

Abstract
Glucose-dependent insulinotropic polypeptide (GIP) and GLP-1 are incretins secreted by respective K and L enteroendocrine cells after eating and amplify glucose-stimulated insulin secretion (GSIS). This amplification has been termed the “incretin response.” To determine the role(s) of K cells for the incretin response and type 2 diabetes mellitus (T2DM), diphtheria toxin–expressing (DT) mice that specifically lack GIP-producing cells were backcrossed five to eight times onto the diabetogenic NONcNZO10/Ltj background. As in humans with T2DM, DT mice lacked an incretin response, although GLP-1 release was maintained. With high-fat (HF) feeding, DT mice remained lean but developed T2DM, whereas wild-type mice developed obesity but not diabetes. Metabolomics identified biochemicals reflecting impaired glucose handling, insulin resistance, and diabetes complications in prediabetic DT/HF mice. β-Hydroxypyruvate and benzoate levels were increased and decreased, respectively, suggesting β-hydroxypyruvate production from d-serine. In vitro, β-hydroxypyruvate altered excitatory properties of myenteric neurons and reduced islet insulin content but not GSIS. β-Hydroxypyruvate–to–d-serine ratios were lower in humans with impaired glucose tolerance compared with normal glucose tolerance and T2DM. Earlier human studies unmasked a neural relay that amplifies GIP-mediated insulin secretion in a pattern reciprocal to β-hydroxypyruvate–to–d-serine ratios in all groups. Thus, K cells may maintain long-term function of neurons and β-cells by regulating β-hydroxypyruvate levels.
Introduction
Glucose-dependent insulinotropic polypeptide (GIP) and GLP-1 are incretins produced predominantly by enteroendocrine K and L cells, respectively, located in the proximal (K cells) and distal (L cells) gut (1,2). Both are released into the circulation immediately after eating in response to nutrients in the gut lumen but not blood (1,2). Orally derived glucose elicits a greater insulin secretory response compared with isoglycemic levels of intravenous glucose. This enhancement of glucose-stimulated insulin secretion (GSIS), or “incretin response,” is thought to be mediated by incretins released from the gut acting on β-cells in the pancreas. Neurotransmitters and neuropeptides also regulate GSIS (3).
Abnormalities in the pancreatic β-cell and insulin secretion play a central role in the pathogenesis of type 2 diabetes mellitus (T2DM) (4). The incretin response, but not GIP or GLP-1 release, is blunted in humans with T2DM (5–7). Nevertheless, exogenously infused GLP-1 remains active in T2DM and forms the rationale for incretin-based pharmacotherapies for T2DM (8). Although effects of exogenous GIP on insulin secretion are reportedly blunted in T2DM (9–11), we recently showed that the magnitude of the augmentation in the insulin secretory response to exogenously infused GIP, but not glucose, is retained in humans with T2DM (12). Thus, the endogenous incretin response may not be mediated by direct action of GIP or GLP-1 on β-cells.
Xenin-25 (Xen) is a 25–amino acid neurotensin-related peptide produced by a subset of K cells (13). In animals, Xen delays gastric emptying (14), reduces food intake (15–17), and increases gut motility (18), gall bladder contractions (19), and exocrine pancreas secretion (20). We (21) and others (22–24) have shown that Xen activates neurotensin receptor-1 (NTSR1) and where known, by activating NTSR1 present on neurons (14,16,17,19,21,25). In mice, Xen, with GIP but not alone, amplifies GSIS via a cholinergic relay, possibly involving myenteric rather than parasympathetic neurons, and not by a direct effect on β-cells (26). Xen also amplifies GIP-mediated insulin secretion in humans—maximally in subjects with impaired glucose tolerance (IGT), less in those with normal glucose tolerance (NGT), but not in those with mild T2DM (12). In humans, NTSR1 is expressed on pancreatic neurons but not islet endocrine cells (27). Thus, neuronal dysfunction may contribute to the development of T2DM, and understanding how neurons and β-cells fail in T2DM is critical.
We previously ablated GIP-producing cells in C57BL/6J mice by expressing a diphtheria toxin transgene (DT) using regulatory elements from the GIP promoter (28). With high-fat (HF) feeding, DT mice (C57BL/6J) remained lean and normoglycemic. However, as in humans with T2DM, the incretin response was severely blunted. To determine if GIP-producing cells are involved in maintaining β-cell and neuron function on a diabetogenic background, DT mice were studied after backcrossing five to eight times with NONcNZO10/Ltj mice, a polygenic model of human T2DM (29). As reported here, these mice represent a novel and powerful model system for studying the development of T2DM.
Research Design and Methods
Animal Studies
Animals
The Washington University Animal Studies Committee approved the studies. Production, housing, and characterization of DT (C57BL/6J) mice were previously described (26,28). Studies were performed with male mice backcrossed with NONcNZO10/Ltj mice (29) (The Jackson Laboratory, Bar Harbor, ME) for the indicated number of times (N). Nontransgenic littermates were controls. Standard chow (PicoLab Rodent Diet 20; Ralston Purina, St. Louis, MO) provided 3.08 kcal/g and 11.9% calories from fat. HF food (TD.88137; Harlan Teklad, Madison, WI) provided 4.5 kcal/g and 42% calories from fat. Hartley guinea pigs (Charles River Laboratories, New York, NY) were euthanized upon arrival.
Metabolic Studies
Body weight and nonfasting blood glucose and hormone levels were determined between 10:00 a.m. and noon. Mice were fasted for 5 h before administration of glucose (33%) plus intralipid (10%) by intragastric gavage (9.1 μL/g). EDTA, a DPPIV inhibitor (Millipore, St. Charles, MO), aprotinin (Sigma-Aldrich, St. Louis, MO), and a protease inhibitor cocktail (Cat. #P8340, Sigma-Aldrich) were added to blood.
Nontargeted Biochemical Profiling
Plasma prepared from nonfasted animals (N8) was shipped frozen to Metabolon (Durham, NC) and extracted and analyzed as described and previously reported (30). Briefly, instrumentation combined three independent platforms (ultra-high-performance liquid chromatography/tandem mass spectrometry optimized for basic species and for acidic species and gas chromatography/mass spectrometry). Metabolites were identified by automated comparison of ion features in samples to a reference library of chemical standards entries, including retention time, molecular weight (m/z), preferred adducts, and in-source fragments as well as associated mass spectrometry spectra. Values below limits of detection were imputed with compound minimum. Statistical analysis of log-transformed data were performed using R open-source software (http://cran.r-project.org/). Two-way ANOVA of log-transformed data with posttest contrasts were performed to compare data between groups. Multiple comparisons were accounted for by estimating false discovery rates.
In Vitro Studies
Insulin content and release assays were previously described (26). Myenteric neurons (MENs) from guinea pig proximal small intestines were isolated and studied as described (21).
Morphological Studies
Tissues were processed and stained as described (28) using mouse antiglucagon (1:50; #G2654, Sigma-Aldrich) and guinea pig anti-insulin (1:100, #PU029-UP; Biogenex Laboratories, Fremont, CA) primary antibodies.
Assays
Insulin (PerkinElmer, Waltham, MA), total GIP (Millipore), intact GLP-1 (Meso Scale Discovery, Rockville, MD), glucagon (Millipore), and leptin (Millipore) were measured by ELISA or AlphaLISA (insulin only) kits. Cholesterol, triglycerides, and free fatty acids were measured using kits (Sigma-Aldrich).
Human Studies
Subjects
Protocols were approved by the Washington University Human Research Protection Office and the U.S. Food and Drug Administration (IND #103,374) and performed in the Washington University Institute of Clinical and Translational Sciences Clinical Research Unit after obtaining written informed consent. Subjects were administered a 75-g oral glucose tolerance test (OGTT) after a 12-h fast during screening for clinical trials NCT00798915 (12,27), NCT00949663 (7), and NCT01434901, as described (7,12), and assigned to groups with NGT, IGT, or T2DM based on 2-h glucose values (NGT: <140 mg/dL, IGT: ≥140 and <200 mg/dL, and T2DM: ≥200 mg/dL). Detailed inclusion/exclusion criteria were reported (7,12). To exclude subjects with advanced β-cell failure, participants had HbA1c ≤9% and had never used insulin or incretin-based medications for treatment of T2DM. Oral diabetes medications were withheld for 48 h before OGTTs. Subjects had no known history of gastroparesis or peripheral neuropathy.
d-Serine Measurement
Plasma was shipped frozen to the Coyle laboratory and analyzed via high-performance liquid chromatography (HPLC) as described (31). Briefly, amino acids were extracted using 10% trichloroacetic acid, derivatized for detection using o-phthaldialdehyde (Alfa Aesar, Ward Hill, MA) and N-tert-butyloxycarbonyl-l-cysteine (Novabiochem, Gibbstown, NJ), and resolved using a Grace Alltima C18 column (3 µm; 150 × 4.6 mm) and a binary gradient of 25 mmol/L sodium acetate (pH 6.5) and acetonitrile. The gradient progressed from 10 to 40% acetonitrile over 40 min. d-serine concentrations (nmol/mL) were calculated by comparing an internal standard, l-homocysteic acid (l-HCA), against standard samples run at the beginning of each analysis using the following formula: (peak height of l-HCA in standard sample/peak height of d-serine in standard sample) × (peak height of d-serine in plasma sample/peak height of l-HCA in plasma sample) × (amount of l-HCA in plasma sample in μmol/amount of plasma in mL).
β-Hydroxypyruvate and Benzoate Measurements
After protein precipitation, extracts were separated by column-switching HPLC on a SecurityGuard Gemini C18 (4 × 3 mm) and XBridge C18 column (3.5 μm, 50 × 3 mm). β-Hydroxypyruvate and benzoate were monitored by an Applied Biosystems Sciex 4000QTRAP tandem mass spectrometer equipped with an electrospray ion source in the negative ion mode and multiple-reaction monitoring detection with precursor → product ion pair of 103 → 59 for β-hydroxypyruvate, 121 → 77 for benzoate, 126 → 82 for d5-benzoate (internal standard for benzoate), respectively. Quality controls were prepared from pooled human plasma. The percent coefficients of variation of assays were <15%. The peak area of β-hydroxypyruvate and the peak area ratio of benzoate to its internal standard (d5-benzoate) are reported.
Assays
Human insulin and HbA1c were measured as previously described (7,12).
Results
DT Mice Remain Lean but Develop Severe Diabetes on an HF Diet
Three-week-old DT and WT littermates (N5) were weaned onto chow or HF diets. Seven weeks later, WT/HF mice weighed 38–46% more than animals in the other three groups (P < 0.0001; Fig. 1A and B). HF feeding caused a progressive and profound increase in nonfasting blood glucose only in DT mice,, reaching 708 ± 129 mg/dL at 10 weeks of age (Fig. 1C and D). Islet size and number of insulin-positive cells were similar in WT/chow and DT/chow mice, profoundly increased in WT/HF mice, and reduced in DT/HF animals (Fig. 1E and F and Fig. 2). Similar numbers of glucagon-producing cells were confined to the islet perimeter in DT/chow and WT/chow mice but were dispersed within islets from WT/HF and DT/HF animals.
Figure 1.

DT mice fed an HF diet develop severe diabetes. Male DT and WT NONcNZO10/Ltj littermates (N5) were fed chow or HF food beginning at 3 weeks of age. Group average body weights and blood glucose levels at the indicated week are shown in panels A and C. Values for individual mice at 10 weeks of age are shown in panels B and D. Values are ± SEM. *P < 0.05, **P = 0.01, ***P = 0.001, and +P = 0.0001, determined by one-way ANOVA using Bonferroni correction for multiple comparisons. At 10 weeks of age, pancreata from WT (panels E and F) and DT (panels G and H) mice fed chow (panels E and G) and the HF diet (panels F and H) were removed, fixed, embedded in paraffin, sectioned, and stained with hematoxylin and eosin. The green arrows point to islets. Photos are representative images and identically cropped and sized. Sections from at least four mice were examined for each group.
Figure 2.

HF feeding reduces β-cell mass in DT mice. Pancreas sections from 10-week-old WT and DT littermates (N5) fed the indicated diet were stained with antibodies to insulin plus glucagon. Images were photographed to show only nuclei (blue) (panels A, E, I, and M), insulin (green) (panels B, F, J, and N), and glucagon (red) (panels C, G, K, and O). Images merged in Photoshop are shown in panels D, H, L, and P. WT mice are shown in panels A–D and I–L. DT mice are shown in panels E–H and M–P. Sections from representative mice fed standard chow or HF food are shown in panels A–H and I–P, respectively. Sections were prepared from mice shown in Fig. 1, and at least four mice were examined for each group. All images were identically cropped and sized.
GIP but Not GLP-1 Levels Are Reduced in DT Mice
To define metabolic changes early in development of T2DM, nonfasted littermates (N7) were characterized 1 week after weaning onto the indicated diet. Plasma GIP levels were 86 ± 18 pg/mL in WT mice fed chow and increased to 1,038 ± 428 pg/mL with 1 week of HF feeding (Fig. 3A). Consistent with ablation of GIP-producing cells, GIP levels were undetectable (<2.7 pg/mL) in DT mice on both diets. Body weight was significantly increased only in WT/HF mice (Fig. 3B). With HF feeding, glucose (Fig. 3C) but not insulin (Fig. 3D) levels significantly increased in DT mice, whereas plasma concentrations of insulin but not glucose increased only in WT mice. Plasma intact GLP-1 (Fig. 3E) and glucagon (Fig. 3F) levels were not statistically different between groups. Importantly, GLP-1 levels were not different in WT and DT mice with HF feeding, although severe diabetes developed rapidly only in the DT animals. Leptin (Fig. 3G) increased with HF feeding in WT mice more than in DT animals. Cholesterol (0.2%) is a component of the HF food, and plasma levels increased with HF feeding in WT and DT mice (Fig. 3H). Diet and genotype did not affect triglyceride (Fig. 3I) and free fatty acid (Fig. 3J) levels.
Figure 3.
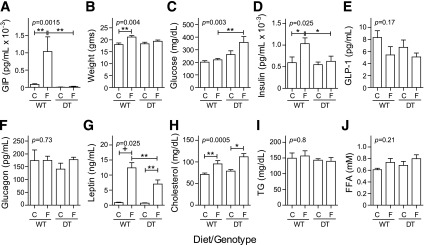
Metabolic markers in nonfasted mice 1 week after diet change. Male DT and WT NONcNZO10/Ltj littermates (N7; 6–12 mice per group) were fed chow (C) or HF food (F) at weaning. Blood was collected for analysis and/or plasma preparation from nonfasted mice between 10:00 a.m. and noon. Group average values ± SEM are shown for plasma total GIP (A), body weight (B), blood glucose (C), plasma insulin (D), active GLP-1 (E), glucagon (F), leptin (G), cholesterol (H), triglyceride (TG) (I), and free fatty acid (FFA) (J). *P< 0.05, **P = 0.01, and +P = 0.0001 determined by one-way ANOVA using Bonferroni correction for multiple comparisons. Pairwise comparisons were limited to DT/chow vs. DT/HF, WT/chow vs. WT/HF, and WT/HF vs. DT/HF.
The Incretin Response Is Absent in DT Mice
Two weeks after weaning onto the indicated diet, plasma GIP (Fig. 4A and B) was detectable only in WT mice and increased 2.4-fold higher with HF feeding compared with chow. Furthermore, GIP levels increased 3.8-fold from their respective fasting levels 15 min after nutrient ingestion, regardless of diet. Fasting glucose (Fig. 4C and D) was slightly elevated in DT versus WT animals being fed the respective diets and increased 2.7- to 3.8-fold after nutrient ingestion. In contrast, plasma insulin (Fig. 4E and F) increased only in WT mice after nutrient administration, indicating that as in humans with T2DM (5), the incretin response is severely blunted in DT mice. The blunted incretin response in DT mice fed chow occurred even though fasting and nutrient-stimulated GLP-1 levels were similar to those in WT animals (Fig. 4G). With HF feeding (Fig. 4H), fasting GLP-1 levels were elevated in DT versus WT mice, whereas postprandial GLP-1 levels were similar. Thus, GLP-1 levels in DT mice are equal to or greater than those in WT mice. Glucagon levels (Fig. 4I) with chow feeding were not different, irrespective of diet and genotype. With HF feeding, glucagon was significantly reduced only in WT mice after nutrient ingestion (Fig. 4J).
Figure 4.

Metabolic response to oral nutrients. Mice from Fig. 3 were characterized 2 weeks after weaning onto standard chow (panels A, C, E, G, and I) or an HF diet (panels B, D, F, H, and J). After a 5-h fast, blood was collected for analysis and preparation of plasma before (0; □) and again 15 min after (15; ▨) administration of glucose plus intralipid by intragastric gavage. Group average values ± SEM are shown for total GIP (panels A and B), blood glucose (panels C and D), plasma insulin (panels E and F), active GLP-1 (panels G and H), and glucagon (panels I and J). For statistics, a mixed random-effects repeated-measures analysis was performed on GIP, glucose, insulin, GLP-1, and glucagon with a single covariance parameter for correlation between time points 0 and 15. For GIP, glucose, and glucagon, a natural log transformation was used to stabilize the variance.
Strategy to Identify Biochemicals That Promote Rather Than Reflect T2DM
Nonbiased biochemical profiling was conducted on plasma prepared from nonfasted animals 1 week after weaning onto chow or HF food (n = 7–9 per group) to potentially identify metabolites that change before the onset of T2DM and thus promote rather than reflect T2DM. Of 343 named biochemicals detected, 47, 176, and 27 exhibited significant genotype, diet, and genotype/diet interaction effects, respectively (Table 1). Thus, 89% of the biochemical changes occurred in response to diet or genotype alone. On the basis of nonfasting glucose levels, T2DM develops as a function of diet and genotype (Fig. 1). Thus, our analysis focused on metabolites exhibiting a genotype/diet interaction. Levels for each metabolite were also plotted versus nonfasting glucose levels to identify metabolites correlated with diabetes rather than a specific genotype (% NONcNZO10/Ltj background or with or without the DT transgene), diet, or other factors. This also controlled for intra- and intergroup outliers and differential rates for developing T2DM within each group. Glucose concentrations measured during sample collection, and biochemical profiling was highly correlated (R2 = 0.63, P < 0.0001; Fig. 5A).
Table 1.
Metabolite changes between groups
| Biochemicals with P ≤ 0.05 |
Biochemicals with P ≤ 0.10 |
|||
|---|---|---|---|---|
| ANOVA contrasts | # Up | # Down | # Up | # Down |
| WT/HF vs. WT/chow | 52 | 82 | 7 | 6 |
| DT/HF vs. DT/chow | 78 | 76 | 11 | 5 |
| DT/chow vs. WT/chow | 8 | 3 | 10 | 2 |
| DT/HF vs. WT/HF | 51 | 10 | 29 | 4 |
| Two-way ANOVA | # Biochemicals with P ≤ 0.05 | # Biochemicals with P ≤ 0.10 |
|---|---|---|
| Genotype effect | 47 | 28 |
| Diet effect | 176 | 17 |
| Genotype-to-diet interaction | 27 | 20 |
After log transformation and imputation with minimum observed values for each compound, a two-way ANOVA with contrasts was used to identify biochemicals that differed between experimental groups. The numbers of metabolites that achieved statistical significance (P ≤ 0.05) and those approaching significance (P < 0.1 to 0.05) are shown.
Figure 5.

Metabolomic studies of mice and humans. Panels A–N: β-Hydroxypyruvate levels increase profoundly early in the pathogenesis of T2DM in mice. Biochemical profiling was conducted on plasma prepared from nonfasting male DT and WT NONcNZO10/Ltj littermates (N8; n = 7–9 mice per group) 1 week after weaning onto the indicated diet. Metabolite levels were plotted as a function of plasma glucose levels for each mouse to allow visualization and control for intra- and intergroup variants as well as for differential rates for developing T2DM. Panel A (y-axis): Blood glucose (mg/dL) was measured at the time of blood collection, whereas all other values represent relative signals for the indicated metabolite as determined by the mass spectrometry platforms. The dotted lines indicate the lower limit of detection. R2 and P values were determined by linear regression with all samples in a single group. P values in panels C and I are only for mice being fed the HF diet because the chow and HF food supply different amounts of 1,5-AG (panel C), and 3-hydroxy 2-ethylpropionate (HEP) (panel I) was not detectable in 10 of 14 animals fed chow. The P value in panel N is for genotype effect. 3-MGC, 3-methylglutarylcarnitine; α-HIV, α-hydroxyisovalerate; DHL, dihomo-linolenate (20:3n3 or n6); Glc, glucose. Panel O: Pathways for β-hydroxypyruvate metabolism. Ala, alanine; DAO, d-amino acid oxidase; HPR, hydroxypyruvate reductase (also d-glycerate dehydrogenase/glyoxylate reductase); Pyr, pyruvate; Ser, serine; SPT, serine-pyruvate transaminase (also alanine:glyoxylate aminotransferase); SR, serine racemase. Panels P–R: β-Hydroxypyruvate–to–d-serine ratios are reduced in humans with IGT. Relative levels of d-serine (panel P) and β-hydroxypyruvate (panel Q) were measured in plasma from fasting humans with NGT (n = 19), IGT (n = 20), and T2DM (n = 20). Ratios for these metabolites (panel R) were determined for each individual. Plots show group values for mean (+) ± SEM, median (bar within the box), upper and lower quartiles (top and bottom of the box, respectively), and outliers (●). Significance was determined using the mixed-effects model with individual variance parameters for each group. Outliers were not excluded from the statistical analyses.
Biochemicals Associated With Insulin Resistance, T2DM, and Diabetes Complications Change Early in Disease Pathogenesis
Plasma mannose is derived via hepatic glycogen breakdown, and fasting levels of glucose and mannose are highly correlated in humans with and without T2DM (32). Mannose and glucose concentrations were highly correlated in all mice (Fig. 5B). The nonmetabolized, diet-derived glucose analog 1,5-anhydroglucitol (1,5-AG) is used clinically to assess glycemic control. Because it competes with glucose for reabsorption by the renal tubules (33), 1,5-AG and glucose levels are inversely related. With HF feeding, 1,5-AG concentrations were decreased in diet-matched DT mice compared with WT mice (Fig. 5C). Diet-genotype interactions were not compared because 1,5-AG contents in chow and HF foods are different. Branched chain amino acids (BCAAs) contribute to neurobehavioral impairment and obesity-related insulin resistance (34,35). BCAA metabolites, including leucine, isoleucine, valine, α-hydroxyisovalerate, 3-methylglutarylcarnitine, and 3-hydroxy-2-ethylproprionate, were increased in DT/HF mice compared with all other groups but were also elevated in WT/HF mice (Fig. 5D–I). Consistent with less mitochondrial oxidation of BCAAs, levels of most carnitine and glycine derivatives (e.g., 2-methylbutyrylcarnitine, isovalerylcarnitine, and isovalerylglycine) were reduced similarly in WT/HF and DT/HF mice (not shown). l-erythrulose is a highly reactive ketose, glycates and crosslinks proteins, and may mediate changes in the lens associated with diabetic cataract formation (36). Sorbitol is generated from glucose by aldose reductase, and increased levels are associated with the onset of diabetic retinopathy and neuropathy (37). l-erythrulose (Fig. 5J) levels were increased in DT/HF mice, whereas sorbitol (Fig. 5K) concentrations increased in DT mice fed both diets (P < 10−6 for genotype effect). As in humans with nonalcoholic fatty liver disease (38), plasma levels of dihomo-linolenate (20:3n3 or 6) were increased in DT/HF mice (Fig. 5L).
Metabolites in the d-Serine Pathway Change Early in the Pathogenesis of T2DM
Plasma β-hydroxypyruvate levels exhibited a diet-genotype interaction (P = 0.03) and increased strikingly with plasma glucose, regardless of diet or genotype (R2 = 0.88, P < 0.0001; Fig. 5M). β-Hydroxypyruvate is produced by serine-pyruvate transaminase (39) and also by d-amino acid oxidase (DAO) acting on d-serine (40) (Fig. 5O). The biochemical profiling was not designed to discriminate between d- and l-stereoisomeric amino acids. However, DAO is inhibited by sodium benzoate (41), and there was a genotype-dependent decrease (P < 0.005) in benzoate levels in DT mice (Fig. 5N) suggesting at least some β-hydroxypyruvate is derived via DAO.
Metabolic Changes in the d-Serine Pathway Occur as Humans Progress From NGT to IGT to T2DM
Targeted assays measured fasting levels of d-serine, β-hydroxypyruvate, and benzoate in plasma samples from humans with NGT (n = 19), IGT (n = 20), and T2DM (n = 20). Group characteristics are in Table 2. Fasting and 2-h glucose levels from OGTTs, HbA1c, fasting insulin, HOMA-insulin resistance (IR), and BMI were progressively higher in groups with NGT versus IGT versus T2DM. Twelve of 20 T2DM subjects were taking oral medications (11 with metformin and 1 with metformin plus glipizide). The remaining subjects had diet-controlled disease. There were no direct correlations between β-hydroxypyruvate, sodium benzoate, or d-serine levels as a function of fasting glucose, 2-h glucose, HbA1c, fasting insulin, HOMA-IR, BMI, subject age, sex, or race, or the length of time samples had been stored at −80°C (not shown). d-serine (Fig. 5P) was highest in the group with IGT (2.96 ± 0.26 μmol/L) compared with NGT (2.55 ± 0.21 μmol/L) and T2DM (2.56 ± 0.22 μmol/L). Conversely, the relative peak area for β-hydroxypyruvate (Fig. 5Q) was lower in those with IGT (3.79 ± 0.43) compared with NGT (5.71 ± 0.97) and T2DM (5.83 ± 0.95). However, group differences in d-serine and β-hydroxypyruvate levels as well as benzoate (not shown) were not significant. There was a small negative correlation (Spearman r = −0.15; P = 0.048; not shown) between β-hydroxypyruvate and benzoate suggesting a potential substrate-product relationship between β-hydroxypyruvate and d-serine. Thus, the β-hydroxypyruvate–to–d-serine ratio was determined for each individual. The group mean for this ratio (Fig. 5R) was twofold lower in IGT compared with NGT (P = 0.031) and T2DM (P = 0.006), suggesting flux through DAO is reduced and/or clearance of β-hydroxypyruvate is increased in IGT.
Table 2.
Group characteristics
| NGT | IGT | T2DM | |
|---|---|---|---|
| (n = 19) | (n = 20) | (n = 20) | |
| Glucose, mg/dL | |||
| 2-h** | 125 ± 14 | 163 ± 20 | 243 ± 46 |
| Fasting** | 91 ± 6.2 | 100 ± 8.4 | 110 ± 8.9 |
| Fasting insulin, μU/mL* | 6.3 ± 4.1 | 12.2 ± 7.4 | 16.0 ± 7.4 |
| HOMA-IR score** | 1.47 ± 1.04 | 2.96 ± 1.80 | 4.39 ± 2.21 |
| HbA1c, %** | 5.5 ± 0.4 | 5.8 ± 0.4 | 6.4 ± 0.7 |
| HbA1c, mmol/mol** | 37 ± 4.4 | 40 ± 4.4 | 46 ± 7.7 |
| BMI, kg/m2** | 28.9 ± 4.9 | 32.0 ± 5.0 | 36.7 ± 6.0 |
| Age, years | 44.6 ± 13.0 | 47.3 ± 8.3 | 51.5 ± 7.3 |
| Race, n | |||
| Caucasian | 12 | 11 | 10 |
| African American | 7 | 9 | 10 |
| Sex, n | |||
| Males | 7 | 10 | 7 |
| Females | 12 | 10 | 13 |
Data are group means ± SD or n. The 2-h glucose values were determined using a standard 75-g OGTT.
*P = 0.0002 indicating group means are significantly different by one-way ANOVA.
**P < 0.0001 indicating group means are significantly different by one-way ANOVA.
β-Hydroxypyruvate Reduces Insulin Content
β-Hydroxypyruvate induces apoptosis of astroglial but not liver or kidney cells (41). Acute β-hydroxypyruvate treatment of islets from NONcNZO10/Ltj mice (not shown) did not affect insulin content or GSIS. However, a 3-day pretreatment with β-hydroxypyruvate (Fig. 6A) reduced insulin content (36%; P = 0.012). When normalized to insulin content, GSIS remained unchanged (Fig. 6B).
Figure 6.

β-Hydroxypyruvate (HP) alters properties of islets and MENs. Insulin content (panel A) and release assays (panel B) were performed on islets from NONcNZO10/Ltj mice after a 3-day pretreatment with HP or without (control [Con]) 1 mmol/L HP. Average values ± SEM from four independent islet preparations are shown. Insulin release in response to 3 mmol/L and 10 mmol/L glucose (Glc) was normalized to the insulin content for each sample. Results from four independent islet preparations (normalized to content) are shown. Panels C–F: MENs were isolated from the proximal small intestine of guinea pigs, cultured for 7 days, and loaded with Fura-2, and calcium imaging studies were done. Some MENs were treated with 1 mmol/L HP only during (acute; green tracings) or for 3 days before but not including (chronic; red tracings), imaging studies. Control cultures for HP treatment were incubated with vehicle alone (phosphate buffer; blue tracings). Neurons were excited with the combination of A/U (100 μmol/L each) or 50 mmol/L KCl. Values are group mean (dark line) ± SEM (light-colored bars).
β-Hydroxypyruvate Alters Excitatory Properties of MENs
Previous studies with DT mice suggested MENs play a role in regulating insulin secretion (26). As previously shown (21), purinergic receptor agonists (ATP plus uridine 5′-triphosphate [A/U]) or potassium chloride (KCl) increased cytosolic free calcium levels in MENs as monitored by the Fura-2 ratio (Fig. 6C–F). Acute treatment with 1 mmol/L β-hydroxypyruvate slightly decreased the peak response to KCl (Fig. 6C and E) but not to A/U. Incubation with β-hydroxypyruvate for 3 days severely blunted excitation in response to KCl but not A/U (Fig. 6D and F).
Discussion
To study the potential role of GIP-producing cells for preventing T2DM, DT mice were backcrossed five to eight times from the nondiabetogenic C57BL/6J strain onto the diabetogenic NONcNZO10/Ltj background. After 7 weeks of HF feeding, DT mice remained lean but developed severe T2DM due to loss of β-cell mass. Conversely, WT mice became obese but nonfasting glucose levels remained comparably low due to islet hyperplasia. Thus, by studying mice at different ages, we have developed a novel and powerful model system that can be used to identify molecules that may promote and/or prevent T2DM and associated complications. Although DT mice do not produce GIP, plasma GLP-1 levels were not reduced compared with WT animals fed their respective diets. Thus, endogenous circulating GLP-1 does not prevent T2DM. This is consistent with human studies showing GLP-1 release remains normal in humans with T2DM (6,7). In contrast, nonfasting GIP levels increased >10-fold in WT mice 1 week after HF feeding, suggesting GIP and/or other K-cell products play important roles in augmenting insulin secretion and maintaining β-cell function in response to HF feeding. Ablating GIP-producing cells for only 2 days does not impair insulin release (42). Thus, long-term production and release of K-cell products are critical for preventing T2DM.
A limitation to our study is that the DT transgene was backcrossed only five to eight times onto the recombinant congenic NONcNZO10/Ltj strain. We previously showed using OGTTs that the glucose area under the curve but not fasting glucose level is elevated in 16- versus 5-week-old in congenic NONcNZO10/Ltj chow-fed mice (26). Thus, it is possible that nonfasting glucose levels in the current study underestimated the degree of glucose intolerance in each group but probably not relative differences between groups. However, T2DM develops due to the interactions between many genes and environmental factors. In the present model, this includes regions from the NONcNZO10/Ltj genome, the presence or absence of GIP-producing cells, diet, the gut microbiota (as evidenced by changes in benzoate levels), and also time. Thus, normalizing metabolite levels to glucose concentrations (Fig. 5) allowed identification of metabolites associated with T2DM rather than the percentage of NONcNZO10/Ltj background in individual N5–N8 mice. Nonfasting glucose levels in chow-fed congenic (not shown) and N5 NONcNZO10/Ltj mice are ∼175 mg/dL. Moreover, the DT transgene has since been backcrossed 20 times with NONcNZO10/Ltj mice, and preliminary studies indicate DT/HF mice remain lean but develop T2DM more rapidly than N5 mice, whereas WT mice become obese and develop T2DM, with diabetes greatly delayed relative to DT mice. Thus, additional genetic modifiers in NONcNZO10/Ltj mice contribute to T2DM and emphasize the value of using this model to study T2DM.
Biochemical profiling identified metabolites that change early in the development of T2DM and thus may promote rather than reflect the disease. Consistent with impaired glucose handling, plasma glucose, mannose, sorbitol, and 1,5-AG levels were altered in DT/HF mice. Sorbitol, erythrulose, dihomo-linolenate, and numerous metabolites in the BCAA pathway were also perturbed, suggesting initiation of metabolic programs promoting insulin resistance, T2DM, and diabetes complications occur before development of T2DM itself. Thus, DT/HF mice exhibit many features of human T2DM and represent a relevant model for studying the pathogenesis of T2DM and testing potential interventions.
Strikingly, the metabolomic study revealed a highly significant correlation between nonfasting glucose and β-hydroxypyruvate levels for mice regardless of diet or genotype, indicating individual intra- and intergroup variants and outliers exhibit an appropriate correlation. β-Hydroxypyruvate induces apoptosis of astroglial but not liver or kidney cells (41). Chronic but not acute β-hydroxypyruvate treatment altered excitatory properties of MENs and reduced insulin content but not GSIS of islets. β-Hydroxypyruvate can be generated from d-serine by DAO (40). Intriguingly, benzoic acid inhibits DAO, is derived predominantly from gut microbes (43), and was significantly reduced in DT mice fed both diets. Thus, alterations in gut microbiota could potentially mediate a decrease in benzoate and concomitant increases in DAO activity and β-hydroxypyruvate levels. Recent studies have emphasized the importance of intestinal microflora for regulating metabolic phenotypes (44). Our results suggest that changes in the gut microbiome promote a permissive environment and that additional factors (e.g., HF feeding) initiate diabetes. Although our mouse data suggest that β-hydroxypyruvate is generated from DAO, β-hydroxypyruvate can also be produced by serine-pyruvate transaminase (Fig. 5O) (39), and defining which pathway(s) increases β-hydroxypyruvate levels will be important. Regardless of how generated, β-hydroxypyruvate alters functional properties of both islets and MENs.
Plasma d-serine, β-hydroxypyruvate, and benzoic acid levels were not statistically different in humans with NGT, IGT, and T2DM. However, there was a highly significant nearly twofold reduction in the β-hydroxypyruvate–to–d-serine ratio for individuals with IGT compared with NGT and T2DM. Moreover, there was a significant, albeit small, negative correlation between β-hydroxypyruvate and benzoate levels in all subjects, suggesting that at least some β-hydroxypyruvate was derived from d-serine.
As reviewed (45,46), d-serine is derived from l-serine via serine racemase, is a coagonist for the N-methyl-d-aspartate (NMDA) receptor, and is released in the brain from neurons. Chronic activation of NMDA receptors on adjacent neurons due to increased d-serine is thought to cause neurotoxicity. Increased levels of d-serine and NMDA receptor activation have been linked to a host of neurological disorders, including amyotrophic lateral sclerosis and Alzheimer disease, whereas insufficient levels have been implicated in schizophrenia and behavioral disorders (45,47,48). Thus, any alteration in DAO activity could reciprocally alter d-serine and β-hydroxypyruvate levels and promote distinct patterns of neurotoxicity. Determining the effects of d-serine on islet and MEN physiology will be important. Intriguingly, metabolic dysfunction is associated with a host of neurodegenerative diseases (49) and it is interesting to speculate that alterations in the d-serine pathway could link T2DM with these disorders.
Xen is produced by a subset of GIP-producing cells (13) and exerts many of its effects by exciting neurons (14,16,17,19,21,25). We previously showed in mice that effects of Xen on insulin secretion are mediated by a neural relay to islets rather than direct action on β-cells (26). In humans, administration of Xen alone during intravenous graded glucose infusions does not affect insulin release but maximally amplifies effects of GIP on insulin secretion in those with IGT compared with NGT and is inactive in individuals with T2DM (12). In the human pancreas, neurotensin receptor type 1 is expressed on neurons but not islet endocrine cells (27) suggesting Xen effects on insulin secretion in humans are also mediated by a neural relay. Thus, previous studies with Xen unmasked a novel neural relay to islets with low activity in NGT when insulin release is sufficient. This relay is activated and amplifies insulin secretion in IGT to potentially compensate for increased insulin demands and fails during the progression from IGT to T2DM, which could lead to insufficient insulin release to maintain glucose homeostasis. Intriguingly, these in vivo responses to Xen (with GIP) are reciprocally related to the β-hydroxypyruvate–to–d-serine ratios in each group and raise the possibility that changes in β-hydroxypyruvate and d-serine levels and/or ratios could 1) mediate the evolving changes in insulin secretion unmasked during the Xen studies and/or 2) independently promote a host of neurological disorders associated with insulin resistance and/or T2DM.
Article Information
Acknowledgments. The authors thank the nurses of the Clinical Research Unit at Washington University for administering the OGTTs and Dr. Daniel Ory of Washington University for helpful discussions and review of the manuscript.
Funding. Portions of this research were supported by funds from National Institutes of Health (grant numbers 1R01-DK-088126, 5RC1-DK-086163, T32-DA-015036, and R01-MH-51290), American Diabetes Association (grant numbers 1-10-CT-58 and 1-13-CE-46), the Blum-Kovler Foundation, the Live Cell Imaging Facility of the Center for Investigation of Membrane Excitability Diseases (CIMED) at Washington University, the Washington University Diabetes Research Center Immunoassay and Metabolomics Cores (grant number P30-DK-020579), the Washington University Nutrition Obesity Research Center grant (P30-DK-056341) from the National Institute of Diabetes and Digestive and Kidney Diseases, and the Washington University Clinical and Translational Science Award (UL1-RR-024992).
Duality of Interest. J.T.C. served as a consultant for Abbvie Laboratories and En Vivo. In addition, a patent owned by Massachusetts General Hospital for the use of d-serine to treat serious mental illness could yield royalties. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. S.Z., S.W., E.L., and M.J.W. researched data. M.D.P., X.J., K.L.H., and K.L.P. researched data and reviewed and edited the manuscript. J.T.C. reviewed and edited the manuscript. B.M.W. conceived, designed, and directed the study; researched data; and wrote the manuscript. B.M.W. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were previously presented as an abstract at the 73rd Scientific Sessions of the American Diabetes Association, Chicago, IL, 21–25 June 2013.
Footnotes
See accompanying article, p. 1099.
References
- 1.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007;132:2131–2157 [DOI] [PubMed] [Google Scholar]
- 2.Fehmann HC, Göke R, Göke B. Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose-dependent insulin releasing polypeptide. Endocr Rev 1995;16:390–410 [DOI] [PubMed] [Google Scholar]
- 3.Ahren B. Neuropeptides and insulin secretion. In International Textbook of Diabetes Mellitus. 3rd ed. DeFronzo RA, Ferrannini E, Keen H, Zimmet P, Eds. Hoboken, New Jersey, John Wiley & Sons, Ltd, 2004, p. 153–163 [Google Scholar]
- 4.Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004;53(Suppl. 3):S16–S21 [DOI] [PubMed] [Google Scholar]
- 5.Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986;29:46–52 [DOI] [PubMed] [Google Scholar]
- 6.Meier JJ, Nauck MA. Is the diminished incretin effect in type 2 diabetes just an epi-phenomenon of impaired beta-cell function? Diabetes 2010;59:1117–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chowdhury S, Reeds DN, Crimmins DL, et al. Xenin-25 delays gastric emptying and reduces postprandial glucose levels in humans with and without type 2 diabetes. Am J Physiol Gastrointest Liver Physiol 2014;306:G301–G309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 2013;17:819–837 [DOI] [PubMed] [Google Scholar]
- 9.Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 1993;91:301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elahi D, McAloon-Dyke M, Fukagawa NK, et al. The insulinotropic actions of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (7-37) in normal and diabetic subjects. Regul Pept 1994;51:63–74 [DOI] [PubMed] [Google Scholar]
- 11.Vilsbøll T, Krarup T, Madsbad S, Holst JJ. Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia 2002;45:1111–1119 [DOI] [PubMed] [Google Scholar]
- 12.Wice BM, Reeds DN, Tran HD, et al. Xenin-25 amplifies GIP-mediated insulin secretion in humans with normal and impaired glucose tolerance but not type 2 diabetes. Diabetes 2012;61:1793–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anlauf M, Weihe E, Hartschuh W, Hamscher G, Feurle GE. Localization of xenin-immunoreactive cells in the duodenal mucosa of humans and various mammals. J Histochem Cytochem 2000;48:1617–1626 [DOI] [PubMed] [Google Scholar]
- 14.Kim ER, Mizuno TM. Xenin delays gastric emptying rate and activates the brainstem in mice. Neurosci Lett 2010;481:59–63 [DOI] [PubMed] [Google Scholar]
- 15.Alexiou C, Zimmermann JP, Schick RR, Schusdziarra V. Xenin—a novel suppressor of food intake in rats. Brain Res 1998;800:294–299 [DOI] [PubMed] [Google Scholar]
- 16.Cooke JH, Patterson M, Patel SR, et al. Peripheral and central administration of xenin and neurotensin suppress food intake in rodents. Obesity (Silver Spring) 2009;17:1135–1143 [DOI] [PubMed] [Google Scholar]
- 17.Leckstrom A, Kim ER, Wong D, Mizuno TM. Xenin, a gastrointestinal peptide, regulates feeding independent of the melanocortin signaling pathway. Diabetes 2009;58:87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feurle GE, Heger M, Niebergall-Roth E, et al. Gastroenteropancreatic effects of xenin in the dog. J Pept Res 1997;49:324–330 [DOI] [PubMed] [Google Scholar]
- 19.Kamiyama Y, Aihara R, Nakabayashi T, Mochiki E, Asao T, Kuwano H. The peptide hormone xenin induces gallbladder contractions in conscious dogs. Neurogastroenterol Motil 2007;19:233–240 [DOI] [PubMed] [Google Scholar]
- 20.Feurle GE, Hamscher G, Kusiek R, Meyer HE, Metzger JW. Identification of xenin, a xenopsin-related peptide, in the human gastric mucosa and its effect on exocrine pancreatic secretion. J Biol Chem 1992;267:22305–22309 [PubMed] [Google Scholar]
- 21.Zhang S, Hyrc K, Wang S, Wice BM. Xenin-25 increases cytosolic free calcium levels and acetylcholine release from a subset of myenteric neurons. Am J Physiol Gastrointest Liver Physiol 2012;303:G1347–G1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feurle GE, Metzger JW, Grudinki A, Hamscher G. Interaction of xenin with the neurotensin receptor of guinea pig enteral smooth muscles. Peptides 2002;23:1519–1525 [DOI] [PubMed] [Google Scholar]
- 23.Kim ER, Mizuno TM. Role of neurotensin receptor 1 in the regulation of food intake by neuromedins and neuromedin-related peptides. Neurosci Lett 2010;468:64–67 [DOI] [PubMed] [Google Scholar]
- 24.Pettibone DJ, Hess JF, Hey PJ, et al. The effects of deleting the mouse neurotensin receptor NTR1 on central and peripheral responses to neurotensin. J Pharmacol Exp Ther 2002;300:305–313 [DOI] [PubMed] [Google Scholar]
- 25.Cline MA, Nandar W, Rogers JO. Xenin reduces feed intake by activating the ventromedial hypothalamus and influences gastrointestinal transit rate in chicks. Behav Brain Res 2007;179:28–32 [DOI] [PubMed] [Google Scholar]
- 26.Wice BM, Wang S, Crimmins DL, et al. Xenin-25 potentiates glucose-dependent insulinotropic polypeptide action via a novel cholinergic relay mechanism. J Biol Chem 2010;285:19842–19853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chowdhury S, Wang S, Patterson BW, Reeds DN, Wice BM. The combination of GIP plus xenin-25 indirectly increases pancreatic polypeptide release in humans with and without type 2 diabetes mellitus. Regul Pept 2013;187:42–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Althage MC, Ford EL, Wang S, Tso P, Polonsky KS, Wice BM. Targeted ablation of glucose-dependent insulinotropic polypeptide-producing cells in transgenic mice reduces obesity and insulin resistance induced by a high fat diet. J Biol Chem 2008;283:18365–18376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leiter EH, Reifsnyder PC. Differential levels of diabetogenic stress in two new mouse models of obesity and type 2 diabetes. Diabetes 2004;53(Suppl. 1):S4–S11 [DOI] [PubMed] [Google Scholar]
- 30.Nieman DC, Shanely RA, Gillitt ND, Pappan KL, Lila MA. Serum metabolic signatures induced by a three-day intensified exercise period persist after 14 h of recovery in runners. J Proteome Res 2013;12:4577–4584 [DOI] [PubMed] [Google Scholar]
- 31.Benneyworth MA, Li Y, Basu AC, Bolshakov VY, Coyle JT. Cell selective conditional null mutations of serine racemase demonstrate a predominate localization in cortical glutamatergic neurons. Cell Mol Neurobiol 2012;32:613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sone H, Shimano H, Ebinuma H, et al. Physiological changes in circulating mannose levels in normal, glucose-intolerant, and diabetic subjects. Metabolism 2003;52:1019–1027 [DOI] [PubMed] [Google Scholar]
- 33.Dungan KM. 1,5-anhydroglucitol (GlycoMark) as a marker of short-term glycemic control and glycemic excursions. Expert Rev Mol Diagn 2008;8:9–19 [DOI] [PubMed] [Google Scholar]
- 34.Coppola A, Wenner BR, Ilkayeva O, et al. Branched-chain amino acids alter neurobehavioral function in rats. Am J Physiol Endocrinol Metab 2013;304:E405–E413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newgard CB, An J, Bain JR, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 2009;9:311–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simpson GL, Ortwerth BJ. The non-oxidative degradation of ascorbic acid at physiological conditions. Biochim Biophys Acta 2000;1501:12–24 [DOI] [PubMed] [Google Scholar]
- 37.Burg MB, Kador PF. Sorbitol, osmoregulation, and the complications of diabetes. J Clin Invest 1988;81:635–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puri P, Wiest MM, Cheung O, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009;50:1827–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sallach HJ. Formation of serine hydroxypryuvate and L-alanine. J Biol Chem 1956;223:1101–1108 [PubMed] [Google Scholar]
- 40.Konno R, Hamase K, Maruyama R, Zaitsu K. Mutant mice and rats lacking D-amino acid oxidase. Chem Biodivers 2010;7:1450–1458 [DOI] [PubMed] [Google Scholar]
- 41.Chung SP, Sogabe K, Park HK, et al. Potential cytotoxic effect of hydroxypyruvate produced from D-serine by astroglial D-amino acid oxidase. J Biochem 2010;148:743–753 [DOI] [PubMed] [Google Scholar]
- 42.Pedersen J, Ugleholdt RK, Jørgensen SM, et al. Glucose metabolism is altered after loss of L cells and α-cells but not influenced by loss of K cells. Am J Physiol Endocrinol Metab 2013;304:E60–E73 [DOI] [PubMed] [Google Scholar]
- 43.Goodwin BL, Ruthven CR, Sandler M. Gut flora and the origin of some urinary aromatic phenolic compounds. Biochem Pharmacol 1994;47:2294–2297 [DOI] [PubMed] [Google Scholar]
- 44.Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013;341:1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jirásková-Vaníčková J, Ettrich R, Vorlová B, et al. Inhibition of human serine racemase, an emerging target for medicinal chemistry. Curr Drug Targets 2011;12:1037–1055 [DOI] [PubMed] [Google Scholar]
- 46.Balu DT, Takagi S, Puhl MD, Benneyworth MA, Coyle JT. D-serine and serine racemase are localized to neurons in the adult mouse and human forebrain. Cell Mol Neurobiol 2014;34:419–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lancelot E, Beal MF. Glutamate toxicity in chronic neurodegenerative disease. Prog Brain Res 1998;116:331–347 [DOI] [PubMed] [Google Scholar]
- 48.Coyle JT. NMDA receptor and schizophrenia: a brief history. Schizophr Bull 2012;38:920–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai H, Cong WN, Ji S, Rothman S, Maudsley S, Martin B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr Alzheimer Res 2012;9:5–17 [DOI] [PMC free article] [PubMed] [Google Scholar]