

Abstract
The details of protein pathways at a structural level provides a bridge between genetics/molecular biology and physiology. The renin-angiotensin system is involved in many physiological pathways with informative structural details in multiple components. Few studies have been performed assessing structural knowledge across the system. This assessment allows use of bioinformatics tools to fill in missing structural voids. In this paper we detail known structures of the renin-angiotensin system and use computational approaches to estimate and model components that do not have their protein structures defined. With the subsequent large library of protein structures, we then created a species specific protein library for human, mouse, rat, bovine, zebrafish, and chicken for the system. The rat structural system allowed for rapid screening of genetic variants from 51 commonly used rat strains, identifying amino acid variants in Angiotensinogen, ACE2, and AT1b that are in contact positions with other macromolecules. We believe the structural map will be of value for other researchers to understand their experimental data in the context of an environment for multiple proteins, providing pdb files of proteins for the renin-angiotensin system in six species. With detailed structural descriptions of each protein, it is easier to assess a species for use in translating human diseases with animal models. Additionally, as whole genome sequencing continues to decrease in cost, tools such as molecular modeling will gain use as an initial step in designing efficient hypothesis driven research, addressing potential functional outcomes of genetic variants with precompiled protein libraries aiding in rapid characterizations.
Keywords: Sequence-to-structure-to-function analysis, rat genetics, comparative modeling, renin-angiotensin system, angiotensin peptides
1. Introduction
Whole genome sequencing is in an age of rapid expansion, with the estimate of wet lab costs to dip below $1000 in the very near future through the release of technology such as the HiSeq X machine. This will require new tools to help in understanding how genetic variants are connected to changes in physiology and how to use the increasing number of sequenced species to better understand the mechanisms of evolution. Molecular modeling and dynamics will provide some of the necessary additions to the toolkit for interpretation of genome variation. Currently whole exome sequencing, an analysis of the protein coding genes, is the least expensive toolset for understanding human genetic variants. Combining the sequencing of protein coding genes with evolutionary analysis of diverse species and molecular modeling tools can move simple identification into functional prediction. More importantly, with the proper tools available, this approach can be done with speed, and improve our ability to address variants in entire protein pathways. Having structural protein libraries of pathways already assembled and publically available is one way to increase the speed in identifying functional outcomes of genetic diversity. Utilizing such an approach, we analyzed the renin-angiotensin system (RAS), building protein libraries for human, mouse, rat, bovine, zebrafish, and chicken. The structural library was then used to analyze 51 rat genomes of strains commonly used as animal models for the genetics of cardiovascular disease.
The RAS is a complex pathway that has important roles in many cardiovascular, renal, and endocrine processes and cell proliferation. The pathway consists of the protein Angiotensinogen (AGT) that is cleaved by various enzymes, generating peptide fragments (Angiotensin, Ang) that bind and activate G-protein coupled receptors (Figure 1). The structures of many of the components in the RAS are known, with several protein-protein or inhibitor bound structures reported (Table S1). Efforts at inhibiting multiple steps of the RAS pathway have been successful in treating hypertension in millions of patients. However, our current knowledge of this pathway at the molecular and DNA sequence variant level is still far from complete and thus many potential mechanisms of the RAS may not be defined. In this paper we integrate all known 3D protein structures for components of the RAS with proteins (and peptides) that do not have a reported structure. For this latter case we use in silico analyses (molecular modeling, AutoDock prediction, and molecular dynamic simulations) to help elucidate a full structural analysis of the RAS (Figure 1). We hypothesize that understanding the complete sequence-to-structure-to-function (SSF) for the RAS will serve as a tool in identifying genetic variants that may alter protein function while also offering a toolset in studying animal models for human diseases.
Figure 1. Multiple components of the renin-angiotensin system.

Structural analysis of the RAS with components that are characterized using the in silico approaches in this paper boxed in red. Biochemically determined structures are known for AGT (reduced/oxidized), Prorenin, Renin, Renin-AGT, Ang I, Ang II, Ang-(1-7), ACE (N/C-terminal domains), and ACE 2. Models were used for the PRR, AT1, AT2, MAS, MRGD, ACE-Ang I, Aminopeptidase AAng II, and ACE2–Ang II. In addition to the models shown here we have generated potential structures for Ang- Neprilysin and Ang-PRCP.
The activation of the RAS begins with the expression of AGT, which can exist in either a reduced or oxidized state, with different production rates of Ang peptides from the two forms (Zhou et al., 2010). The oxidized AGT is rapidly processed by the enzyme Renin to produce a ten amino acid fragment known as Angiotensin I (Ang I). Renin is first translated as an inactive protein (zymogen) with a propeptide (Sealey et al., 1980). This propeptide disrupts beta sheet packing within Renin causing a steric block of the active site (Morales et al., 2012) resulting in low levels of catalytic activity in cleaving AGT (Heinrikson et al., 1989). Activation of the zymogen’s propeptide through cleavage by various enzymes (Reudelhuber et al., 1994), low pH (Danser and Deinum, 2005), cold temperatures (Danser and Deinum, 2005), or binding of prorenin to the (pro)renin receptor (PRR) can all increase the catalytic activity of Renin on AGT, resulting in the release of Ang I. Analysis of the concentrations of Renin vs. Prorenin circulating in the blood has suggested upwards of a 10 fold higher level of Prorenin, implicating potential tissue specific roles of prorenin activation by PRR (Zhuo, 2011). In addition, Prorenin binding to PRR activates intracellular pathways such as ERK 1/2 with proposed roles in cranial RAS signaling (Nguyen, 2011; Nguyen et al., 2002). The process of activation of Renin and the impact of oxidized and reduced AGT can be seen at https://www.youtube.com/watch?v=RsEEs5WFkSQ&feature=youtu.be.
Once Ang I is formed, it is cleaved by various enzymes to produce peptides of additional sizes. These enzymes include ACE (PDB files 2c6f and 1o8a), ACE2 (1r42), Neprilysin (NEP, 1r1h), and the Lysosomal Pro-X carboxypeptidase (PRCP, 3n2z). ACE contains two catalytic active sites (Soubrier et al., 1988) highly documented for the production of the Ang II peptide. The two domains show different Cl− ion concentration in activation (Wei et al., 1991) with an additive effect when both domains are present (Marcic et al., 2000). Ang II can further be processed in two directions. Aminopeptidase A converts Ang II into Ang III by cleaving off the first amino acid (Asp1), which is further processed by Aminopeptidase N into Ang IV with removal of Arg2. A homologous enzyme to ACE, known as ACE2, efficiently cleaves Ang II to make Ang-(1-7) (Rice et al., 2004). Ang-(1-7) can be converted into the peptide Alamandine by cleavage of the amino acid 1 side chain resulting in an alanine at this site (Lautner et al., 2013).
The production of these peptides contributes to the two arms of the RAS pathway, that of Ang II eliciting vasoconstriction or that of Ang-(1-7) functioning in opposition. Numerous studies have addressed the balance between these two arms of the pathway in diseases from hypertension (Brosnihan et al., 2005) to cancer (Ager et al., 2008). Ang II activates the AT1 receptor in a two-step process eliciting increase in vasoconstriction, angiogenic, proliferative effects (Hines et al., 2003; Holloway et al., 2002; Hunyady et al., 2003; Kobilka and Deupi, 2007; Noda et al., 1996; Prokop et al., 2013; Vauquelin and Van Liefde, 2005). Ang III preferentially binds AT2 over AT1 (Bosnyak et al., 2011) and has been suggested as the primary agonist of AT2 in the kidney (Kemp et al., 2012; Padia and Carey, 2013; Padia et al., 2008). Ang IV is suggested to bind to the AT4 receptor (Chai et al., 2004), but much less is known of the functional outcomes of this binding. Ang-(1-7) activates the MAS receptor (Santos et al., 2003) eliciting antihypertensive, antiangiogenic, antifibrotic and antiproliferative (Tallant et al., 2005) effects. The newly identified Ang peptide, Alamandine, was shown to be a ligand of MrgD (Lautner et al., 2013). The question thus remains, is it possible to develop methods to allow for systematic studies of evolution and genetic variants that may perturb function of a protein system? In this paper we develop a working structural map of the renin-angiotensin system, providing the tools to rapidly screen genetic variants for potential perturbation of protein interactions.
2. Methods
2.1 Analysis of Prorenin, Renin, and Angiotensinogen
The structure of Renin interacting with AGT (pdb 2x0b) was used in molecular dynamic simulations. To simplify the simulations and identify amino acids that contribute to interaction between the catalytic active site and the substrate, AGT was shortened to contain only a small stretch that fits into the active site. To determine amino acid stabilities, additional simulations were run containing only Renin or only the fragment of AGT. Simulations were run for 5 nanoseconds (ns) with all structures providing a level root-mean squared deviation (RMSD) and energy values throughout, suggesting equilibrated simulations. With the majority of focus on amino acid side chain stability with and without AGT on Renin, 5 ns provides a reasonable simulation time to assess these outcomes. The simulation trajectories were analyzed with the md_analyze and md_analyzeres macros (www.yasara.org/macros) and the RMSD values compared for the complex to each individual protein’s trajectory. These macros enable the determination of how individual amino acids move throughout the simulation. Regions with different RMSD values were visually inspected for interaction and hydrogen bonding between the two proteins.
2.2 Propeptide of renin: cloning, purification, and pull down assays
The DNA sequence for the propeptide of renin (amino acids 24–66) was codon optimized and synthesized by DNA2.0 (www.dna20.com) into pJ416 with an N-terminal ATG and C-terminal thrombin cleavage site, 6x-His tag, and stop codon. This was sub-cloned into pGEX4T using L-BamHIProRen (5′TCC CAA GGA TCC ATG CTT CCC ACG GAT ACC ACA) and R4T-NotIProRen (5’TTG GGA GCG GCC GCT CAA TGA TGG TGG TGA TGA TGA GAA C). The resulting protein contained an N-terminal GST-tag and a C-terminal 6x-His-tag that are both removable with thrombin cleavage. After sequence confirmation with BigDye Sanger sequencing, the construct was transformed into BL21(DE3) E. coil cells (NEB). A single colony was inoculated into 10 mol of LB containing 50 μg/mL ampicillin and grown overnight. This culture was then added to 500 mol of terrific broth containing 50 μg/mL ampicillin and grown to an OD600 of 0.5 at 37°C and induced with 0.5 mM IPTG at 23°C overnight. Cells were spun down and resuspended in PBS, sonicated and centrifuged at 16,000*g for 10 min. The lysate was passed over 3mL of glutathione sepharose and washed with 40 mol of PBS, 5 mol PBS + 10 mM DTT, 5 mol of 2 M NaCl, and 10 mol PBS. Protein was eluted in 1 mol fractions with 10 mM glutathione suspended in PBS. Each fraction was run on a 13.5% Tris-glycine SDS PAGE and the fractions containing the GST-propeptide were then collected and run on a 3 mol Ni-Sepharose prepacked column, washed with 60 mol phosphate buffered imidazole, and eluted in 2 mol fractions with 500 mM imidazole. Digests of the purified propeptide were performed using varying concentrations of purified thrombin (BD Bioscience) at 0.125 to 4 units per 42 μL digest to confirm the proper peptide size.
Pull down assays were performed by freshly binding lysate of the GST-propeptide to 40 μL glutathione sepharose in a 50:50 BB500 solution for three samples. Samples were incubated for 1 hour at room temperature, followed by five washes with BB750 and one wash with BB500. The beads were resuspended in 500 μL of BB500. One sample had nothing added to the pulled down GST-propeptide (negative control), to the second sample 4 μg of purchased recombinant renin (Lee Biosolutions catalogue number 510-11R) was added (positive control), and the third sample had 4 μg of recombinant Renin and 10 μg of synthesized shortened propeptide (GenScript, DTTTFKRIFLKRMPSIRESLKER). Samples were mixed for one hour at room temperature and washed two times with BB500. Beads were then resuspended in 20 μL of 5X sample buffer and run on 15% Tris-Tricine SDS PAGE.
2.3 AutoDocking of Ang peptides to ACE, ACE2, Neprilysin, and PRCP
Three of the Ang peptides have solution structures determined: Ang I (pdb 1n9u), Ang II (pdb 1n9v), and Ang-(1-7) (pdb 2jp8). These structures were aligned using the Mustang algorithm (Konagurthu et al., 2006). Simulations were then performed on each of the peptides in a 50Å cubed simulation square for 10 ns. The structures for the ACE N-terminus (pdb 2c6f), ACE C-terminus (1o8a), ACE2 (1r42), Neprilysin (pdb 1r1h), and PRCP (pdb 3n2z) were used to study potential mechanisms of Ang peptide docking. Sequence-to-structure comparisons were first performed between the two domains of ACE and ACE2 by aligning the sequences with CLUSTALW default settings (Gonnet matrix), saving this as a FASTA file, and then coloring the conserved and variant amino acids on a red (conserved) to gray (divergent) color scale in YASARA (Krieger et al., 2002). Based on the structure of Ang I, additional Ang peptides were created (Ang-(1-9), Ang III, Ang IV, and Ang-(1-5)) by cleaving off the amino acids of each and fixing the terminus to which the deletions were made (adding a hydrogen to the N-terminus or a hydroxyl group to the C-terminus). AutoDock (Morris et al., 2009) was performed on each of the enzymes with all of the Ang peptides (Ang I, Ang-(1-9), Ang II, Ang III, Ang IV, Ang-(1-7) and Ang-(1-5)) using five ensembles of the receptor (enzyme) side chains and 20 peptide predictions on each ensemble for a total of 100 docking results per run. With a total of seven peptides and five enzymes, this resulted in 3,500 peptide binding predictions. The simulation square for each docking was 35Å cubed and place within the active site of each enzyme.
2.4 G-protein coupled receptors (GPCRs)
Ang III receptor docking was created by removing the first amino acid in the structure of Ang II bound to either AT1 or AT2. This was energy minimized and molecular dynamic simulations run for 5 ns. The energy minimized structure of MAS in a lipid membrane (Prokop et al., 2013) was used to perform AutoDock analysis (Morris et al., 2009) in YASARA to predict how Ang-(1-7) interacts with MAS. The structure of Ang-(1-7) as determined in pdb file 2jp8 (Lula et al., 2007) was docked into the MAS receptor with the dock_runensemble macro (http://www.yasara.com/macros.htm). The top docking conformation was energy minimized with the AMBER03 force field (Duan et al., 2003). The MRGD protein was modeled using the same approaches taken with AT1, AT2, and MAS (Prokop et al., 2013). The docked model of Ang-(1-7) into MAS was then aligned to the model of MRGD (Mas-related GPCR member D) and MAS removed. Ang-(1-7) was converted into Alamandine and energy minimized.
2.5 Rat Genome analysis
Following the previously mentioned modeling for each protein, all of the proteins were modeled for human, mouse, rat, zebrafish, and chicken. To begin, known structures for RAS components were identified and updated until December 1, 2013 (Table S1). Even when the structure was known for a species, models were still generated for that species to remove crystal packing forces and fill in missing loops of the structure. To create all of the models, YASARA (Krieger et al., 2002) homology modeling was employed using the template models and sequences shown in Table 1. The multiple rat genomes were then analyzed using the Variant Visualizer of the Rat Genome Database (Laulederkind et al., 2013) for all available strains on April 28, 2014. The rat protein structure was aligned (using Mustang (Konagurthu et al., 2006)) to the predicted protein-protein interactions, followed by energy minimizations using the YASARA2 force field. Variants were manually mapped to each protein and sequence alignments were performed using ClustalW (Larkin et al., 2007).
Table 1. Models created for components of the RAS for human, mouse, and rat.
The sequence used for modeling the protein is shown for each with the UniProt code shown and amino acids listed below. The Z-score of each model was calculated using the YASARA2 force field. The % homology and carbon alpha (CA) RMSD were calculated following model alignment with the Mustang algorithm.
| Protein | Model made with |
Sequences used |
Z- score |
% Homology |
CA RMSD (Å) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Human | Mouse | Rat | Human | Mouse | Rat | Mouse to Human |
Rat to Human |
Rat to Mouse |
Mouse to Human |
Rat to Human |
Rat to Mouse |
||
| ACE N-Terminal | pdb 2c6f |
P12821 30-630 |
P094703 5-635 |
P47820 36-636 |
0 | 0.05 | 0.01 | 87.69 | 88.52 | 94.01 | 0.369 | 0.337 | 0.315 |
|
| |||||||||||||
| ACE C-Terminal | pdb 1o8a |
P12821 631-1232 |
P094706 36-1237 |
P47820 637-1238 |
0.34 | 0.33 | 0.31 | 81.66 | 82.73 | 93.62 | 0.373 | 0.34 | 0.335 |
|
| |||||||||||||
| ACE2 | pdb 1r42 |
Q9BYF1 18-708 |
Q8R0I01 8-740 |
Q5EGZ1 18-708 |
0.33 | 0.43 | 0.2 | 84.25 | 84.28 | 90.76 | 0.334 | 0.379 | 0.385 |
|
| |||||||||||||
| AGT | pdb 2x0b |
P01019 34-485 |
P118592 5-477 |
P01015 25-477 |
−1.79 | −2.03 | −1.76 | 62.47 | 66.42 | 86.06 | 1.111 | 1.368 | 1.311 |
|
| |||||||||||||
| AT1a | I-TASSER |
P30556 1-359 |
P29754 1-359 |
P25095 1-359 |
−2.23 | −1.74 | −1.78 | 94.38 | 95.21 | 98.61 | 0.871 | 0.837 | 0.321 |
|
| |||||||||||||
| AT1b | I-TASSER | - |
P29755 1-359 |
P29089 1-359 |
−2.23 | −1.81 | −1.71 | 93.24 | 94.89 | 96.29 | 0.884 | 0.909 | 0.442 |
|
| |||||||||||||
| AT2 | I-TASSER |
P50052 1-363 |
P35374 1-364 |
P35351 1-365 |
−2.3 | −2 | −1.99 | 91.57 | 92.39 | 98.89 | 0.968 | 0.933 | 0.481 |
|
| |||||||||||||
| MAS | I-TASSER |
P04201 1-325 |
P30554 1-325 |
P12526 1-325 |
−1.71 | −1.46 | −1.59 | 90.12 | 91.02 | 98.45 | 1.124 | 0.981 | 0.752 |
|
| |||||||||||||
| MRGD | I-TASSER |
Q8TDS7 1-321 |
Q91ZB8 1-321 |
Q7TN41 1-319 |
−1.96 | −1.52 | −1.4 | 59.93 | 60.07 | 85.57 | 1.474 | 1.39 | 1.178 |
|
| |||||||||||||
| NEP | pdb 1r1h |
P08473 52-750 |
Q61391 52-750 |
P07861 52-750 |
0.27 | 0.2 | 0.15 | 93.97 | 94.13 | 98.42 | 0.357 | 0.314 | 0.323 |
|
| |||||||||||||
| PRCP | pdb 3n2z |
P42785 46-496 |
Q7TMR0 44-491 |
D4AA31 43-488 |
−0.3 | −0.31 | −0.29 | 80.27 | 80.49 | 90.7 | 0.308 | 0.333 | 0.315 |
|
| |||||||||||||
| Prorenin | pdb 4amt |
P00797 24-406 |
P06281 22-402 |
P08424 27-402 |
−1.04 | −1.24 | −1.11 | 72.36 | 69.86 | 86.08 | 0.562 | 0.52 | 0.547 |
|
| |||||||||||||
| PRR | I-TASSER |
O75787 |17-350 |
Q9CYN9 18-350 |
Q6AXS4 18-350 |
−3.01 | −3.15 | −3.02 | 95.06 | 94.44 | 97.9 | 0.47 | 0.628 | 0.374 |
|
| |||||||||||||
| Renin1 | pdb 2x0b |
P00797 67-406 |
P06281 72-402 |
P08424 65-402 |
−0.88 | −0.81 | −1.05 | 71.73 | 67.67 | 85.54 | 0.625 | 0.479 | 0.608 |
|
| |||||||||||||
| Renin2 | pdb 2x0b | - |
P00796 64-401 |
- | - | −0.62 | - | - | - | - | - | - | - |
3. Results and Discussion
3.1 Generation of protein models and docking for RAS components
To begin understanding what genetic variants could alter the RAS, the components of the system were compiled, and structural models generated for components in the human, mouse, and rat (Table 1, Supplemental folder of models). Models were generated for ACE (both N- and C-terminal domains), ACE2, AGT, AT1 (a form for all species, and b form known in rodents), AT2, MAS, MRGD, NEP, PRCP, Prorenin, Renin 1, Renin 2 (only in the mouse), and PRR. A Z-score for structural accuracy of our models relative to all known protein structures in the protein databank (Table 1) was used, with values <−4.0 considered bad, those >−4.0 considered fair, and anything >−2.0 considered a good protein model. All protein models had values >−4, with all but PRR also having values >−2.0 in the rat. This suggests a high level of confidence in the protein modeling techniques for the system in all species. Additional protein alignments between species show a high sequence homology (>60% for all) with low deviations of the carbon alpha alignments (Table 1) indicating the models align well between species. For comparative display of each protein, we have added a new track to the Rat Genome database that compares side by side the protein models of the various components in human, mouse, and rat (for example AT1a, http://rgd.mcw.edu/rgdweb/jsmol/rgd.jsp?d=AT1a). These new displays can be found in the gene page for each gene in the RGD under the protein structures tab.
Having the protein models, it was next possible to predict the binding of various proteins and peptides. This was started with an analysis of Renin (REN) and AGT binding. The known structures revealed that the propeptide of Prorenin results in an allosteric modification of the activation domain to block binding and cleavage of AGT (Figure 2A). Surprisingly, purified propeptide is able to bind and pull Renin out of solution, which is blocked by a larger version of the human handle region peptide (HRP) we call the ProR peptide. This ability to pull Renin out of solution suggests strong amino acid contacts in the beta sheet region of Renin. The HRP has been suggested as a new therapeutic to inhibit the activation of Prorenin through interaction with PRR (Batenburg et al., 2013); however recent evidence for use as a dual inhibitor of the RAS is cautioned based on its agonist potentials on PRR (Riet et al., 2014). The HRP (blue) is composed of the beta-sheet packing of the Prorenin structure (Figure 2A), further suggesting that PRR critical contacts have the potential to alter the allosteric modifications required to move the activation domain (yellow). Having the propeptide removed, creating the functional Renin, allows for binding and cleavage of AGT. Several of the loops on Renin stabilize AGT contacts (Figure 2C) as determined through use of differential molecular dynamic simulations of Renin alone or bound to AGT (Figure 2D).
Figure 2. Role of cleavage of the propeptide in activation of Renin and binding of AGT.

A) Addition of the propeptide (red) of Prorenin results in the activation sequence (yellow) to cover the active site as well as steric hindrance with the contact points between Renin and AGT. Cleavage of the propeptide results in the movement of the activation domain, allowing for Ang I (magenta) proper cleavage and additional contact points between Renin and AGT. The proposed potential therapeutic RAS peptide, known as the handle region peptide (HRP, blue), is identified to contribute to the beta sheet packing that allosterically modifies the activation sequence. B) Pull down experiments of Renin with the GST-Propeptide on glutathione sepharose. Sample 1 is a control of GST-Propeptide pull down, 2 is the pull down with additional Renin added in for co-pull down, 3 has Renin and the shorter ProR peptide (longer version of the human HRP) added resulting in a loss of Renin co-pull down, and the final lane contains the purified Renin protein. C) Interaction between a segment of the N-terminus of AGT (red) in the active site of Renin (gray) used for molecular dynamics simulations. D) Molecular dynamics simulation comparing renin alone (red) or complexed with the AGT fragment (blue) showing the carbon alpha RMSD differences for each amino acid on Renin. For areas with large differences in dynamics, shown above with a green box are the amino acids contacts with AGT in the loops of Renin which are highlighted in green on the structure of Renin in C.
Once bound to Renin, AGT is cleaved to produce Ang I, which is further processed to produce various Ang peptides. Three of the Ang peptides (Ang I, Ang II, and Ang-(1-7)) have known structures. Structural alignment shows a similar backbone (Figure 3A), with higher variation of side chain positions (Figure 3B). To study whether the structures are stable and what amino acids contribute to the structural organization, molecular dynamic simulations were performed. All three peptides had a low RMSD for amino acids 4–6 (Tyr, Ile, and His) likely due to hydrophobic packing (Figure 3C). Overall, Ang-(1-7) provided the most stability, with decreased movement in amino acids 1–3 (Asp, Arg, and Val). Using the known structure of the Ang peptides, additional Ang peptide metabolites (Figure 3D) were modeled, removing the corresponding amino acids for each (red, Figure 3D).
Figure 3. Known and modeled structures of the Ang peptides.

A–B) Structural alignment of Ang I (green), Ang II (blue), and Ang-(1-7) (red) aligned together showing just the backbone (A) or the side chains (B). C) Molecular dynamic simulations on each peptide for 10 nanoseconds showing the average movement of the carbon alpha for each of the amino acids of the peptide. D) Sequences of the various Ang peptides used for Autodock experiments. Amino acids in red were removed from Ang I to produce the peptide sequence shown in black.
Each of the modeled peptides was then docked to the various enzymes using the automated ligand docking program AutoDock, and the relative binding energies determined (Table 2). Manual inspection of the top binding conformations for each enzyme confirmed the Ang peptide location relative to the active site residues in ACE (Zn), ACE2 (Zn), NEP (Zn), and PRCP (Ser active site). The top conformation of Ang I bound to ACE or Ang II bound to ACE2 puts the peptide close to the active Zn ion (Figure 4A). Both are in the correct orientation for the amino acids expected to be cleaved in each case (yellow Figure 4A), with cleavage of amino acids 9–10 in ACE binding resulting in Ang II, and cleavage of amino acid 8 in ACE2 binding resulting in Ang-(1-7). Looking at the top conformation of Ang peptides bound to ACE with amino acids conserved (blue) or variant (gray) between the N- and C-terminal domains shows several amino acids that contact Ang II that vary between the two domains (Lys, Arg, Ser; Figure 4B), corresponding to data previously suggested to be important to domain specificity and targeted inhibitors (Ehlers et al., 2013). Most amino acids of either Ang I (ACE) or Ang II (ACE2) had a lower RMSD in molecular dynamic simulations when bound to the enzyme (Figure 4C) than when they were free in solution (Figure 3C). Comparing Ang I binding to the C- (blue, Figure 4C) or N-terminus (red, Figure 4C) shows some similarity in dynamics, but large differences can be seen in amino acid 1 (Asp), 7 (Pro), 8 (Phe), and 9 (His) between Ang I bound to the two domains. This result is in agreement with the known biochemical data suggesting the C-terminus to have a higher processing rate for Ang I to Ang II than the N-terminal domain (Deddish et al., 1998). The increased coordination of amino acids 8 and 9 by the C-terminus of ACE likely allows for short term stability and alignment to the Zn ion to increase the cleavage of Ang I to produce Ang II.
Table 2. AutoDock of Ang peptides to various receptors.
Binding energies (kcal/mol) from AutoDock experiments for N- and C-terminal domains of ACE, ACE2, Neprilysin, and PRCP to the various Ang peptides with the top five binding energies shown for each docking run. The top binding energy for each is shown in the top line.
| Ang I | Ang-(1-9) | Ang II | Ang III | Ang IV | Ang-(1-7) | Ang-(1-5) | |
|---|---|---|---|---|---|---|---|
| ACE C-term | 10.11 | 10.18 | 10.26 | 10.61 | 10.1 | 8.29 | 8.32 |
| 9.1 | 9.54 | 9.91 | 10.35 | 9.43 | 6.99 | 7.29 | |
| 8.07 | 8.72 | 9.55 | 9.98 | 8.53 | 6.98 | 7.25 | |
| 7.88 | 8.56 | 9.32 | 9.86 | 8.33 | 6.68 | 7.11 | |
| 7.36 | 8.51 | 9.24 | 9.86 | 8.31 | 6.51 | 6.52 | |
|
| |||||||
| ACE N-term | 9.73 | 7.5 | 9.63 | 9.77 | 9.14 | 8.56 | 6.58 |
| 8.54 | 7.23 | 8.61 | 8.07 | 8.74 | 6.79 | 6.45 | |
| 7.87 | 7.16 | 8.43 | 7.46 | 8.69 | 6.14 | 6.42 | |
| 7.65 | 7.12 | 8.15 | 6.89 | 8.47 | 6.13 | 6.16 | |
| 7.53 | 6.97 | 8.05 | 6.87 | 8.45 | 6.09 | 5.96 | |
|
| |||||||
| ACE2 | 3.4 | 4.58 | 5.94 | 6.51 | 7.22 | 2.4 | 5.82 |
| 2.69 | 4.41 | 4.97 | 4.67 | 6.75 | 1.98 | 5.58 | |
| 2.25 | 3.42 | 3.51 | 4.62 | 6.61 | 0.95 | 5.41 | |
| 1.6 | 3.26 | 3.37 | 4 | 6.05 | 0.57 | 4.93 | |
| 1.55 | 3.26 | 3.21 | 3.93 | 5.85 | 0.49 | 4.79 | |
|
| |||||||
| Neprilysin | 4.43 | 8.03 | 9.5 | 8.4 | 8.23 | 10.32 | 7.61 |
| 3.69 | 6.89 | 6.92 | 7.02 | 7.71 | 6.61 | 7.44 | |
| 3.57 | 5.18 | 5.34 | 6.6 | 7.4 | 6.38 | 5.87 | |
| 3.2 | 4.66 | 5.04 | 6.31 | 7.27 | 6.27 | 5.67 | |
| 3.14 | 4.5 | 4.54 | 6.12 | 7.09 | 5.95 | 5.34 | |
|
| |||||||
| PRCP | 5.73 | 6.54 | 5.74 | 7.21 | 6.4 | 1.9 | 4.82 |
| 5.4 | 6.51 | 5.49 | 6.69 | 6.18 | 0.82 | 4.32 | |
| 4.69 | 5.83 | 5.28 | 6.32 | 6.15 | 0.76 | 4.3 | |
| 4.65 | 5.52 | 4.9 | 6 | 6.09 | 0.7 | 4.25 | |
| 4.54 | 5 | 4.59 | 5.76 | 5.98 | 0.54 | 4.15 | |
Figure 4. Docking of Ang peptides to ACE and ACE2.

A) Structures of the top docking of Ang I to ACE (left) or Ang II to ACE 2 (right). The Ang peptides are shown in red with the amino acids cleaved off in yellow. The Zn active site is magenta and the Cl ions are green. B) Variant amino acids (grey to light blue) between the N and Cterminus showing contributing amino acids to variant binding of Ang I. C) Molecular dynamics simulations showing the movement of Ang I to either the C-terminus (blue) or the N-terminus (red) of ACE or Ang II to ACE 2 (green).
To finalize our sequence-to-structure-to-function (SSF) analysis of the RAS, we focused on the various receptors of Ang peptides. Structures of many GPCRs are currently being determined, with AT1 as a target for structure determination (http://gpcr.scripps.edu/tracking_status.htm); however, this protein determination is still not in physiological conditions of the lipid membrane. Using our previously created models for AT1, AT2 and MAS receptors in a lipid membrane (Prokop et al., 2013), additional models were generated for the docking of Ang III to AT2 (Figure 5A), Ang-(1-7) to MAS (Figure 5B), and Alamandine to MRGD (Figure 5C). The AT2-Ang III model was created by taking the model of Ang II bound to AT2, converting Ang II to Ang III, followed by several rounds of energy minimizations. The model for MAS–Ang-(1-7) is previously described (Prokop et al., 2013). Using the MAS-Ang-(1-7) model, the model for MRGD was aligned to MAS, MAS removed, Ang-(1-7) converted to Alamandine, and several rounds of energy minimizations performed.
Figure 5. Docking of Ang peptides into various uncharacterized receptors.
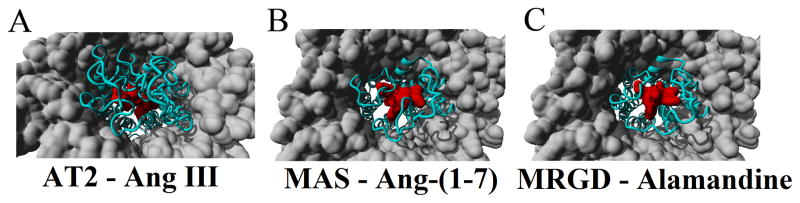
A) The AT2 receptor model (cyan) interacting with Ang III (red) in a lipid membrane (gray). B) The MAS (cyan) model in a lipid membrane (gray) with Ang 1-7 (red) docked into the binding site as determined by the best conformation in 100 Autodock predictions. C) Model of MRGD (cyan) with Alamandine (red) bound into the binding pocket based on the docking results of MAS/Ang-(1-7).
3.2 Analysis of genetic variants in the rat
Having the largest compiled analysis of protein structural work for the RAS it is thus possible to study genetic variants in components of the pathway with speed and precision for prediction of alteration of protein function. Analysis of Ren (Renin), Agt (AGT), Ace (ACE), Ace2 (ACE2), Agtr1a (AT1a), Agtr1b (AT1b), Agtr2 (AT2), Mas1 (MAS), Mrgprd (MRGD), Mme (NEP), Prcp (PRCP), and Atp6ap2 (PRR) in the genomes of 51 sequenced rats (Atanur et al., 2013; Gibbs et al., 2004) was performed. The analysis revealed nonsynonymous mutations in the various rat genomes (Figure 6A). Genes that did not contain any nonsynonymous mutations included Ren, Agtr1a, Agtr2, Mas1, and Atp6ap2. Total nonsynonymous mutations for the other genes were; Agt = 4, Ace = 2, Ace2 = 3, Agtr1b = 2, Mrgprd = 1, Mme = 1, and Prcp = 2. Mapping of all the variants onto the seven protein structures with nonsynonymous mutations was then performed (Figures S1–S7), revealing several variants located near contact points with other macromolecules (Figure 6). A variation in AGT at amino acid 154 is located near the interaction with Renin (Figure 6B), variations at amino acid 90 and 509 in ACE2 are located at functioning sites (Figure 6C), and amino acid 40 in AT1b is located at a lipid membrane contact (Figure 6D).
Figure 6. Variation of RAS proteins in the multiple rat genomes.

A) Nonsynonymous mutations identified in the multiple rat genomes (left) for several of the RAS genes (top). B) Based on structural assessment of all rat variants, the amino acid 154 (red) variant of AGT (gray) is in close proximity to contacts with Renin (cyan). C) Amino acid 90 (red) variant of ACE2 (gray) is a cite of known site for N-linked glycosylation (blue) while amino acid 509 variant is located close to Ang II (cyan) binding. D) Amino acid 40 (red) variant in AT1b (gray) is located at a membrane (cyan) contact point on a helix with known contacts with the Ang II peptide (blue).
Amino acid 154 of AGT is located near a contact point in the known structure of AGT interacting with Renin, and this variant is found in the sequenced SHR/Olalpcv rat (Figure S1) at 100% frequency with a depth of 19 reads. However, the amino acid change is from a V to I, maintaining functional properties of the amino acids. In agreement with functional properties, Polyphen (Adzhubei et al., 2010) analysis based on conservation and properties of this variant predicts it to be benign. Of the three nonsynonymous variants in ACE2, two fall within the catalytic domain (N90D and D509Y). Substitution D509Y is located near the docked Ang II peptide (Figure S3); however, it is only found in the SHRSP/Gcrc rat genome with a 29% allele frequency and with a low sequence depth of only 7 reads, suggesting the impact of this change is negligible. In contrast, the amino acid change N90D falls at a known N-linked glycosylation site in a diverse range of rat strains (from FHH/FHL to WKY) with 100% allelic frequency (Figure S3), suggesting a potential altered posttranslational modification in these strains. Finally, two variants were found in AT1b at amino acids 2 and 40, both found in the same strains with 100% allele frequency (Figure S4). Amino acid 2 is not highly conserved and the variant seen matches that of human AT1, suggesting a nonfunctioning role. Contrary, amino acid 40 is found at a highly conserved amino acid that is in the middle of the hydrophobic membrane interaction. Changes from the shorter Val to the longer Met could potentially perturb membrane packing and dynamics.
Once a base map of structural components is created, one can easily model homology in the protein system for additional species of interest. For example, we created the system map for bovine, zebrafish and chicken based on the human map in only a few hours of computation demands (Supplemental Folder of models). Although the modeling approaches cannot definitively answer the question of how a genetic variant will change a proteins function, they provide testable hypotheses to reduce the burden of numerous potential candidate variants of interest. For every genome sequenced, hundreds to thousands of protein coding variants are found. With proposals now underway to sequence hundreds of thousands of human genomes and the completion of more species genomes, methods are needed to functionally characterize the many protein coding variants. Tools such as PolyPhen, though powerful and high throughput, only suggest if a variant has the potential to be damaging based on evolution. These tools predict hundreds to thousands of variants to be probably or possibly damaging, a value suggesting a high rate of false discovery. In addition they do not assess the mechanisms by which a variant could be pathogenic. This inhibits hypothesis oriented testing of genetic variants. In theory, if large macromolecular structural databases for individual species existed, these could be combined with the advancing sequence based prediction tools such as PolyPhen to deploy a program that is both high throughput and results in hypothesis driven experiments to test functional outcomes at a molecular level.
4. Conclusions
The structures and interactions of several proteins of the RAS have not been structurally determined, such as how Ang peptides bind to enzymes and receptors. Much of the lack of Ang peptide-protein structures is due to difficulty in interpretation and capture of structural transition states of peptide binding. However, techniques such as AutoDock and molecular dynamic simulations can give predictions for these details and allow for the modeling of these different components of the RAS. Therefore, we sought to build a more complete structural view of the RAS in this paper. Having the structural dataset for the system allows for the generation of the protein components in a species of interest, for example in the human, mouse or rat This then allows for in silico assessment of an animal model system to study human disease. In addition, these datasets can allow for computational screening of drug compounds before animal studies in order to validate the animal model for testing of the human phenotype. These protein system based molecular structures thus provide tools for future researchers to carefully choose the animal model to comparatively study human diseases. In addition, it is also possible to rapidly identify variants found in sequenced genomes for components of these protein systems, such as the RAS, to understand the potential outcomes of the variants. Here we identify amino acid variations in AGT, ACE2, and AT1b in commonly used rat strains that have the potential to alter contacts with other macromolecules. This work highlights the use of protein modeling at a system level, showing the need for the development of macromolecular modeling libraries for dissemination of structure based interpretation of protein coding genetic variants in the age of whole genome and exome sequencing to come.
Supplementary Material
Highlights.
Modeling and docking of unknown structures for the renin-angiotensin system
Creation of a structural map of the renin-angiotensin system
Structural library of renin-angiotensin system ligands and receptors for 6 species
Analysis of variants for the renin-angiotensin system in 51 sequenced rat strains
Acknowledgments
We thank Marek Tutaj and Jeffrey De Pons for help in setting up the human, mouse, and rat protein structure visualizer for components of the RAS in the Rat Genome Database (RGD). Funding for this study was provided by the American Heart Association (award 11PRE7380033 to JWP), Ohio Board of Regents Choose Ohio First Bioinformatics initiative award (to JWP), The University of Akron, NIH (award 5R01HL064541-15 to HJJ for the Rat Genome Database), Brazilian National Institute of Science and Technology in Hormones and Women’s Health (To RASS), and the Federal University of Minas Gerais. The funding sources had no contribution to design, collection, analysis, interpretation of data, drafting of the manuscript, or decision to publish this study.
Abbreviations
- RAS
renin-angiotensin system
- AGT
angiotensinogen
- GPCRs
G-protein coupled receptors
- SSF
sequence-to-structure-to-function
- Ang
angiotensin
- PRR
(pro)renin receptor
- pdb
Protein Data Bank
- ACE
angiotensin converting enzyme
- md
molecular dynamic
- PRCP
Lysosomal Pro-X carboxypeptidase
- GST
glutathione S-transferase
- RMSD
root-mean squared deviation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ager EI, Neo J, Christophi C. The renin-angiotensin system and malignancy. Carcinogenesis. 2008;29:1675–1684. doi: 10.1093/carcin/bgn171. [DOI] [PubMed] [Google Scholar]
- Atanur SS, Diaz AG, Maratou K, Sarkis A, Rotival M, Game L, Tschannen MR, Kaisaki PJ, Otto GW, Ma MCJ, Keane TM, Hummel O, Saar K, Chen W, Guryev V, Gopalakrishnan K, Garrett MR, Joe B, Citterio L, Bianchi G, McBride M, Dominiczak A, Adams DJ, Serikawa T, Flicek P, Cuppen E, Hubner N, Petretto E, Gauguier D, Kwitek A, Jacob H, Aitman TJ. Genome Sequencing Reveals Loci under Artificial Selection that Underlie Disease Phenotypes in the Laboratory Rat. Cell. 2013;154:691–703. doi: 10.1016/j.cell.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batenburg WW, van den Heuvel M, van Esch J, van Veghel R, Garrelds I, Leijten F, Danser A. The (pro)renin receptor blocker handle region peptide upregulates endothelium-derived contractile factors in aliskiren-treated diabetic transgenic (mREN2)27 rats. J Hypertens Febr 2013. 2013;31:292–302. doi: 10.1097/HJH.0b013e32835c1789. [DOI] [PubMed] [Google Scholar]
- Bosnyak S, Jones ES, Christopoulos A, Aguilar MI, Thomas WG, Widdop RE. Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin Sci Lond Engl 1979. 2011;121:297–303. doi: 10.1042/CS20110036. [DOI] [PubMed] [Google Scholar]
- Brosnihan KB, Neves LAA, Chappell MC. Does the Angiotensin-Converting Enzyme (ACE)/ACE2 Balance Contribute to the Fate of Angiotensin Peptides in Programmed Hypertension? Hypertension. 2005;46:1097–1099. doi: 10.1161/01.HYP.0000185149.56516.0a. [DOI] [PubMed] [Google Scholar]
- Chai SY, Fernando R, Peck G, Ye SY, Mendelsohn FAO, Jenkins TA, Albiston AL. The angiotensin IV/AT4 receptor. Cell Mol Life Sci CMLS. 2004;61:2728–2737. doi: 10.1007/s00018-004-4246-1. [DOI] [PubMed] [Google Scholar]
- Danser AHJ, Deinum J. Renin, Prorenin and the Putative (Pro)renin Receptor. Hypertension. 2005;46:1069–1076. doi: 10.1161/01.HYP.0000186329.92187.2e. [DOI] [PubMed] [Google Scholar]
- Deddish PA, Marcic B, Jackman HL, Wang HZ, Skidgel RA, Erdös EG. N-domain-specific substrate and C-domain inhibitors of angiotensin-converting enzyme: angiotensin-(1-7) and keto-ACE. Hypertension. 1998;31:912–917. doi: 10.1161/01.hyp.31.4.912. [DOI] [PubMed] [Google Scholar]
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- Ehlers MR, Abrie JA, Sturrock ED. C domain-selective inhibition of angiotensin-converting enzyme. J Renin Angiotensin Aldosterone Syst. 2013;14:189–192. doi: 10.1177/1470320313489206. [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, Okwuonu G, Hines S, Lewis L, DeRamo C, Delgado O, Dugan-Rocha S, Miner G, Morgan M, Hawes A, Gill R, Celera Holt RA, Adams MD, Amanatides PG, Baden-Tillson H, Barnstead M, Chin S, Evans CA, Ferriera S, Fosler C, Glodek A, Gu Z, Jennings D, Kraft CL, Nguyen T, Pfannkoch CM, Sitter C, Sutton GG, Venter JC, Woodage T, Smith D, Lee H-M, Gustafson E, Cahill P, Kana A, Doucette-Stamm L, Weinstock K, Fechtel K, Weiss RB, Dunn DM, Green ED, Blakesley RW, Bouffard GG, De Jong PJ, Osoegawa K, Zhu B, Marra M, Schein J, Bosdet I, Fjell C, Jones S, Krzywinski M, Mathewson C, Siddiqui A, Wye N, McPherson J, Zhao S, Fraser CM, Shetty J, Shatsman S, Geer K, Chen Y, Abramzon S, Nierman WC, Havlak PH, Chen R, Durbin KJ, Egan A, Ren Y, Song X-Z, Li B, Liu Y, Qin X, Cawley S, Worley KC, Cooney AJ, D’Souza LM, Martin K, Wu JQ, Gonzalez-Garay ML, Jackson AR, Kalafus KJ, McLeod MP, Milosavljevic A, Virk D, Volkov A, Wheeler DA, Zhang Z, Bailey JA, Eichler EE, Tuzun E, Birney E, Mongin E, Ureta-Vidal A, Woodwark C, Zdobnov E, Bork P, Suyama M, Torrents D, Alexandersson M, Trask BJ, Young JM, Huang H, Wang H, Xing H, Daniels S, Gietzen D, Schmidt J, Stevens K, Vitt U, Wingrove J, Camara F, Mar Albà M, Abril JF, Guigo R, Smit A, Dubchak I, Rubin EM, Couronne O, Poliakov A, Hübner N, Ganten D, Goesele C, Hummel O, Kreitler T, Lee Y-A, Monti J, Schulz H, Zimdahl H, Himmelbauer H, Lehrach H, Jacob HJ, Bromberg S, Gullings-Handley J, Jensen-Seaman MI, Kwitek AE, Lazar J, Pasko D, Tonellato PJ, Twigger S, Ponting CP, Duarte JM, Rice S, Goodstadt L, Beatson SA, Emes RD, Winter EE, Webber C, Brandt P, Nyakatura G, Adetobi M, Chiaromonte F, Elnitski L, Eswara P, Hardison RC, Hou M, Kolbe D, Makova K, Miller W, Nekrutenko A, Riemer C, Schwartz S, Taylor J, Yang S, Zhang Y, Lindpaintner K, Andrews TD, Caccamo M, Clamp M, Clarke L, Curwen V, Durbin R, Eyras E, Searle SM, Cooper GM, Batzoglou S, Brudno M, Sidow A, Stone EA, Venter JC, Payseur BA, Bourque G, López-Otín C, Puente XS, Chakrabarti K, Chatterji S, Dewey C, Pachter L, Bray N, Yap VB, Caspi A, Tesler G, Pevzner PA, Haussler D, Roskin KM, Baertsch R, Clawson H, Furey TS, Hinrichs AS, Karolchik D, Kent WJ, Rosenbloom KR, Trumbower H, Weirauch M, Cooper DN, Stenson PD, Ma B, Brent M, Arumugam M, Shteynberg D, Copley RR, Taylor MS, Riethman H, Mudunuri U, Peterson J, Guyer M, Felsenfeld A, Old S, Mockrin S, Collins F Rat Genome Sequencing Project Consortium. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Heinrikson RL, Hui J, Zürcher-Neely H, Poorman RA. A structural model to explain the partial catalytic activity of human prorenin. Am J Hypertens. 1989;2:367–380. doi: 10.1093/ajh/2.5.367. [DOI] [PubMed] [Google Scholar]
- Hines J, Fluharty SJ, Yee DK. Structural determinants for the activation mechanism of the angiotensin II type 1 receptor differ for phosphoinositide hydrolysis and mitogen-activated protein kinase pathways. Biochem Pharmacol. 2003;66:251–262. doi: 10.1016/s0006-2952(03)00257-0. [DOI] [PubMed] [Google Scholar]
- Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, Southwell BR, Lew MJ, Thomas WG. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol. 2002;61:768–777. doi: 10.1124/mol.61.4.768. [DOI] [PubMed] [Google Scholar]
- Hunyady L, Vauquelin G, Vanderheyden P. Agonist induction and conformational selection during activation of a G-protein-coupled receptor. Trends Pharmacol Sci. 2003;24:81–86. doi: 10.1016/S0165-6147(02)00050-0. [DOI] [PubMed] [Google Scholar]
- Kemp BA, Bell JF, Rottkamp DM, Howell NL, Shao W, Navar LG, Padia SH, Carey RM. Intrarenal angiotensin III is the predominant agonist for proximal tubule angiotensin type 2 receptors. Hypertension. 2012;60:387–395. doi: 10.1161/HYPERTENSIONAHA.112.191403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM. MUSTANG: A multiple structural alignment algorithm. Proteins Struct Funct Bioinforma. 2006;64:559–574. doi: 10.1002/prot.20921. [DOI] [PubMed] [Google Scholar]
- Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Laulederkind SJF, Hayman GT, Wang SJ, Smith JR, Lowry TF, Nigam R, Petri V, de Pons J, Dwinell MR, Shimoyama M, Munzenmaier DH, Worthey EA, Jacob HJ. The Rat Genome Database 2013--data, tools and users. Brief Bioinform. 2013;14:520–526. doi: 10.1093/bib/bbt007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, Jankowski J, Jankowski V, Sousa F, Alzamora A, Soares E, Barbosa C, Kjeldsen F, Oliveira A, Braga J, Savergnini S, Maia G, Peluso AB, Passos-Silva D, Ferreira A, Alves F, Martins A, Raizada M, Paula R, Motta-Santos D, Klempin F, Kemplin F, Pimenta A, Alenina N, Sinisterra R, Bader M, Campagnole-Santos MJ, Santos RAS. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res. 2013;112:1104–1111. doi: 10.1161/CIRCRESAHA.113.301077. [DOI] [PubMed] [Google Scholar]
- Lula I, Denadai ÂL, Resende JM, de Sousa FB, de Lima GF, Pilo-Veloso D, Heine T, Duarte HA, Santos RAS, Sinisterra RD. Study of angiotensin-(1–7) vasoactive peptide and its β-cyclodextrin inclusion complexes: Complete sequence-specific NMR assignments and structural studies. Peptides. 2007;28:2199–2210. doi: 10.1016/j.peptides.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Marcic B, Deddish PA, Jackman HL, Erdös EG, Tan F. Effects of the N-Terminal Sequence of ACE on the Properties of Its C-Domain. Hypertension. 2000;36:116–121. doi: 10.1161/01.HYP.36.1.116-a. [DOI] [PubMed] [Google Scholar]
- Morales R, Watier Y, Böcskei Z. Human prorenin structure sheds light on a novel mechanism of its autoinhibition and on its non-proteolytic activation by the (pro)renin receptor. J Mol Biol. 2012;421:100–111. doi: 10.1016/j.jmb.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen G. Renin, (pro)renin and receptor: an update. Clin Sci Lond Engl 1979. 2011;120:169–178. doi: 10.1042/CS20100432. [DOI] [PubMed] [Google Scholar]
- Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer J. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–1427. doi: 10.1172/JCI200214276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda K, Feng YH, Liu XP, Saad Y, Husain A, Karnik SS. The active state of the AT1 angiotensin receptor is generated by angiotensin II induction. Biochemistry (Mosc) 1996;35:16435–16442. doi: 10.1021/bi961593m. [DOI] [PubMed] [Google Scholar]
- Padia SH, Carey RM. AT2 receptors: beneficial counter-regulatory role in cardiovascular and renal function. Pflüg Arch Eur J Physiol. 2013;465:99–110. doi: 10.1007/s00424-012-1146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padia SH, Kemp BA, Howell NL, Fournie-Zaluski MC, Roques BP, Carey RM. Conversion of Renal Angiotensin II to Angiotensin III Is Critical for AT2 Receptor–Mediated Natriuresis In Rats. Hypertension. 2008;51:460–465. doi: 10.1161/HYPERTENSIONAHA.107.103242. [DOI] [PubMed] [Google Scholar]
- Prokop J, Santos RA, Milsted A. Differential mechanisms of activation of the Ang peptide 1 receptors AT1, AT2, and MAS: Using in 2 silico techniques to differentiate the three receptors. PLoS ONE. 2013 doi: 10.1371/journal.pone.0065307. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reudelhuber TL, Ramla D, Chiu L, Mercure C, Seidah NG. Proteolytic processing of human prorenin in renal and non-renal tissues. Kidney Int. 1994;46:1522–1524. doi: 10.1038/ki.1994.435. [DOI] [PubMed] [Google Scholar]
- Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383:45–51. doi: 10.1042/BJ20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Riet L, van den Heuvel M, Peutz-Kootstra CJ, van Esch JHM, van Veghel R, Garrelds IM, Musterd-Bhaggoe U, Bouhuizen AM, Leijten FPJ, Danser AHJ, Batenburg WW. Deterioration of Kidney Function by the (Pro)renin Receptor Blocker Handle Region Peptide in Aliskiren-treated Diabetic Transgenic (mRen2)27 Rats. Am J Physiol - Ren Physiol. 2014 doi: 10.1152/ajprenal.00010.2014. [DOI] [PubMed] [Google Scholar]
- Santos RAS, Simoes e Silva AC, Maric C, Silva DMR, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SVB, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, Walther T. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sealey JE, Atlas SA, Laragh JH. Prorenin and Other Large Molecular Weight Forms of Renin. Endocr Rev. 1980;1:365–391. doi: 10.1210/edrv-1-4-365. [DOI] [PubMed] [Google Scholar]
- Soubrier F, Alhenc-Gelas F, Hubert C, Allegrini J, John M, Tregear G, Corvol P. Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc Natl Acad Sci U S A. 1988;85:9386–9390. doi: 10.1073/pnas.85.24.9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallant E, Ferrario C, Gallagher P. Angiotensin-(1-7) inhibits growth of cardiac myocytes through activation of the mas receptor. Am J Physiol-Heart Circ Physiol. 2005;289:H1560–H1566. doi: 10.1152/ajpheart.00941.2004. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I. G protein-coupled receptors: a count of 1001 conformations. Fundam Clin Pharmacol. 2005;19:45–56. doi: 10.1111/j.1472-8206.2005.00319.x. [DOI] [PubMed] [Google Scholar]
- Wei L, Alhenc-Gelas F, Corvol P, Clauser E. The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J Biol Chem. 1991;266:9002–9008. [PubMed] [Google Scholar]
- Zhou A, Carrell RW, Murphy MP, Wei Z, Yan Y, Stanley PLD, Stein PE, Pipkin FB, Read RJ. A redox switch in angiotensinogen modulates angiotensin release. Nature. 2010;468:108–111. doi: 10.1038/nature09505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo JL. Augmented intratubular renin and prorenin expression in the medullary collecting ducts of the kidney as a novel mechanism of angiotensin II-induced hypertension. Am J Physiol - Ren Physiol. 2011;301:F1193–F1194. doi: 10.1152/ajprenal.00555.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.