buffer (pH 9.0) | 1 min on high, 7.5 min on 10% |
| Rabbit anti–p-ERK1/2 | Cell Signaling Technology | 1:100/4°C/ON | 1× Na citrate (10 mmol/L; pH 6.0) | Pressure cooker: 15 min on high, 45 min in heat |
| Rabbit anti-Tph1 | Abcam | 1:150/4°C/ON | 1× Na citrate (10 mmol/L; pH 6.0) | Pressure cooker: 15 min on high, 45 min in heat |
| Mouse anti-BrdU | BD Pharmingen | 1:100/4°C/ON | 1× TEG buffer (pH 9.0) | (20) |
| Rat anti-BrdU | ACSC | 1:250/4°C/ON | 1× TEG buffer (pH 9.0) | (20) |
Na citrate, sodium citrate; ON, overnight; RT, room temperature; TEG, Tris-EGTA.
β-Cell Mass
Ten to 12 slides per animal (1 to 2% of entire pancreas) were immunolabeled for insulin, visualized via the DAB Peroxidase Substrate Kit (Vector Laboratories), and counterstained with eosin. Quantification of β-cell mass was performed as in Golson et al. (21). Briefly, one pancreatic section per slide was scanned using a ScanScope CS slide scanner (Aperio Technologies, Inc.). Images from each experiment were processed identically with the ImageScope Software (Aperio Technologies, Inc.). β-Cell mass was measured by obtaining the ratio of insulin-positive (ins+) area to total pancreas area of all scanned sections per animal and multiplied by the pancreatic wet weight.
β-Cell Proliferation
Five slides (at least 250 µm apart) per animal were selected and immunolabeled for insulin and Ki67. A minimum of 4,000 cells were counted using Metamorph 6.1 software (Molecular Devices). The percentage of proliferating cells was determined by dividing the number of Ki67/insulin double-positive cells by the total number of ins+ cells.
β-Cell Size
Number of β-cells in each islet was quantified. Five slides at least 250 µm apart per animal were selected. Average β-cell size was determined by dividing the area of each islet by β-cell number. β-Cells from ≥125 islets per animal were assessed.
Islet Number and Size
Sections were imaged and analyzed for insulin. All ins+ clusters were counted and binned by size (<8 or ≥8 cells, ≥200 islets).
Islet Microvascular Density
Sections were immunolabeled for insulin and CD31. Platelet endothelial cell adhesion molecule (PECAM)-positive area of at least 100 islets was measured. Percentage of blood vessel area was determined by dividing total PECAM area by total ins+ area.
Proliferating β-Cell E-cadherin Expression
Sections were immunolabeled for insulin, E-cadherin, and Ki67. The number of Ki67/E-cadherin/insulin triple-positive cells and Ki67/insulin double-positive cells was quantified.
E-cadherin and p16 Expression Analysis
A total of 50 ng cDNA was prepared from islet RNA using the SuperScript III First Stand Synthesis System according to the manufacturer (Invitrogen). Real-time reactions were with iQ SYBR Green Supermix (Bio-Rad) (E-cadherin: forward, 5′-AGGCGGGAATCGTGGC-3′ and reverse, 5′-AAGGATTCCGAGGATGGCA-3′; and p16: forward, 5′-CCGTCGTACCCCGATTCAG-3′ and reverse, 5′-GCACCGTAGTTGAGCAGAAGAG-3′).
Collagen Deposition
Collagen staining and quantification was completed as described (15).
β-Cell Death
β-Cell apoptosis and necrosis was quantified via ApoAlert DNA fragmentation kits according to the manufacturer (Clontech Laboratories, Inc.). Slides were colabeled with insulin.
β-Cell Maturity
Sections were immunolabeled for insulin, MafA, MafB, and Ki67. Percentage of mature and immature β-cells was determined by dividing number of MafA/insulin double-positive cells or MafB/insulin double-positive cells by total number of ins+ cells, respectively. To assess maturity of proliferating β-cells, the number of triple-positive cells was divided by total number of MafA/insulin double-positive cells. The percentage of immature proliferating β-cells was assessed by dividing the number of Ki67+/MafA−/ins+ cells by total number of MafA−/ins+ cells.
β-Cell Refractory Period
Mice were treated with CldU for 2 days after diptheria toxin (DT), followed by a 1- or 3-week washout period, and then 5 days' IdU treatment. Doxycycline (Dox) was administered from the last DT injection until sacrifice. Sections were immunolabeled for insulin or MafA, with CldU and IdU, as in Teta et al. (20).
Ex Vivo β-Cell Proliferation Assay
Islets were isolated from 10-week-old female C57Bl/6J mice and cultured at 37°C in RPMI containing 11 mmol/L glucose and 10% horse serum. Approximately 20 islet equivalents were cultured per well in a 96-well plate overnight. Islets were treated with vehicle or recombinant human CTGF (22). β-Cell proliferation was assayed as in Mosser and Gannon (23), with a minimum of 2,500 cells quantified per animal.
Gene Expression Analysis
Islets were isolated from 10-week-old females and immediately prepared for RNA isolation by being placed in TRIzol reagent. RNA was isolated using the RNeasy Mini kits (Qiagen). A total of >250 ng cDNA was prepared from islet RNA using the SuperScript III First Stand Synthesis System (Invitrogen). Primers are available on request. TaqMan low-density arrays were conducted on a 7900HT Fast Real-Time PCR system. Data were analyzed with SDS RQ Study software (Applied Biosystems, Life Technologies).
Western Blotting
Islets were isolated from 8-week-old animals as described above. Following isolation, islets were immediately lysed in RIPA buffer, and protein content was quantified using the Bio-Rad DC protein assay (Bio-Rad). A total of 3.5 μg of protein per sample was electrophoresed on 4–12% Bis-Tris gels under denaturing conditions and blotted onto polyvinylidene difluoride membrane using the NuPAGE Western blotting system (Invitrogen). Blots were blocked and probed with the following primary antibodies diluted in 5% nonfat milk in 1x Tris-buffered saline with Tween and incubated overnight at 4°C: rabbit anti–phospho-Smad3 (1:1,000; Abcam), rabbit anti-Smad2/3 (1:500; Cell Signaling Technology), and rabbit anti–β-tubulin (1:5,000; Santa Cruz Biotechnology). Horseradish peroxidase–conjugated rabbit secondary antibody (1:5,000; Jackson ImmunoResearch Laboratories) was used for protein detection and facilitated by an ECL Prime detection system (Amersham) using Kodak X-Omat Blue film. Protein levels were quantified using ImageJ software.
Statistics
Results are expressed as mean ± SEM. Statistical significance was calculated by Student t test, one-way or two-way ANOVA, and Tukey post hoc analysis where applicable. P values ≤0.05 were considered significant.
Results
β-Cell Ablation and CTGF Induction
We hypothesized that CTGF would promote β-cell mass regeneration following β-cell loss. To assess this, a DT-mediated model of β-cell ablation was used (17). In this model, a transgene containing the RIP driving DTR expression was targeted to the hprt locus on the X chromosome. We used the less severe model of 50% β-cell ablation afforded by hemizygous RIP-DTR female mice (which express DTR in 50% of β-cells on average due to random X-inactivation). β-Cell mass was not affected in control animals (RIP-rtTA or TetO-CTGF) after DT administration, and α-cell mass was unaffected in all treatments (Supplementary Fig. 1A–C). No changes in blood glucose clearance were observed after 50% β-cell ablation, as assessed by intraperitoneal glucose tolerance test (Supplementary Fig. 1D). This was anticipated, as 50% β-cell mass is sufficient to maintain glucose tolerance under normal conditions.
To assess if CTGF promotes β-cell mass regeneration, a β-cell–specific Dox-inducible CTGF bitransgenic model was used (RIP-rtTA;TetO-CTGF). Specificity of CTGF induction has been previously confirmed (15). Homozygous RIP-DTR females were interbred with RIP-rtTA;TetO-CTGF males. Hemizygous RIP-DTR;RIP-rtTA;TetO-CTGF females and hemizygous RIP-DTR;RIP-rtTA female littermate controls were used. Since CTGF upregulation is sometimes associated with increased fibrosis (24), collagen deposition was quantified. No increase in peri-islet collagen was observed, even after 8 weeks of CTGF overexpression (Supplementary Fig. 2). This agrees with our previously published results (15) and demonstrates that islets can be exposed to continuous CTGF for several weeks with no deleterious effects on fibrosis.
CTGF Induces β-Cell Regeneration After 50% β-Cell Ablation via Increased Proliferation
DT was administered at 8 weeks of age to RIP-DTR;RIP-rtTA controls (Ablation) and RIP-DTR;RIP-rtTA;TetO-CTGF experimental animals (Ablation+CTGF) and CTGF induced for 2 days, 2 weeks, or 4 weeks after DT injection. Non-DT–injected animals included controls (Control) and those in which CTGF was induced without ablation (CTGF) (Fig. 1A). Neither 50% β-cell ablation nor CTGF induction alone had an effect on glucose homeostasis (Supplementary Fig. 3A–C). CTGF induction under normal conditions in adult islets elicited no increase in either β-cell mass expansion or β-cell proliferation (Fig. 1B and C) in agreement with our previous observations (15). Likewise, 50% β-cell ablation alone did not induce β-cell regeneration or proliferation (Fig. 1B and C). This finding was unsurprising, as these animals maintain euglycemia and thus have no physiological impetus to expand β-cell mass.
Figure 1.

CTGF promotes β-cell mass regeneration and proliferation. (A) Experimental outline. Mice were administered 2 mg/mL of Dox in 2% Splenda in drinking water. DT (126 ng; Sigma-Aldrich) was given intraperitoneally three times at 8 weeks of age. β-Cell mass (B–B″) and proliferation (C–C″). (D) Representative images of β-cell mass at 4 weeks. (E) Representative images of β-cell proliferation 2 days after ablation with or without CTGF. Nuclei were visualized with DAPI (Molecular Probes). Primary antibodies were detected by species-specific donkey secondary antibodies conjugated to either Alexa 488 or Cy3 fluorophores (1:400; Jackson ImmunoResearch Laboratories). Pink arrowheads: proliferating β-cells. (F) β-Cell proliferation precedes β-cell mass recovery (group 4 data). Two-day time point, n = 6; 2- and 4-week time points, n = 8. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. N.S., not significant.
CTGF overexpression after ablation resulted in partial restoration of β-cell mass at 2 weeks and reached 50% mass recovery at 4 weeks (Fig. 1B and D). The α-cell/β-cell ratio of the Ablation+CTGF cohort improved after both 2 and 4 weeks of CTGF induction, indicative of an increase in β-cell number (Fig. 2). α-Cell mass was unaltered with any treatment, and 50% β-cell ablation did not stimulate α-cell transdifferentiation to a β-cell fate (F. Thorel and P.L. Hererra, unpublished observations). β-Cell proliferation was enhanced after only 2 days of CTGF treatment and maintained at all time points, although the degree of enhanced β-cell proliferation declines at 4 weeks of CTGF overexpression (Fig. 1C and E). Thus, the increase in β-cell proliferation precedes β-cell mass recovery (Fig. 1F). Eight weeks of continuous CTGF treatment after 50% β-cell ablation elicited no further improvement in β-cell mass recovery, suggesting a limit of restoration had been reached (data not shown). Thus, although adult β-cells are unresponsive to CTGF under nonstimulatory conditions, CTGF can induce replication and β-cell mass expansion in the unique microenvironment of β-cell destruction.
Figure 2.
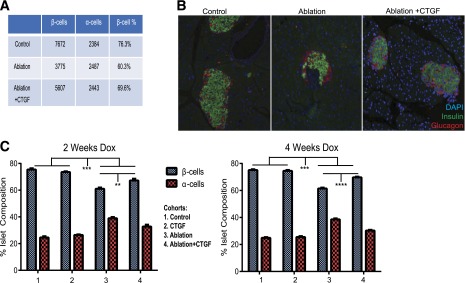
CTGF treatment after 50% β-cell destruction elicits improved α-/β-cell ratios. A and B: Example of actual raw data obtained: the numbers of ins+ (green) and glucagon+ (red) cells were quantified. Under normal conditions, β-cells constitute ∼75% of the total number of counted cells (Control). After DT injection, 50% of β-cells are ablated, while α-cells remained unchanged (Ablation). This results in β-cells accounting for ∼60% of counted cells. In Ablation+CTGF animals, β-cells account for ∼70% of counted cells. C: Quantification of α-/β-cell ratios after 2 (left) and 4 (right) weeks of Dox administration. n = 8. **P < 0.01, ***P < 0.001, ****P < 0.0001.
No Evidence for Other Modes of β-Cell Mass Recovery in Response to CTGF
We examined whether other compensatory mechanisms also contributed to regeneration. Individual β-cell size remained constant regardless of treatment (Fig. 3A), indicating that β-cell mass expansion was not due to β-cell hypertrophy. As surrogates for islet neogenesis, we evaluated total islet number and percentage of small ins+ clusters. No change in either parameter was observed (Fig. 3B and C). Overall, CTGF does not appear to promote β-cell neogenesis in this model of β-cell regeneration, although in the absence of a specific marker for neogenesis, we cannot conclusively rule this out. This was not surprising as neogenesis has been primarily described in more severe models of massive combined acinar and islet cell ablation (25).
Figure 3.

CTGF does not mediate β-cell regeneration via hypertrophy, neogenesis, increased vascularization, or E-cadherin. 1. Control; 2. CTGF; 3. Ablation; and 4. Ablation+CTGF. β-Cell size after either 2 (A) or 4 (A′) weeks of CTGF treatment. Average number of islets per animal after 2 (B) or 4 (B′) weeks of CTGF induction. Number of small ins+ clusters after CTGF induction for 2 (C) or 4 (C′) weeks. (D and E) Islet vascularization quantification, as assessed by immunolabeling for blood vessels (PECAM; red) within islets (insulin; green). n = 8. (F) No change in E-cadherin mRNA expression as assessed by qRT-PCR. Real-time reactions were carried out in technical duplicates on a CFX Real-Time PCR Detection system (Bio-Rad). (G) No alteration in the percentage of proliferating β-cells either completely (gray bars) or incompletely surrounded (white bars) by E-cadherin. (H) Representative images of Ablation+CTGF islets at 2 days Dox. Immunolabeling for insulin (green), E-cadherin (red), and Ki67 (yellow). Yellow arrowheads indicate a proliferating β-cell surrounded by E-cadherin localized to the membrane. White arrowheads indicate a proliferating β-cell with incomplete E-cadherin membrane localization. A cell was considered E-cadherin positive if >75% of the cell membrane displayed E-cadherin immunolabeling. For qRT-PCR, n = 3 for CTGF and n = 4 for Control, Ablation, and Ablation+CTGF. For proliferation analysis, n = 4. *P < 0.05, **P < 0.01, ***P < 0.001.
Pancreatic islets are highly vascularized, and their endothelium produces factors such as hepatocyte growth factor (HGF), which induce β-cell mass expansion (26,27). As CTGF is an angiogenic factor (28), it could promote β-cell regeneration indirectly via enhanced vascularization. CTGF did not elicit an increase in islet vascular density, as assessed by PECAM+ area (Fig. 3D and E), in agreement with our previously published results (15), although it remains possible that CTGF induces production of endothelial-derived β-cell growth factors.
Based on a recent study showing that E-cadherin knockdown enhances β-cell proliferation (29), we hypothesized that 50% β-cell ablation might result in decreased cell–cell contacts and decreased E-cadherin expression, contributing to increased β-cell proliferation. We assessed E-cadherin expression via quantitative RT-PCR (qRT-PCR) after 2 days of CTGF induction (the peak of β-cell proliferation). E-cadherin levels (Fig. 3F) and the percentage of proliferating β-cells that were E-cadherin+ or E-cadherin− was the same across all cohorts (Fig. 3G). β-Cell replication did not correlate with decreased E-cadherin protein membrane localization (Fig. 3H).
CTGF Does Not Enhance β-Cell Survival in the Setting of DT
We next tested whether CTGF improves β-cell mass regeneration by enhancing cell survival. CTGF expression was induced for 1 week prior to, during, and for 2 days following β-cell destruction (Fig. 4A). No difference in β-cell death (TUNEL) was observed between Ablation and Ablation+CTGF, indicating that CTGF cannot protect against β-cell death in this particular model (Fig. 4B and C), although this does not exclude the possibility that CTGF may have prophylactic affects in other models of β-cell death. Additionally, no impairment to blood glucose homeostasis was observed (Supplementary Fig. 3D). “Primed” animals displayed no improvement in either β-cell mass (Fig. 4D) or proliferation (Fig. 4E) compared with 2 days' CTGF treatment.
Figure 4.
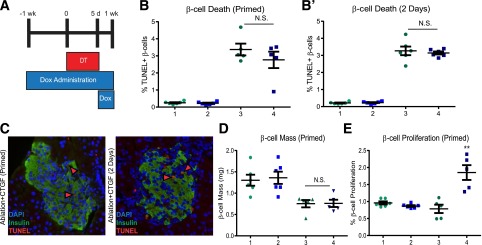
Priming islets with CTGF does not improve β-cell survival or enhance β-cell proliferation and mass. A: Experimental outline. Mice were administered 2 mg/mL of Dox in 2% Splenda in drinking water. DT (126 ng) was given intraperitoneally three times at 8 weeks of age. Cohorts are as follows: 1. Control; 2. CTGF; 3. Ablation; and 4. Ablation+CTGF. B and B′: β-Cell survival as assessed by quantifying the percentage of TUNEL+ β-cells. The percentage of apoptotic or necrotic β-cells was determined by dividing the number of TUNEL/insulin double-positive cells by the total number of insulin cells. A minimum of 4,000 cells were counted. C: Representative images of β-cell death in prophylactic- (left) and therapeutic-treated (right) islets (green, insulin; red, TUNEL; blue, DAPI; red arrowheads, proliferating ins+ cells). D: β-Cell mass. E: β-Cell proliferation. For priming time point, n = 5. For 2-day time point, n = 6. **P = 0.0027. N.S., not significant.
Taken together, our findings suggest that adult β-cell ablation results in an islet microenvironment that facilitates CTGF-induced β-cell regeneration solely through increased replication of existing β-cells. Thus, following ablation, changes intrinsic to β-cells themselves or in the islet microenvironment allow for responsiveness to CTGF. β-Cell intrinsic changes that we considered in this study include alterations in the maturation state, replication refractory period, and expression of genes involved in key signaling pathways.
CTGF Promotes Proliferation in Mature and Immature β-Cells After β-Cell Destruction
There is controversy about whether β-Cell maturity is maintained during replication (30–32). The MafA transcription factor is associated with β-cell maturity and optimal function (30,33,34). Previous studies from our laboratory demonstrated that CTGF overexpression in embryonic β-cells resulted in increased proliferation of MafA−/ins+ cells; MafA+ cells did proliferate, but their proliferation was unaltered with CTGF (14). CTGF may specifically enhance β-cell proliferation in immature (MafA−) β-cells following β-cell ablation. Approximately 15–20% of adult β-cells are normally MafA− (33) (Fig. 5A); this population of cells may be the most responsive to CTGF treatment.
Figure 5.

β-Cell proliferation characteristics in response to ablation and CTGF. Cohorts: 1. Control; 2. CTGF; 3. Ablation; and 4. Ablation+CTGF. β-Cell maturation (A) and proliferative state (B). Mature β-cells (red bars) and immature β-cells (yellow bars). Detection of Ki67 required direct conjugation to a Cy5 fluorophore using the Zenon conjugation kit according to the manufacturer’s instructions (Life Technologies). A minimum of 4,000 cells were counted. (C–C″) Representative images of Ablation+CTGF islets at 2 days' CTGF. Insulin (green), Ki67 (yellow), and MafA (red). (C) Yellow arrowheads, proliferating β-cells. (C′) Red arrowheads, mature β-cell; white arrowhead, immature β-cell. (C″) Orange arrowhead, mature proliferating β-cell; yellow arrowhead, immature proliferating β-cell. (D) Experimental outline for double uridine analog labeling. Mice were administered 2 mg/mL of Dox in 2% Splenda in drinking water. DT (126 ng) was given intraperitoneally three times at 8 weeks of age. Uridine analogs (CldU or IdU; both from Sigma-Aldrich) were administered at 1 mg/mL in Dox-treated drinking water. β-Cell replication during the first 2 days (red bars), last 5 days (green bars), and both labeling periods (yellow bars) at 2 (E) and 4 (F) weeks was determined. A minimum of 4,000 cells were counted. The percentage of β-cells undergoing replication during both labeling periods was obtained by dividing the number of CldU/IdU/Insulin triple-positive cells by the total number of ins+ cells. The percentage of dual-labeled β-cells at 4 weeks was significantly higher than at 2 weeks (demarked by #). Ratios between subgroups (i.e., CldU+:IdU+, CldU:CldU+;IdU+, and IdU:CldU+; IdU+) were determined. (G–G″) Representative images at 4 weeks. Replicating β-cells in the first 2 days incorporated CldU (red arrowheads) and in the last 5 days incorporated IdU (green arrowheads). Replicating cells in both periods incorporated CldU and IdU (yellow arrowheads). n = 6. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, #P = 0.0414. N.S., not significant.
To assess whether β-cell immaturity or partial dedifferentiation is associated with enhanced permissiveness to CTGF, we examined MafA expression in ins+ cells at 2 days (Fig. 5A). β-Cell ablation alone resulted in a significantly higher percentage (∼28%) of immature (MafA−) β-cells compared with controls (∼20%). CTGF treatment enhanced the increase in MafA− β-cells after ablation (∼35%), but had no effect on the number of MafA−/ins+ cells under normal conditions. Additionally, MafB expression in ins+ cells was assayed at 2 days. In mice, the MafB transcription factor is normally expressed in embryonic and early neonatal β-cells and is indicative of an immature β-cell (33). Correlating with the decrease in MafA+ cells (Fig. 5A), a significant increase (∼4%) in immature (MafB+) β-cells was detected in Ablation alone or Ablation+CTGF cohorts as compared with controls (∼1%) (Supplementary Fig. 4A).
To determine whether CTGF specifically promotes replication of immature β-cells in the setting of regeneration, mature (MafA+;Ki67+/MafA+;ins+) and immature (MafA−;Ki67+/MafA−;ins+) proliferating β-cells were quantified (Fig. 5B and C). The 50% β-cell destruction alone did not provoke an increase in proliferation in immature β-cells. However, CTGF expression after ablation yielded an increase in proliferation of both mature and immature β-cells. While the increase in mature β-cell proliferation was significant, the percentage of immature β-cells proliferating was strikingly increased threefold as compared with all other cohorts. Overall, these data indicate that in the setting of β-cell destruction, CTGF stimulates proliferation in both mature and immature β-cells. The increase in immature β-cells after β-cell destruction is particularly exploited by CTGF as a mechanism for β-cell regeneration.
CTGF Induction Shortens the Replicative Refractory Period
Between self-renewal cycles, β-cells undergo an extensive refractory period and are unable to re-enter the cell cycle (4,20). This refractory period is labile, shortening in periods of increased demand, such as pregnancy and 50% partial pancreatectomy (20). To determine if 50% β-cell ablation and/or CTGF treatment induces changes in the replicative refractory period, we used a dual labeling system involving two distinct uridine analogs (Fig. 5D and G) (20). This process allows for labeling of newly synthesized DNA, marking actively proliferating β-cells, at two distinct time points. β-Cell nuclei incorporating both labels are indicative of two rounds of replication during the labeling period.
We quantified the number of single-positive (CldU+ or IdU+) and dual-positive (CldU+;IdU+) β-cells after 2 or 4 weeks of CTGF treatment (Fig. 5E and F). At both time points, islets from Ablation+CTGF animals displayed an increase in the percentage of single-labeled β-cells. Additionally, in this cohort a greater number of β-cells underwent replication in the first 2 days (CldU+) compared with the last 5 days of labeling (IdU+) (red vs. green bars). A prominent increase in the number of dual-labeled β-cells was observed in the Ablation+CTGF cohort, indicative of a shortened β-cell replicative refractory period. An increase in dual-labeled β-cells was observed with 4 weeks of CTGF induction after β-cell ablation as compared with the 2-week time point (Fig. 5E and F; P = 0.0414). This increase is not surprising given the longer washout period. We calculated the ratio of single-labeled cells versus dual-labeled cells to ensure the increase in dual-labeled cells was not simply proportional to the overall increase in proliferation. In the Ablation+CTGF cohort, there was a statistically significant decrease in the average ratio of single-labeled β-cells to dual-labeled β-cells (4.2:1 for CldU and 2.9:1 for IdU) as compared with control animals (8.2:1 for CldU and 8.4:1 for IdU).
As our previous studies indicated that immature β-cells proliferate at higher percentages than mature β-cells in our Ablation+CTGF cohort (Fig. 5B), we assessed whether immature β-cells were more likely to be dual-labeled (and thus have undergone two cycles of replication). We quantified the number of mature (MafA+;CldU+;IdU+) and immature (MafA−;CldU+;IdU+) β-cells at the 2-week time point (Supplementary Fig. 4B). In all cohorts, dual-labeled β-cells were not specific to the immature (MafA−) phenotype, as both mature and immature β-cells underwent multiple rounds of replication in the 2-week time course. Most (∼60%) of the dual-labeled β-cells in the Control, CTGF-treated, and Ablation cohorts were mature. However, in congruence with our findings in Fig. 5B, the Ablation+CTGF cohort had an increase in the number of dual-labeled immature β-cells (∼60%) with a shortened refractory period. Therefore, in the setting of β-cell destruction, CTGF elicits β-cell mass regeneration both by increasing the number of proliferating β-cells and by reducing the period of time before these cells can re-enter the cell cycle.
CTGF Protein Induces Adult β-Cell Proliferation Ex Vivo
To determine whether CTGF can induce adult β-cell proliferation directly, in the absence of other pancreatic cell types or systemic factors, we treated wild-type islets ex vivo for 4 days with different concentrations of recombinant human CTGF (rhCTGF). An increase in β-cell proliferation was observed with increasing doses of rhCTGF (Fig. 6A–C), with an approximate twofold increase in proliferation observed at 250 ng/mL rhCTGF compared with vehicle control. These results indicate that the mitogenic effects of CTGF on adult β-cells are not unique to our ablation model.
Figure 6.

CTGF induces β-cell proliferation ex vivo and expression of genes involved in key signaling pathways and cell-cycle regulators. Dispersed islets labeled for insulin and Ki67, with representative images of the vehicle-treated (0) (A) and 250 ng/mL rhCTGF-treated (B) islets. Pink arrowheads: proliferating β-cells. (C) Quantification of β-cell proliferation (dual insulin/Ki67-positive cells) for various concentrations of rhCTGF. (D–F) Gene expression analysis on whole islets using TaqMan Universal PCR Master Mix (with UNG; Applied Biosystems). Islets isolated from animals with/without β-cell ablation ± CTGF treatment for 2 days. (E′ and F′) Islets from wild-type animals treated ex vivo with rhCTGF for 4 days. Islet cell markers (D), cell-cycle regulators (E and E′), and signaling pathways and growth factors (F and F′). All samples were run in duplicate. n = 4 for D–F and n = 3 for E′ and F′. *Compared with Control, #compared with CTGF, ^compared with ablation. *,^,#P < 0.5; **,##P < 0.01; ***,###P < 0.001; ****,^^^^P < 0.0001.
CTGF Upregulates Cell-Cycle Regulators, Proproliferative Growth Factors, and TGF-β Signaling
To gain insight into signaling pathways altered by CTGF with or without β-cell ablation, gene expression analysis was conducted on islets isolated from animals following 2 days of CTGF induction in vivo. We specifically probed changes in genes associated with β-cell functional maturity, proliferation, and growth factor signaling pathways (Fig. 6D–F). Gene expression alterations specific to CTGF treatment, β-cell ablation, and CTGF treatment after β-cell ablation were determined and compared with changes in islets treated ex vivo with rhCTGF for 4 days.
There were several changes in key cell-cycle regulators in response to ablation and/or CTGF treatment (Fig. 6E and E′). CTGF induction under normal conditions elicited an increase in cyclinD3 expression, while ablation alone induced an increase in cyclinB1, cyclinD1, cyclinD2, and Ki67. β-Cell ablation also upregulated Foxm1, a key transcriptional regulator of β-cell proliferation (35–38). These results are intriguing as no increase in β-cell proliferation is detected in the Ablation cohort (Fig. 1C). CyclinD3, Ki67, and PCNA mRNAs were strikingly upregulated in the Ablation+CTGF cohort, in agreement with the highly significant increase in β-cell proliferation. Upregulation of the cell-cycle inhibitor Cdkn1a (p21) was also observed only in the Ablation+CTGF cohort, suggesting the increased β-cell proliferation triggers an inhibitory feedback loop. However, other cell-cycle inhibitors, Cdkn1b (p27) and Cdkn2a (p16), displayed no alteration in gene transcription (Fig. 6E, data not shown). Wild-type islets treated with rhCTGF ex vivo showed significant upregulation of cyclinD1 and cdk2 and a trend toward an increase in Cdkn1a (p21) and Foxm1.
With regard to growth factor signaling pathways, CTGF treatment after 50% ablation resulted in an increase in TGF-β (TGF-β2, TGF-β3, and TGF-βR2), BMP (BMP2, BMP4, and BMP7), and Wnt-associated (β-catenin and Lrp5) genes (Fig. 6F and F′). CTGF promotes, and is promoted by, TGF-β signaling in other tissues, yet no significant changes in phospho-SMAD3 protein levels were detected in any experimental cohort (Supplementary Fig. 5) (8,39). However, in other tissues, CTGF expression is promoted by SMAD-independent TGF-β signaling via extracellular signal–related kinase (ERK) (40–42). Thus, in this model of β-cell regeneration, CTGF may be promoted by TGF-β signaling in an ERK-dependent manner. Additionally, BMP is a known inhibitor of CTGF (43), indicating CTGF expression regulatory networks are activated in response to CTGF upregulation. An increase in expression of tryptophan hydroxylase (Tph1), the serotonin (5-HT) synthesizing enzyme, was observed specifically in islets of the Ablation+CTGF cohort as compared with all other cohorts (Fig. 7A–D). Several additional genes were upregulated only in the presence of both 50% β-cell ablation and CTGF treatment, including HGF, its receptor c-Met, and integrins α5 (Itgα5) and β1 (Itgβ1). Immunohistochemistry revealed an increase in phosphorylated ERK1/2 (p-ERK1/2), a known downstream mediator of activated integrin β1 (44,45), in islets of the Ablation+CTGF cohort as compared with all other cohorts (Fig. 7E–H). In addition, this strengthens the hypothesis that CTGF may be activating TGF-β signaling in an ERK-dependent fashion. As 5-HT, integrin β1, and HGF each promote β-cell proliferation (27,45), these findings indicate that CTGF-mediated β-cell regeneration involves recruitment of other proproliferative factors. Ex vivo incubation with rhCTGF upregulated endogenous CTGF expression and other genes involved in TGF-β signaling; Itgα5 and Mmp2 showed a trend in increased expression. No change in BMP-associated gene expression was observed in wild-type mouse islets treated with rhCTGF ex vivo (data not shown).
Figure 7.

Alterations in Tph1 expression and ERK1/2 signaling in response to CTGF and/or ablation. A–H: Representative images of islets at 2 days of CTGF.A and E: Control. B and F: CTGF. C and G: Ablation. D and H: Ablation+CTGF. Tph1: Primary antibodies were visualized via a DAB Peroxidase Substrate Kit (Vector Laboratories) and counterstained with hematoxylin. p-ERK1/2: Primary antibodies for p-ERK1/2 and insulin were visualized via a DAB Peroxidase Substrate Kit (Vector Laboratories) and an alkaline phosphatase Vector Blue Substrate Kit (Vector Laboratories), respectively. Brown and black arrowheads demark β-cells or other islet cells with activated ERK1/2 signaling, respectively. White arrowheads demark endothelial cells with activated ERK1/2 signaling. n = 4 for Tph1 and n = 3 for p-ERK1/2.
Gene expression analysis revealed no significant alteration to several key β-cell functional identity genes (Fig. 6D). This was unsurprising as all cohorts remain euglycemic at all time points and suggests that the surviving 50% of β-cells in the ablation cohorts upregulate expression of these genes to wild-type levels. Intriguingly, upon 50% β-cell ablation, an increase in glucokinase, involved in both β-cell proliferation and function (46), is observed. Ngn3 expression increased with CTGF treatment, and/or ablation, supporting the concept that these treatments promote a less differentiated state. Finally, upregulation of MafB, a gene expressed in α-cells and immature β-cells, is observed only in the Ablation+CTGF cohort. This correlates with the observed increase in the percentage of MafB+/ins+ cells in the Ablation+CTGF cohort (Supplementary Fig. 4). No alteration in MafA expression was observed in any cohort. This is not unexpected as only an 8 or 15% decrease in MafA+ cells was observed in our Ablation or Ablation+CTGF cohorts, respectively (Fig. 5), and changes in MafA protein may be disconnected from changes in MafA gene expression (30). There were no significant changes in β-cell function genes in response to exogenous CTGF in culture (data not shown).
Discussion
In this study, we investigated the potential of CTGF to promote adult β-cell proliferation and mass expansion in the face of reduced functional β-cell mass. Since diabetes results from insufficient β-cell mass, identification of signaling pathways promoting adult β-cell mass expansion could translate into therapies for endogenous β-cell mass regeneration. In this study, we show that CTGF, a critical regulator of embryonic β-cell proliferation, induces adult β-cell mass proliferation and regeneration, and this is accompanied by increased expression of cell-cycle regulators, TGF-β signaling components, and other known β-cell proliferative stimuli (e.g., HGF and serotonin synthesis).
Responsiveness to CTGF in vivo seems to be restricted to periods of increased functional demand: late embryogenesis, pregnancy, HFD, and reduced β-cell mass, in which the strain on individual β-cells has increased. The effects of CTGF on β-cell mass recovery do not appear to be due to factors other than increased β-cell proliferation. Importantly, CTGF treatment of adult β-cells does not result in uncontrolled growth or hypoglycemia. Ex vivo, CTGF treatment activates genes involved in cell-cycle progression and induces adult β-cell replication in the absence of systemic factors.
During embryonic development, both MafA−/ins+ and MafA+/ins+ cells proliferate; however, CTGF enhances proliferation of only MafA− β-cells (14). The lack of responsiveness of MafA+/ins+ cells to CTGF under normal conditions might explain the inability of CTGF to induce replication in adult β-cells in vivo under conditions of normal β-cell mass. In the setting of β-cell ablation, we find that CTGF induction enhanced proliferation of both mature and immature β-cells. However, CTGF consistently elicits a greater increase in proliferation of immature β-cells. Ablation alone also significantly increased the number of MafA− β-cells, although these show no increase in proliferation. β-Cells normally have a refractory period on the order of months but this period is labile and was shortened specifically with β-cell ablation plus CTGF. Thus, CTGF appears to mediate β-cell regeneration via enhanced β-cell proliferation, particularly of immature β-cells, and by shortening the β-cell replicative refractory period. It may be that cells that are slightly less mature are more responsive to proliferative stimuli, such as CTGF. Whether these effects are unique to CTGF or are a property of other β-cell proliferative factors remains to be tested.
We have begun to dissect the pathways through which CTGF elicits β-cell proliferation and regeneration. Increased expression of TGF-β, BMP, and Wnt genes was observed only upon induction of CTGF after β-cell ablation, while rhCTGF specifically induced genes involved in TGF-β signaling. CTGF and TGF-β are in a positive-feedback loop (39). Increases in BMP and Wnt components point toward an attempt to negatively regulate the effects of CTGF induction (47). The roles these signaling pathways play in β-cell proliferation are unclear, although TGF-β signaling may be required for β-cell regeneration (48), and Wnt signaling is involved downstream of GLP-1–mediated β-cell proliferation (49).
A specific receptor for CTGF has not been identified—rather, CTGF interacts with integrins (11). Increased expression of integrins α5 and β1 was observed in Ablation+CTGF animals. It was recently shown that integrin β1 is absolutely critical for β-cell proliferation and mass expansion at all ages (45). Additionally, an increase in phosphorylated ERK1/2, a known downstream effector of integrin β1–mediated β-cell proliferation (44,45), was observed solely in islets from the Ablation+CTGF cohort. Thus, it is highly likely that CTGF promotes β-cell proliferation through integrin signaling. Additionally, CTGF induction after β-cell ablation increases expression of Hgf and the serotonin synthesizing enzyme Tph1, indicating that CTGF-mediated β-cell regeneration involves the recruitment of other proproliferative factors (27,50,51).
β-Cell ablation and CTGF induction are each necessary, but not sufficient, to induce in vivo β-cell proliferation in this model. However, rhCTGF can induce adult β-cell proliferation in islets removed from their endogenous environment. Adult β-cell cell-cycle re-entry may therefore require combinatorial stimuli to successfully initiate. All forms of diabetes are characterized by insufficient functional β-cell mass. Thus, strategies to promote the replication, and subsequent mass regeneration, of pre-existing β-cells are critical. The ability to elicit β-cell mass regeneration is a novel role for CTGF and suggests that manipulation of CTGF signaling may serve as a therapeutic for diabetes. Additionally, our studies highlight the vital role the islet microenvironment plays in β-cell responsiveness to proliferative stimuli.
Supplementary Material
Article Information
Acknowledgments. The authors thank Drs. Adolfo Garcia-Ocaña (Mt. Sinai School of Medicine) and Vincent Poitout (Montreal Diabetes Research Center) for critical reading of the manuscript. They also thank Bethany Carboneau and Chris Wilson for technical assistance, Anastasia Coldren for islet isolations, and Dr. Alvin C. Powers (all at Vanderbilt University) for reagents.
Funding. This research involved use of the Islet Procurement & Analysis Core of the Vanderbilt Diabetes Research and Training Center for all islet isolations. This core is supported by National Institutes of Health grant DK-20593, and the VANTAGE facility is supported by the Vanderbilt-Ingram Cancer Center (P30-CA-58648), the Vanderbilt Vision Research Center (P30-EY-08126), and National Institutes of Health/National Center for Research Resources (G20-RR-030956). This work was supported by Vanderbilt Molecular Endocrinology Training Program grant T32-DK-07563 (to K.G.R. and R.C.P.), the National Center for Research Resources R25-RR-024261 (to J.P.), JDRF International (1-2011-592), and Department of Veterans Affairs Merit Review Award 1BX00090-01A1 (to M.G).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. K.G.R. designed and conducted experiments and wrote the manuscript. R.C.P. designed and conducted experiments. M.F.M. and J.P. assisted with experiments. F.T. and P.L.H. provided a mouse line, contributed to experimental design, and edited the manuscript. D.R.B. provided reagents and help with experimental design. M.G. helped with experimental design and wrote the manuscript. M.G. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-1195/-/DC1.
References
- 1.Stolovich-Rain M, Hija A, Grimsby J, Glaser B, Dor Y. Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J Biol Chem 2012;287:27407–27414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes 2009;58:1365–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H, Gu X, Liu Y, et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature 2011;478:349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes 2005;54:2557–2567 [DOI] [PubMed] [Google Scholar]
- 5.Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006;443:453–457 [DOI] [PubMed] [Google Scholar]
- 6.Migliorini A, Bader E, Lickert H. Islet cell plasticity and regeneration. Mol Metab 2014;3:268–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katsube K, Sakamoto K, Tamamura Y, Yamaguchi A. Role of CCN, a vertebrate specific gene family, in development. Dev Growth Differ 2009;51:55–67 [DOI] [PubMed] [Google Scholar]
- 8.de Winter P, Leoni P, Abraham D. Connective tissue growth factor: structure-function relationships of a mosaic, multifunctional protein. Growth Factors 2008;26:80–91 [DOI] [PubMed] [Google Scholar]
- 9.Nguyen TQ, Roestenberg P, van Nieuwenhoven FA, et al. CTGF inhibits BMP-7 signaling in diabetic nephropathy. J Am Soc Nephrol 2008;19:2098–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mercurio S, Latinkic B, Itasaki N, Krumlauf R, Smith JC. Connective-tissue growth factor modulates WNT signalling and interacts with the WNT receptor complex. Development 2004;131:2137–2147 [DOI] [PubMed] [Google Scholar]
- 11.Lau LF, Lam SC. The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res 1999;248:44–57 [DOI] [PubMed] [Google Scholar]
- 12.Charrier A, Brigstock DR. Regulation of pancreatic function by connective tissue growth factor (CTGF, CCN2). Cytokine Growth Factor Rev 2013;24:59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford LA, Guney MA, Oh YA, et al. Connective tissue growth factor (CTGF) inactivation leads to defects in islet cell lineage allocation and beta-cell proliferation during embryogenesis. Mol Endocrinol 2009;23:324–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guney MA, Petersen CP, Boustani A, et al. Connective tissue growth factor acts within both endothelial cells and beta cells to promote proliferation of developing beta cells. Proc Natl Acad Sci USA 2011;108:15242–15247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gunasekaran U, Hudgens CW, Wright BT, Maulis MF, Gannon M. Differential regulation of embryonic and adult β cell replication. Cell Cycle 2012;11:2431–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milo-Landesman D, Surana M, Berkovich I, et al. Correction of hyperglycemia in diabetic mice transplanted with reversibly immortalized pancreatic beta cells controlled by the tet-on regulatory system. Cell Transplant 2001;10:645–650 [PubMed] [Google Scholar]
- 17.Thorel F, Népote V, Avril I, et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010;464:1149–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henley KD, Gooding KA, Economides AN, Gannon M. Inactivation of the dual Bmp/Wnt inhibitor Sostdc1 enhances pancreatic islet function. Am J Physiol Endocrinol Metab 2012;303:E752–E761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golson ML, Maulis MF, Dunn JC, et al. Activated FoxM1 attenuates streptozotocin-mediated β-cell death. Mol Endocrinol 2014;28:1435–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell 2007;12:817–826 [DOI] [PubMed] [Google Scholar]
- 21.Golson ML, Bush WS, Brissova M. Automated quantification of pancreatic β-cell mass. Am J Physiol Endocrinol Metab 2014;306:E1460–E1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao P, Roccisana J, Takane KK, et al. Gene transfer of constitutively active Akt markedly improves human islet transplant outcomes in diabetic severe combined immunodeficient mice. Diabetes 2005;54:1664–1675 [DOI] [PubMed] [Google Scholar]
- 23.Mosser RE, Gannon M. An assay for small scale screening of candidate β cell proliferative factors using intact islets. Biotechniques 2013;55:310–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.di Mola FF, Friess H, Martignoni ME, et al. Connective tissue growth factor is a regulator for fibrosis in human chronic pancreatitis. Ann Surg 1999;230:63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Criscimanna A, Speicher JA, Houshmand G, et al. Duct cells contribute to regeneration of endocrine and acinar cells following pancreatic damage in adult mice. Gastroenterology 2011; 141:1451–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lammert E, Gu G, McLaughlin M, et al. Role of VEGF-A in vascularization of pancreatic islets. Curr Biol 2003;13:1070–1074 [DOI] [PubMed] [Google Scholar]
- 27.Alvarez-Perez JC, Ernst S, Demirci C, et al. Hepatocyte growth factor/c-Met signaling is required for β-cell regeneration. Diabetes 2014;63:216–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 2002;5:153–165 [DOI] [PubMed] [Google Scholar]
- 29.Wakae-Takada N, Xuan S, Watanabe K, Meda P, Leibel RL. Molecular basis for the regulation of islet beta cell mass in mice: the role of E-cadherin. Diabetologia 2013;56:856–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo S, Dai C, Guo M, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest 2013;123:3305–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinberg N, Ouziel-Yahalom L, Knoller S, Efrat S, Dor Y. Lineage tracing evidence for in vitro dedifferentiation but rare proliferation of mouse pancreatic beta-cells. Diabetes 2007;56:1299–1304 [DOI] [PubMed] [Google Scholar]
- 32.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012;150:1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Artner I, Hang Y, Mazur M, et al. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes 2010;59:2530–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishimura W, Kondo T, Salameh T, et al. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Dev Biol 2006;293:526–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Ackermann AM, Gusarova GA, et al. The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol Endocrinol 2006;20:1853–1866 [DOI] [PubMed] [Google Scholar]
- 36.Ackermann AM, Gannon M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J Mol Endocrinol 2007;38:193–206 [DOI] [PubMed] [Google Scholar]
- 37.Zhang H, Zhang J, Pope CF, et al. Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes 2010;59:143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golson ML, Misfeldt AA, Kopsombut UG, Petersen CP, Gannon M. High Fat Diet Regulation of β-Cell Proliferation and β-Cell Mass. Open Endocrinol J 2010;4:66–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnott JA, Nuglozeh E, Rico MC, et al. Connective tissue growth factor (CTGF/CCN2) is a downstream mediator for TGF-beta1-induced extracellular matrix production in osteoblasts. J Cell Physiol 2007;210:843–852 [DOI] [PubMed] [Google Scholar]
- 40.Leask A, Holmes A, Black CM, Abraham DJ. Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor-beta 2 in fibroblasts. J Biol Chem 2003;278:13008–13015 [DOI] [PubMed] [Google Scholar]
- 41.Phanish MK, Wahab NA, Hendry BM, Dockrell ME. TGF-beta1-induced connective tissue growth factor (CCN2) expression in human renal proximal tubule epithelial cells requires Ras/MEK/ERK and Smad signalling. Nephron, Exp Nephrol 2005;100:e156–e165 [DOI] [PubMed] [Google Scholar]
- 42.Pannu J, Nakerakanti S, Smith E, ten Dijke P, Trojanowska M. Transforming growth factor-beta receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J Biol Chem 2007;282:10405–10413 [DOI] [PubMed] [Google Scholar]
- 43.Ren W, Sun X, Wang K, et al. BMP9 inhibits the bone metastasis of breast cancer cells by downregulating CCN2 (connective tissue growth factor, CTGF) expression. Mol Biol Rep 2014;41:1373–1383 [DOI] [PubMed] [Google Scholar]
- 44.Saleem S, Li J, Yee SP, Fellows GF, Goodyer CG, Wang R. beta1 integrin/FAK/ERK signalling pathway is essential for human fetal islet cell differentiation and survival. J Pathol 2009;219:182–192 [DOI] [PubMed] [Google Scholar]
- 45.Diaferia GR, Jimenez-Caliani AJ, Ranjitkar P, et al. β1 integrin is a crucial regulator of pancreatic β-cell expansion. Development 2013;140:3360–3372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terauchi Y, Takamoto I, Kubota N, et al. Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest 2007;117:246–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo Q, Kang Q, Si W, et al. Connective tissue growth factor (CTGF) is regulated by Wnt and bone morphogenetic proteins signaling in osteoblast differentiation of mesenchymal stem cells. J Biol Chem 2004;279:55958–55968 [DOI] [PubMed] [Google Scholar]
- 48.El-Gohary Y, Tulachan S, Wiersch J, et al. A smad signaling network regulates islet cell proliferation. Diabetes 2014;63:224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Z, Habener JF. Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. J Biol Chem 2008;283:8723–8735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim H, Toyofuku Y, Lynn FC, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 2010;16:804–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohara-Imaizumi M, Kim H, Yoshida M, et al. Serotonin regulates glucose-stimulated insulin secretion from pancreatic β cells during pregnancy. Proc Natl Acad Sci USA 2013;110:19420–19425 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.