

SUMMARY
AdaSGC binds Hsc70s to inhibit ATPase activity. Using single-turnover assays, adaSGC, a soluble SGC mimic, preferentially inhibited Hsp40-activated Hsc70 ATP hydrolysis (Ki ~ 10 μM) to reduce C-terminal Hsc70-peptide binding and, potentially, chaperone function. ERAD of misfolded ΔF508 CFTR requires Hsc70-Hsp40 chaperones. In transfected baby hamster kidney (BHK) cells, adaSGC increased ΔF508CFTR ERAD escape, and after low-temperature glycerol rescue, maturation, and iodide efflux. Inhibition of SGC biosynthesis reduced ΔF508CFTR but not wtCFTR expression, whereas depletion of other glycosphingolipids had no affect. WtCFTR transfected BHK cells showed increased SGC synthesis compared with ΔF508CFTR/mock-transfected cells. Partial rescue of ΔF508CFTR by low-temperature glycerol increased SGC synthesis. AdaSGC also increased cellular endogenous SGC levels. SGC in the lung, liver, and kidney was severely depleted in ΔF508CFTR compared with wtCFTR mice, suggesting a role for CFTR in SGC biosynthesis.
INTRODUCTION
The cystic fibrosis transmembrane regulator (CFTR) is a multi-spanning membrane ATP-regulated chloride channel (Riordan et al., 1989). Defects in the activity and cell surface trafficking of CFTR result in the loss of chloride transport and the development of cystic fibrosis (CF). Although many mutations in CFTR have been described in CF patients, the ΔF508CFTR mutation is of particular interest because this is the most common CF mutation and the protein retains chloride transport activity (Denning et al., 1992). Cell surface trafficking and stability are impaired due to misfolding and subsequent degradation by the cellular quality control machinery. It is speculated that interventive procedures that allow the cell surface trafficking of ΔF508CFTR will be sufficient to ameliorate the CF phenotype (Farmen et al., 2005; Amaral, 2005). Hsp70-related chaperones are an important component of the endoplasmic reticulum associated degradation (ERAD) quality control mechanism responsible for the elimination of misfolded proteins such as ΔF508CFTR (Brodsky, 2001). In addition, a significant fraction of wild-type (wt) CFTR is degraded by the ERAD pathway (Lukacs et al., 1994). Hsp70 chaperone function is primarily involved in nascent polypeptide folding, whereas the constitutive homolog, Hsc70, appears to play a more significant role in ERAD (Hohfeld et al., 2001). It has been shown that wtCFTR and ΔF508CFTR are degraded by the ubiquitin-proteasome pathway (Ward et al., 1995). Other studies have implicated members of the Hsp70 family in the cell surface turnover of CFTR (and other proteins) via the ubiquitination pathway (Zhang et al., 2001). Members of the Hsp70 family are also prominently induced following ΔF508CFTR expression (Singh et al., 2006; Xu et al., 2006).
Our laboratory has been interested in the cell surface receptor function of sulfogalactosyl ceramide (SGC) and has shown that members of the Hsp70 chaperone family specifically bind this glycosphingolipid (GSL) (Mamelak et al., 2001a) within the N-terminal ATPase domain (Mamelak and Lingwood, 2001), such that cell surface Hsp70s primarily expressed on bacterial pathogens can mediate binding to host cell SGC (Lingwood et al., 1990; Huesca et al., 1996; Hartmann et al., 2001). AdamantylSGC (adaSGC) was developed as a water-soluble mimic of membrane SGC (Mamelak et al., 2001b), which retained receptor function. The fatty acid of SGC is replaced with an adamantane frame. This results in little change in hydrophobicity but a large increase in water partitioning (Whetstone and Lingwood, 2003).
AdaSGC (or SGC) binding to the Hsc70 ATPase domain was shown to inhibit Hsc70 ATPase activity (Whetstone and Lingwood, 2003). AdaSGC retains membrane permeability and was considered, therefore, as a potential inhibitor of cellular Hsc70-mediated chaperone function. Hsc70 inhibition has been found to augment ΔF508 ERAD escape and cell surface expression (Rubenstein et al., 1997; Rubenstein and Zeitlin, 2000).
AdaSGC might, therefore, modulate ΔF508CFTR expression by inhibiting the chaperone function of Hsp70 family members. We also investigated endogenous SGC expression in wtCFTR and ΔF508CFTR mutant cells and mice. Our results indicate that AdaSGC increased ΔF508CFTR degradation escape to augment low-temperature/glycerol maturation of ΔF508CFTR. SGC expression is increased in cells and mice expressing wtCFTR.
RESULTS
AdaSGC Inhibits Hsc70 ATPase Activity
The soluble analog of SGC, adaSGC (Figure 1C), inhibits bovine Hsc70 ATPase activity in steady-state assays (Whetstone and Lingwood, 2003). To specifically monitor the effects of adaSGC on the ATP hydrolytic step, single-turnover assays were performed using yeast Hsc70 (Ssa1p) in the presence or absence of J domain containing SV40 T antigen (TAg). In this case, the TAg was used as an Hsp40 mimic (i.e., a protein containing a J domain), as previously described (Srinivasan et al., 1997; Fewell et al., 2004). As expected, TAg stimulated Hsc70 ATPase activity. When 300 μM adaSGC was added in the presence of TAg, ATPase activity was reduced 2–4-fold. We found that the inhibition of ATPase activity for the ATP preloaded chaperone in the presence of 300 μM adaSGC was dependent on the presence of Hsp40 (Figure 1A). No inhibition of endogenous Hsc70 ATPase activity in the absence of TAg was observed. Within this single ATPase cycle, 70% of ADP formation was prevented by adaSGC. The dose response for Hsp40-activated Hsc70 ATPase inhibition (Figure 1B) showed a Ki of ~10 μM for adaSGC.
Figure 1. AdamantylSGC Inhibition of Yeast Hsc70 ATPase Activity-Single Turnover Assay.

(A) Yeast Hsc70 (Ssa1p) was preloaded with [32P] ATP ± SV40 T antigen (TAg, J-domain-containing protein) on ice and 32P release from the purified complex ± adaSGC (300 μM added at 50 s) monitored with incubation at 30°C. Means of triplicate determinations ± standard error are shown. ○ Yeast Hsc70 (Ssa1p); ● Yeast Hsc70 (Ssa1p) + 300 μM adaSGC; □ Yeast Hsc70 (Ssa1p) + TAg; ■Yeast Hsc70 (Ssap1) + TAg + 300 μM adaSGC.
(B) Inhibition of T antigen stimulated Hsc70 single-turnover ATPase activity was monitored as a function of adaSGC concentration.
(C) Structure of adamantylSGC (adaSGC).
Inhibition of Hsc70 Peptide Binding by AdaSGC
Hsp70 and Hsc70 chaperones bind and release peptide substrates concomitant with cycles of ATP binding/hydrolysis, and this activity is essential for their function. Therefore, to investigate whether adaSGC could inhibit Hsc70-peptide binding, 125I-carboxylmethylated α-lactalbumin (CMLA), a permanently unfolded Hsc70 chaperone substrate (McClellan et al., 1998), and Hsc70 were incubated in the presence or absence of 100 μM adaSGC (Figure 2). CMLA-Hsc70 complexes were detected (Figure 2, lanes 3 and 4), but the presence of 100 μM adaSGC largely prevented the formation of these CMLA-Hsc70 complexes, irrespective of the presence of Hsp40 (Figure 2, lanes 5 and 6). This result suggests that adaSGC inhibits Hsc70-peptide complex formation or increases peptide dissociation in vitro.
Figure 2. AdamantylSGC Inhibits Hsc70-Peptide Binding.

Yeast Hsc70 (Ssa1p) was incubated ± Hsp40 (Ydj1p), ± 100 μM adaSGC in the presence of 125I-CMLA, a permanently unfolded, polypeptide substrate for Hsp70s. The complex was separated by nondenaturing gel electrophoresis and detected by autoradiography. AdaSGC prevented 125I-CMLA-Hsc70 complex formation both in the presence and absence of Hsp40.
Effect of AdamantylSGC on ΔF508CFTR Expression
ΔF508CFTR, the most common disease-causing CFTR mutation, is subject to ERAD (Ward et al., 1995). A major step during the ERAD of misfolded ΔF508CFTR might be the prolonged binding to chaperone(s). This, in turn, might trigger subsequent protein ubiquitination (Ward et al., 1995). Because adaSGC inhibits Hsc70 ATPase activity and Hsc70-CMLA complex formation, we hypothesized that ΔF508CFTR might be rescued from ERAD by adaSGC. Treatment of wtCFTR cells with 50 μM adaSGC showed no effect, but adaSGC treatment had a selective augmentary effect on the levels of ΔF508CFTR. An increase in the immature form (band b) of ΔF508CFTR was observed (Figure 3A), suggesting increased escape from ERAD. Increased maturation, however, was not observed for ΔF508CFTR. Only when adaSGC treated cells were rescued by low-temperature (26°C–27°C) 10% glycerol treatment for 24 hr was a major increase in the level of mature, lactosamine-glycosylated ΔF508CFTR observed. Dose-dependent experiments showed that up to 100 μM adaSGC could increase the level of fully glycosylated ΔF508CFTR under rescuing conditions (Figure 3B), with maximum effect between 25 and 50 μM. The levels of both immature and mature ΔF508CFTR were elevated as compared with nontreated cells. Immunoprecipitation with antihemagglutinin followed by immunoblot with anti-CFTR also showed the increased expression of fully glycosylated ΔF508CFTR (band c) by adaSGC (Figure 3C). No effect on wtCFTR cells was evident.
Figure 3. AdamantylSGC Promotes ΔF508CFTR ERAD Escape.

(A) WtCFTR and ΔF508CFTR-expressing BHK cells were cultured with 50 μM adaSGC for 48 hr. ΔF508CFTR BHK cells were rescued under low-temperature (26°C–27°C) 10% glycerol. ΔF508CFTR expression was monitored by western blot (ΔF508). AdaSGC treatment enhances ΔF508CFTR expression under rescuing condition (ΔF508R). One of four similar experiments is shown.
(B) Concentration dependent rescue effect of adaSGC. Up to 100 μM adaSGC treatment increased the fully glycosylated, mature form (band c) of ΔF508CFTR.
(C) Immunoprecipitation (IP) of rescued ΔF508CFTR BHK cells treated with 50 μm adaSGC. IP was performed with anti-HA and then an immunoblot was performed using anti-CFTR. IP assay confirmed the increased ΔF508CFTR degradation escape by adaSGC treatment.
(D) Comparison of iodide efflux in rescued ΔF508CFTR BHK cells treated with or without 50 μM adaSGC. AdaSGC treatment markedly increases iodide efflux, suggesting increased functional ΔF508CFTR in the plasma membrane. Mean of triplicate determinations ± standard error are shown.
AdamantylSGC Increases the Level of Iodide Efflux in ΔF508CFTR-Transfected Cells
To confirm that rescued ΔF508CFTR is functional at the plasma membrane, an iodide efflux assay was performed. Consistent with the effect on ΔF508CFTR expression levels, adaSGC treatment of ΔF508CFTR-transfected cells alone had no effect on iodine efflux (data not shown). However, the treatment of ΔF508CFTR-transfected cells with adaSGC under low-temperature glycerol treatment significantly stimulated iodide efflux, confirming the increased level of functional plasma membrane ΔF508CFTR in these cells (Figure 3D).
Effect of CFTR Transfection on SGC Synthesis
Based on our findings that both SGC and adaSGC bind to Hsc70 and inhibit Hsc70 ATPase activity to reduce peptide binding, we compared SGC expression in wtCFTR and ΔF508CFTR-expressing cells. BHK cells stably transfected with the gene encoding hemagglutinin (HA)-tagged wtCFTR or HA-tagged ΔF508CFTR were used. The SGC content was assessed by GSL extraction and thin-layer chromatography (TLC) immunostaining using monoclonal anti-SGC (Fredman et al., 1988). SGC was barely detectable in mock-transfected cells but markedly increased in wtCFTR-transfected cells (Figure 4A). This increase was not observed for ΔF508CFTR-transfected cells (Figure 4A). However, when such cells were “rescued” by low-temperature glycerol conditions established to increase the maturation and plasma membrane chloride transport by ΔF508CFTR, SGC synthesis was elevated, similar to wtCFTR-expressing cells (Figure 4A). The difference in SGC expression between wtCFTR and ΔF508CFTR BHK cells was confirmed by immunofluorescence staining using anti-SGC antibody (Figure 4B). The staining of permeabilized cells showed extensive perinuclear SGC, consistent with ER and thereby appropriately located to affect ERAD.
Figure 4. Functional CFTR Expression Correlates with SGC Expression in BHK Cell Transfectants and IB3/S9 Cells.

(A) BHK cells transfected with HA-tagged wtCFTR or ΔF508CFTR were tested for SGC content by anti-SGC-TLC immunostaining of total lipid extract. ΔF508CFTR cells were also tested after low-temperature (26°C–27°C) 10% glycerol rescue. SGC content was increased after wtCFTR transfection and after rescue of ΔF508CFTR-transfected cells (presumably due to ΔF508CFTR maturation). IB3 cells (CF) contain lower levels of SGC than S9 cells (wtCFTR corrected IB3). For upper panel TLC, ECL was used for detection. For lower panel TLC, 4-chloro-1-naphthol and hydrogen peroxide were used to detect HRP-conjugated goat anti-mouse IgG.
(B) Immunofluorescence staining of SGC expression in wtCFTR and ΔF508CFTR BHK cells. Cells were fixed, permeabilized, and then stained with anti-SGC. SGC expression, particularly around the nucleus, is increased in wtCFTR-expressing cells.
GSL Modulation of CFTR Expression
The change of SGC expression in wild-type, ΔF508CFTR, and rescued ΔF508CFTR suggests a possible relationship between CFTR and SGC levels. To further examine this possibility, SGC synthesis was modulated and the level of HA-tagged wtCFTR and ΔF508CFTR in the transfected BHK cells was monitored by western blot. Incubation of cells with the glucosyl ceramide inhibitor P4 for 10 days had virtually no effect on wtCFTR or ΔF508CFTR expression (Figure 5A). P4 depletes glucosyl ceramide-based GSLs (~95% of total GSLs). The synthesis of SGC and its precursor, galactosyl ceramide, are unaffected (Emam et al., 2006). Cell treatment with sodium chlorate, a general sulfation inhibitor that depletes SGC, showed a selective inhibitory effect on ΔF508CFTR expression (Figure 5B). To prevent total GSL synthesis, including SGC, ceramide synthase was inhibited with fumonisin B1 (Soriano et al., 2005). Fumonisin B1 treatment slightly increased the fully glycosylated form of ΔF508CFTR, but, like sodium chlorate, decreased ΔF508CFTR after rescue (Figure 5C). These GSL inhibitor experiments indicate wtCFTR is unaffected by GSL depletion and that only changes in SGC levels affect ΔF508CFTR expression, particularly during rescue. Thus, decreased level of SGC could be one of the consequences of, and contribute to, the cellular phenomena associated with CF.
Figure 5. Effect of Inhibitors of GSL Synthesis on wtCFTR and ΔF508CFTR Expression.

Cells were treated with 2 μM P4 for 10 days, or 20 μM fumonisin B1 for 48 hr, or 30 mM sodium chlorate for 48 hr. Western blots were performed to analyze any changes in immature form (band b) and fully glycosylated mature form (band c) of wtCFTR and ΔF508CFTR. ΔF508R (rescued) represents that ΔF508CFTR cells were incubated under 26°C–27°C with 10% glycerol for 24 hr to rescue the protein and recovered for 3 hr at 37°C. Densitometric results using Image J are plotted to show relative levels of bands b and c.
(A) P4 is a glucosyl ceramide synthase inhibitor that prevents the synthesis of most GSLs except those based on galactosyl ceramide such as SGC. P4 inhibition of GSL synthesis has no effect on ΔF508CFTR expression.
(B) Sodium chlorate is a sulfation inhibitor and inhibits SGC synthesis. Incubation with 30mM sodium chlorate for 48hrs reduces ΔF508CFTR expression.
(C) Fumonisin B1 is an inhibitor of ceramide synthase and prevents the synthesis of all GSLs. Fumonisin B1 slightly increases ΔF508CFTR expression but reduced rescue efficacy.
The effect of adaSGC on BHK cell SGC metabolism was assessed. Treatment of cells with 50 μM adaSGC for 48hr resulted in a significant increase in endogenous SGC. In addition, a species corresponding to sulfolactosyl ceramide, immunoreactive with anti-SGC, was now detected (Figure 6). In contrast, SGC was undetectable after cell treatment with sodium chlorate. Thus, adaSGC can enter cells to modify endogenous sulfoglycolipid metabolism. Detection of sulfolactosyl ceramide after treatment implies adaSGC inhibition of desulfation as a mechanism of SGC accumulation.
Figure 6. Effect of AdaSGC on SGC Metabolism.
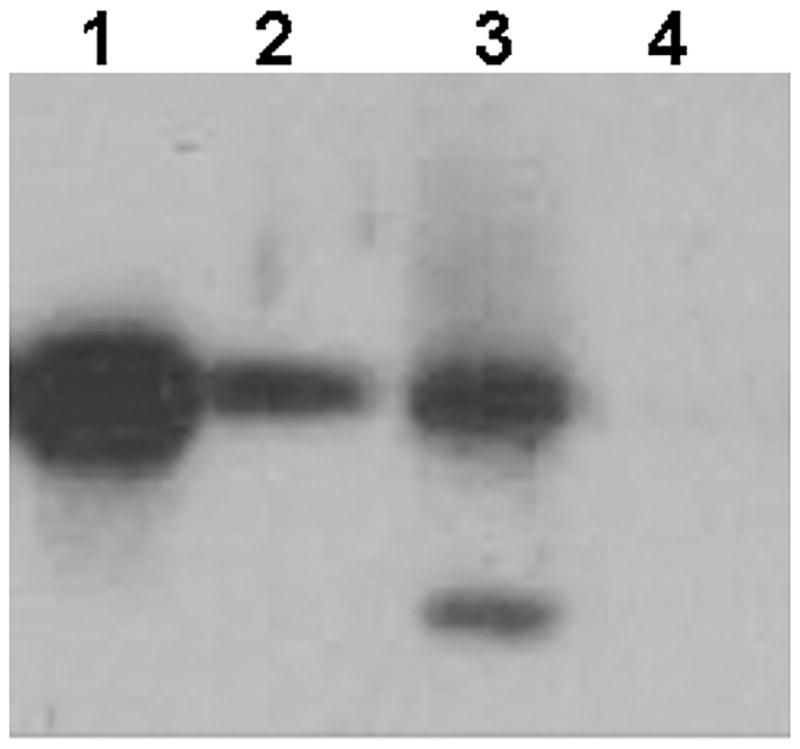
WtCFTR-transfected BHK cells were cultured for 48 hr in the presence of 50 μM adaSGC or 30 mM sodium chlorate and the SGC content of the lipid extract determined by anti-SGC-TLC immunostaining. Lane 1, SGC standard; lane 2, untreated cells; lane 3, adaSGC-treated cells; lane 4, sodium-chlorate-treated cells. AdaSGC treatment increased SGC and a new, more slowly migrating species corresponding to sulfolactosyl ceramide was immunodetected. SGC was undetectable in sodium-chlorate-treated cells.
Effect of CFTR on GSL Expression in Mouse Tissues
Our studies on CFTR-transfected cells indicated that functional CFTR can stimulate SGC synthesis. A reduced SGC content could therefore be a consequence of, and contribute to, the phenotype in CF cells. To begin to study the effect of CFTR on GSL synthesis in general and on SGC in particular, we compared the SGC content of several tissues of ΔF508CFTR transgenic and CFTR wild-type mice. Figure 7 shows that lung tissues in ΔF508CFTR-transgenic mice express very low levels of SGC as assessed by TLC immunostaining compared with wild-type mice (Figure 7A). SGC is also a significant component of mouse gastrointestinal epithelial cells and kidney cells. Similar to lung, in the ΔF508CFTR mice, reduced level of SGC in the ileum and kidney is seen by immunocytochemistry (Figure 7B). In the ileal sections, SGC is expressed in the villus crypts from normal mice. SGC is a major GSL in the mouse kidney (Lingwood et al., 1981), but ΔF508CFTR-transgenic mouse kidney showed no SGC. These results showed that there are dramatic changes of SGC expression in CFTR defective mice.
Figure 7. SGC Content of Wild-Type and ΔF508CFTR Mouse Tissues.

(A) TLC immunostaining. Total lipids were extracted from lung. Anti-SGC was used to detect SGC levels. Bovine SGC was used as a standard. Total lipid was assessed by iodine staining.
(B) Tissue immunohistochemistry. Frozen kidney and ileum sections from wild-type (WT) and ΔF508CFTR mice were stained with anti-SGC. SGC is found in WT ileal crypts and villus tips (arrows) but ΔF508CFTR ileum showed little SGC expression. In WT kidney, most tubules but no glomeruli expressed SGC. No SGC was detected in ΔF508CFTR kidney. Two or three animals were tested in each group.
DISCUSSION
These studies were initiated from our work showing that the Hsp70 chaperone family members specifically bind SGC (Mamelak et al., 2001a) and that the SGC binding site was within the N-terminal ATPase domain (Mamelak and Lingwood, 2001). Our initial studies suggested that the endogenous, steady-state ATPase activity of Hsc70 was inhibited by this compound. Interestingly, the results presented in this report indicate that adaSGC only inhibited the Hsp40-stimulated ATPase activity of the chaperone, as assessed in single-turnover assays. The simplest interpretation of these observations is that the KCAT for ATP hydrolysis is unaffected by adaSGC (which is measured in single-turnover studies) but that an alternative step (such as nucleotide binding or release) is impacted by the lipid. Moreover, unlike other inhibitors of Hsc70 ATPase activity (Fewell et al., 2001), adaSGC prevents peptide binding, which occurs in the C-terminal domain of Hsp70s. We therefore believe that adaSGC association with the N-terminal domain directly hinders Hsp40 binding (which also occurs in the ATPase domain). In addition, adaSGC most likely impacts a conformational cascade that is required to “open” the peptide-binding site, an event that is known to require ATP binding.
The Hsc70 cognate examined in these studies is a member of the Hsp70 family and is involved in the ERAD of misfolded proteins such as ΔF508CFTR (Meacham et al., 2001; Arndt et al., 2005). Prevention of ΔF508CFTR ERAD degradation is a major focus of CF research because a relatively minor increase in ΔF508CFTR maturation and cell surface expression is considered of potential therapeutic benefit (Farmen et al., 2005). Indeed, several studies examining inhibition of Hsc70 have been carried out. These have been met with variable success. Deoxyspergualin (DSG) is an Hsc70 inhibitor (Nadler et al., 1992), effective to partially correct ΔF508CFTR in vitro (Jiang et al., 1998), but Farinha et al. (2002) reported that DSG only slightly stabilizes the immature form of wtCFTR. Although used widely as an immunosuppressant, clinical studies in CF have yet to be reported. Sodium phenyl butyrate (4-PBA) reduces Hsc70 expression in cell culture to promote ΔF508CFTR maturation (Rubenstein et al., 1997; Rubenstein and Lyons, 2001), but the effect on chloride transport has been questioned (Loffing et al., 1999) and the increased instability of ΔF508 by 4-PBA was also reported (Farinha et al., 2002). 4-PBA treatments have yielded initial encouraging results in vivo (Rubenstein and Zeitlin, 1998; Zeitlin et al., 2002), but the selectivity of action is broad (Singh et al., 2006; Wright et al., 2004) and early clinical studies have not been followed up. Finally, improved short chain fatty acid analogs have been reported to be effective in vitro (Nguyen et al., 2006), but have yet to be tested in vivo. We were unable to demonstrate any change in ΔF508CFTR processing in transfected BHK cells in vitro using these butyric acid derivatives (data not shown).
Accumulating evidence indicates that wtCFTR and ΔF508CFTR are subjected to differential ERAD regulation (Farinha and Amaral, 2005; Brodsky, 2007). SGC depletion affects only ΔF508CFTR, not wtCFTR, supporting wtCFTR and ΔF508CFTR participation in different ERAD pathways. AdaSGC protection against ΔF508CFTR ERAD is consistent with, but does not prove, cellular Hsc70 inhibition.
Our studies infer that in both the BHK transfectants and CFTR-defective transgenic mice, CFTR plays a tissue selective role in regulating the biosynthesis of SGC (and other GLS). Hyperacidification of the Golgi and trans-Golgi network has been ascribed as the basis for altered glycosylation in CF (Poschet et al., 2002). Extrapolating from our studies indicating that the SGC mimic, adaSGC can attenuate Hsc70-mediated ERAD, endogenous SGC could be a component of the physiological mechanism by which cells can regulate Hsp70 family chaperone activity. In the case of CF associated with the ΔF508CFTR protein, this could be of double importance: the reduced SGC levels in CF tissue might compound the efficacy of ΔF508CFTR removal by ERAD. AdaSGC is membrane soluble and could be endocytosed and trafficked to the Golgi as other similar derivatized GSLs (Pagano, 2003). However, amphipathic adaSGC might also access cytosolic Hsc70, whereas SGC would be expected to interact only with Hsp70 family members within the ER. The adaSGC-increased endogenous SGC cell content might play a role in the ΔF508CFTR protection, but it remains to be shown whether CFTR-dependent changes in endogenous SGC levels are more than coincidental. The bulk of intracellular SGC is, appropriately, consistent with an ER location. CFTR can function as a nucleotide channel for membrane transport of adenosine 3′phosphate 5′-phosphosulfate (PAPS) (Pasyk and Foskett, 1997). Because PAPS is the universal sulfate donor used for SGC synthesis (Sakac et al., 1992), this provides a potential basis of the CFTR-regulated SGC synthesis we report.
Recent work highlighted the phenotypic similarity between CF and Niemann-Pick disease type C (White et al., 2004, 2007; Gentzsch et al., 2007), in that both show a defect in cholesterol metabolism. In many GSL-associated lysosomal storage diseases, a link between GSL and cholesterol trafficking has been established (Pagano, 2003). Defects in GSL metabolism result in altered intracellular trafficking of both GSL and cholesterol (Pagano et al., 2000). Similarly, defects in cholesterol metabolism, as in Niemann-Pick type C, result in aberrant GSL trafficking. Correction of the cholesterol defect also corrects GSL trafficking, and vice versa (Choudhury et al., 2002). Indeed, it has been proposed that the primary defect in Niemann-Pick type C is not in cholesterol but in GSL metabolism, and that cholesterol accumulation is a secondary effect (Gondre-Lewis et al., 2003). Thus, cholesterol and GSL homeostasis are functionally linked. If defects in CFTR affect cholesterol trafficking, we might expect changes in GSL in CF. Initial studies indicate that loss of CFTR has significant tissue-restricted effects on the GSL profile. Whether these turn out to be related to cholesterol (lipid raft) trafficking, as in Niemann-Pick type C, will be of interest. Thorough GSL analysis of the tissues of CFTR knockout and ΔF508CFTR mice is required. Our studies in progress indicate that not only are selective GSLs missing, but also alternative GSLs are made (data not shown). Recent work showed age-dependent ceramide accumulation in CFTR-deficient mice, implicating changes in glycosphingolipid homeostasis (Teichgraber et al., 2008). In our study, fumonisin B1, a ceramide synthase inhibitor, slightly increased the amount of fully mature CFTR in ΔF508CFTR cells. Fumonisin B1 treatment decreases not only SGC but also ceramide levels, which might favor the expression of the mature glycosylated form of ΔF508CFTR. Nevertheless, both fumonisin and sodium chlorate depletion of SGC inhibited low-temperature glycerol rescue of ΔF508CFTR, whereas depletion of other GSLs was without effect, implying a protective role for endogenous SGC.
The present studies have assessed SGC synthesis in relation to CFTR. The reduced renal SGC content of CFTR-deficient mice correlates with renal CFTR (and a renal-specific variant) expression. CFTR is in proximal and distal tubules but not glomeruli (Devuyst et al., 1996), which corresponds to the expression pattern of SGC (Trick et al., 1999). In addition, significant tissues-elective changes in other GSLs are also found in CFTR-defective mice (studies in progress) such that differential GSL biosynthesis might be a previously unrecognized phenotype of CF. Defects in CFTR have been associated with altered GSL content in terms of GM1 and gangliotetraosyl ceramide (Gg4) content. Defective CFTR-mediated change in Golgi pH was reported to block sialylation and hence decrease GM1 and increase asialoGM1 (Gg4) (Poschet et al., 2001). However, this is more complex, because Gg4 is not the precursor to GM1. CFTR might play additional roles in the regulation of GSL biosynthesis.
The efficacy of adaSGC to increase ΔF508CFTR degradation escape but only promote maturation/iodine efflux during low-temperature glycerol rescue suggests that ERAD escape alone is insufficient to promote ΔF508CFTR cell surface trafficking. Unknown additional signals within the ER must be necessary for the further anterograde trafficking of ΔF508CFTR.
Our study shows wtCFTR is associated with increased SGC synthesis, which might restrict Hsc70 chaperone activity. SGC depletion in ΔF508CFTR-expressing cells might increase ΔF508CFTR ERAD. Because adamantylSGC, a soluble SGC analog, can induce ΔF508CFTR ERAD escape, SGC or SGC analogs might augment therapeutic approaches for CF.
SIGNIFICANCE
These studies indicate that adamantylSGC inhibition of Hsc70 ATPase and peptide binding can reduce cellular Hsc70 chaperone function within ERAD, allowing partial ΔF508CFTR escape and increasing the potential for therapeutic rescue of this misfolded protein. Lower SGC levels in ΔF508CFTR cells might contribute to rapid ΔF508CFTR ER degradation.
EXPERIMENTAL PROCEDURES
Cell Culture
BHK cells (either mock or HA-tagged wtCFTR or ΔF508CFTR transfected) were a generous gift from Dr. G. Lukacs (McGill University, Montreal, Canada). Cells were maintained in DMEM/Ham’s F12 (Wisent) in 5% FBS, 1% antibiotics (10,000 μg streptomycin ml−1 and 10,000 IU penicillin; Wisent) and 250 μg methotrexate ml−1 (Sharma et al., 2001, 2004). The cystic fibrosis cell line IB3-1 (ΔF508/W1282X) and the S9 (IB3 cells transfected with wtCFTR) were maintained in LHC-8 serum free medium supplemented with 5% FBS and 1% antibiotics as described previously (Emam et al., 2006). AdamantylSGC was synthesized from SGC as previously described (Whetstone and Lingwood, 2003).
Single-Turnover Hsc70 ATPase Assay
Single-turnover ATPase assays employing a constitutively expressed form of the yeast Hsp70 chaperone, Ssa1p (Hsc70), and a 2-fold molar excess of a J domain-containing cochaperone, T antigen, were performed as described elsewhere (Fewell et al., 2004) in the absence or presence of increasing concentrations of adaSGC or 300 μM adaSGC.
Hsc70-Peptide Binding Assay
125I-labeled carboxymethylated α-lactalbumin (CMLA) was prepared and binding to the yeast Hsp70 chaperone, Ssa1p, was performed as previously described (McClellan et al., 1998; McClellan and Brodsky, 2000). Where indicated, adaSGC (100 μM final concentration) or an equivalent volume of water was preincubated during peptide-Ssa1p complex formation at 4°C in the presence or absence of an equimolar concentration of a J domain-containing Hsp40 from yeast, Ydj1p. CMLA/Hsc70 complexes were detected using non-denaturing gel electrophoresis by band shift assay.
AdamantylSGC Treatment and Modulation of GSL Synthesis
Cells were treated with 25 μM or 50 μM adaSGC, unless otherwise stated, for 48 hr. To “rescue” ΔF508CFTR, one set of ΔF508CFTR cells were incubated at 26°C–27°C/10% glycerol (Denning et al., 1992; Brown et al., 1996) for another 24 hr after 1 day incubation with adaSGC. Cells were then washed thoroughly and recovered for 3 hr at 37°C. For inhibition of GSL biosynthesis, cells were treated with 20 μM fumonisin B1 (Sigma) or 30 mM sodium chlorate (Sigma) for 48 hr or 2 μM P4 (1-phenyl-2-palmitoylamino-3-pyrrolidino-1-propanol, a gift from Dr. Shayman (University of Michigan) for 10 days (Lee et al., 1999). After the treatment, either total lipid or cell lysates were prepared for further analysis.
GSL Extraction
GSLs were extracted from exponentially growing cells. The cells were pelleted by centrifugation at 1000 × g for 5 min. The GSLs were extracted by Folch partition (chloroform: methanol: water, 2:1:0.6, v/v/v). For tissue extraction, tissues were homogenized in phosphate-buffered saline (PBS) and extracted overnight with 20 volumes of chloroform: methanol (2:1, v/v). The suspension was filtered by glasswool column and partitioned by the Folch method. The lower phase was collected and dried down under a mild N2 gas stream. Samples were saponified with 1 N NaOH in methanol for 2 hr at 37°C or overnight at room temperature (RT). Five milliliters of water was added, and the mixture applied onto a C-18 column (Sep-Pak Plus C-18; Waters). The column was washed with water, and glycolipids were eluted with methanol, dried and resuspended in chloroform:methanol (2:1, v/v).
TLC Immunostaining
Equivalent GSL aliquots, based on cell number, were separated by TLC and plates were dried and blocked with 1% bovine serum albumin (BSA) in TBS (50 mM Tris, 154 mM NaCl [pH 7.4]) at RT for 1 hr. Plates were incubated with monoclonal anti-SGC, SULF-1, which was a gift from Dr. P Fredman (Göteborg University, Sweden) (Fredman et al., 1988). After three washes with TBS, plates were incubated with peroxidase-conjugated goat anti-mouse antibody. Antibody binding was visualized with enhanced chemiluminescent system (ECL, Pierce) or 4-chloro-1-naphthol (Sigma; 3 mg/ml methanol) and hydrogen peroxide (Nutikka et al., 2003).
Western Blot and Immunoprecipitation
Cells were grown on six-well plates and treated with 25 μMor 50 μM adaSGC, unless otherwise stated, for 48 hr. To rescue ΔF508CFTR, one set of ΔF508CFTR cells was incubated at 26°C–27°C/10% glycerol (Denning et al., 1992; Brown et al., 1996) for another 24 hr after 1 day incubation with adaSGC. Cells were then washed thoroughly and recovered for 3 hr at 37°C. For inhibition of GSL biosynthesis, cells were treated with 20 μMfumonisin B1 (Sigma) or 30 mM sodium chlorate (Sigma) for 48 hr or 2 μM P4 (1-phenyl-2-palmitoylamino-3-pyrrolidino-1-propanol, a gift from Dr. Shayman) for 10 days (Lee et al., 1999). Cells were lysed with TNTE lysis buffer (10 mM Tris [pH 7.4], 154 mM NaCl, 1mM EDTA, 1% Triton X-100, protease inhibitor cocktail; Sigma) and incubated for 30 min at 4°C in a shaker. Samples were centrifuged at 12,000 × g for 20 min at 4°C and the supernatant was collected. Protein concentration was quantified with BCA method (Pierce). Equal amounts (50 μg) of total protein were analyzed by 6% SDS-PAGE, unless otherwise stated. CFTR expression was detected using anti-HA (Covance) or anti-CFTR (Chemicon). For immunoprecipitation, 500 μg total lysate was incubated with anti-HAovernight. Immune complexes were captured with precleared recombinant ProteinA agarose (Exalpha). Beads were washed three times with lysis buffer without Triton X-100 and resuspended with 2X sample buffer. Proteins were detected with anti-CFTR. Control samples were incubated with isotype immunoglobin G (IgG) instead of anti-HA.
Immunofluorescence Microscopy
BHK cells were grown on glass coverslips for 24 hr. Cells were fixed with 4.0% paraformaldehyde for 15 min at 4°C and permeabilized with 0.2% Triton X-100 for 2 min at 20°C. Cells were blocked with PBS-10% FBS overnight at 4°C, incubated with 1:400 dilution of anti-SGC antibody. After three washes with PBS, the cells were incubated with TRICTC-labeled goat anti-mouse antibody for 1 hr. Fluorescent images were taken using a Zeiss LSM510 confocal microscope under a 63× oil-immersion objective.
Iodide Efflux Assay
Iodide efflux assay was performed as previously described (Mohamed et al., 1997). Briefly, cells grown in six-well culture dishes were incubated with loading buffer (136 mM NaI, 3mM KNO3, 2 mM Ca[NO3]2, 11 mM glucose, and 20 M HEPES [pH 7.4]) for 1 hr at RT. After washing away extracellular NaI thoroughly with efflux buffer containing 136 mM NaNO3, 1 ml efflux buffer was loaded three times for 1 min to establish a baseline. Efflux buffer containing agonists (10 μMforskolin [Sigma], 0.2mMCTP-cAMP [Sigma], and 0.2mM isobutylmethylxanthine [Sigma]) were added (time zero) for 1 min and collected. Three or four additional 1 ml efflux buffer aliquots containing agonists were incubated for 1 min and collected. The standard curve was generated with a known NaI solution each time and the amount of iodide in each sample was measured with an iodide-selective electrode (Orion).
ΔF508CFTR Mice
All mouse studies were approved by the animal care committee at Hospital for Sick Children, Toronto. Wild-type mice (C57Bl/6J, Cftr+/+) and matched littermate ΔF508 (Cftrtm1kth) mice were maintained on a liquid diet (Peptamen, Nestle Nutrition). At age 10 weeks, lung, ileum, and kidney were harvested. Tissues were either frozen and stored at −80°C until lipid extraction or embedded in OTC (Tissue Tek), frozen in liquid nitrogen, and stored at −80°C. Tissues were cryosectioned (5 μM) and stored at −80°C.
Immunohistochemistry
Serial 5 μM cryosections were thawed and dried overnight at RT. Procedures was performed with M.O.M.™ Peroxidase Kit according to manufacturer’s protocol except for a few modifications (Vector Laboratories). Throughout all incubation steps, slides were kept in a humidified chamber at RT. Briefly tissue sections were blocked with endogenous peroxidase blocker (Universal Block, KPL) for 30 min. After washing with PBS, tissues were incubated with Avidin solution for 15 min, followed by 15 min with biotin solution (avidin/biotin blocking kit, Vector Laboratories). Tissue sections were further blocked with M.O.M.™ IgG blocking solution. Slides were incubated with anti-SGC antibody (1:200) diluted with M.O.M.™ diluent for 30 min. After a wash with PBS, the slides were incubated with M.O.M.™ biotinylated anti-mouse IgG working solution for 10 min. Following the wash step, Vectastain ABC reagents were applied for 5 min. Sections were developed using DAB (3,3′-diaminobenzidine) substrates for 1–2 min. To stop the DAB reaction, sections were dipped in distilled water for 5 min. Hematoxylin counterstain was applied for 30 s and excess staining was removed by immersion in tap water for 10 min. Sections were dehydrated twice for 3 min in, sequentially, 70%, 95%, and 100% ethanol, cleared in xylene for 5 min, and mounted in Permount (Fisher Scientific). Images were taken using an Olympus BX60 microscope.
Acknowledgments
This work was supported by grants from the Sellers Foundation (to C.A.L.) and by Cystic Fibrosis Foundation Therapeutics grant BRODSK 8XX0 and by NIH grant GM 75061 (to J.L.B.).
References
- Amaral MD. Processing of CFTR: traversing the cellular maze–how much CFTR needs to go through to avoid cystic fibrosis? Pediatr Pulmonol. 2005;39:479–491. doi: 10.1002/ppul.20168. [DOI] [PubMed] [Google Scholar]
- Arndt V, Daniel C, Nastainczyk W, Alberti S, Hohfeld J. BAG-2 acts as an inhibitor of the chaperone-associated ubiquitin ligase CHIP. Mol Biol Cell. 2005;16:5891–5900. doi: 10.1091/mbc.E05-07-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL. Chaperoning the maturation of the cystic fibrosis transmembrane conductance regulator. Am J Physiol Lung Cell Mol Physiol. 2001;281:L39–L42. doi: 10.1152/ajplung.2001.281.1.L39. [DOI] [PubMed] [Google Scholar]
- Brodsky JL. The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation) Biochem J. 2007;404:353–363. doi: 10.1042/BJ20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Hong-Brown LQ, Biwersi J, Verkman AS, Welch WJ. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones. 1996;1:117–125. doi: 10.1379/1466-1268(1996)001<0117:ccctmp>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury A, Dominguez M, Puri V, Sharma DK, Narita K, Wheatley CL, Marks DL, Pagano RE. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J Clin Invest. 2002;109:1541–1550. doi: 10.1172/JCI15420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Devuyst O, Burrow CR, Schwiebert EM, Guggino WB, Wilson PD. Developmental regulation of CFTR expression during human nephrogenesis. Am J Physiol. 1996;271:F723–F735. doi: 10.1152/ajprenal.1996.271.3.F723. [DOI] [PubMed] [Google Scholar]
- Emam A, Yu A, Park HJ, Mahfoud R, Kus J, Burrows L, Lingwood C. Laboratory and clinical Pseudomonas aeruginosa strains do not bind glycosphingolipids in vitro or during type IV pili-mediated initial host cell attachment. Microbiology. 2006;152:2789–2799. doi: 10.1099/mic.0.28863-0. [DOI] [PubMed] [Google Scholar]
- Farinha CM, Amaral MD. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol Cell Biol. 2005;25:5242–5252. doi: 10.1128/MCB.25.12.5242-5252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha CM, Nogueira P, Mendes F, Penque D, Amaral MD. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem J. 2002;366:797–806. doi: 10.1042/BJ20011717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmen SL, Karp PH, Ng P, Palmer DJ, Koehler DR, Hu J, Beaudet AL, Zabner J, Welsh MJ. Gene transfer of CFTR to airway epithelia: low levels of expression are sufficient to correct Cl− transport and overexpression can generate basolateral CFTR. Am J Physiol Lung Cell Mol Physiol. 2005;289:L1123–L1130. doi: 10.1152/ajplung.00049.2005. [DOI] [PubMed] [Google Scholar]
- Fewell SW, Day BW, Brodsky JL. Identification of an inhibitor of hsc70-mediated protein translocation and ATP hydrolysis. J Biol Chem. 2001;276:910–914. doi: 10.1074/jbc.M008535200. [DOI] [PubMed] [Google Scholar]
- Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, Brodsky JL. Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J Biol Chem. 2004;279:51131–51140. doi: 10.1074/jbc.M404857200. [DOI] [PubMed] [Google Scholar]
- Fredman P, Mattsson L, Andersson K, Davidsson P, Ishizuka I, Jeansson S, Mansson JE, Svennerholm L. Characterization of the binding epitope of a monoclonal antibody to sulphatide. Biochem J. 1988;251:17–22. doi: 10.1042/bj2510017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch M, Choudhury A, Chang XB, Pagano RE, Riordan JR. Misassembled mutant ΔF508 CFTR in the distal secretory pathway alters cellular lipid trafficking. J Cell Sci. 2007;120:447–455. doi: 10.1242/jcs.03350. [DOI] [PubMed] [Google Scholar]
- Gondre-Lewis MC, McGlynn R, Walkley SU. Cholesterol accumulation in NPC1-deficient neurons is ganglioside dependent. Curr Biol. 2003;13:1324–1329. doi: 10.1016/s0960-9822(03)00531-1. [DOI] [PubMed] [Google Scholar]
- Hartmann E, Lingwood C, Reidl J. Heat inducible surface stress protein (Hsp70) mediates sulfatide recognition of the respiratory pathogen Haemophilus influenzae. Infect Immun. 2001;69:3438–3441. doi: 10.1128/IAI.69.5.3438-3441.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohfeld J, Cyr DM, Patterson C. From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep. 2001;2:885–890. doi: 10.1093/embo-reports/kve206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huesca M, Borgia S, Hoffman P, Lingwood CA. Acidic pH changes receptor binding of Helicobacter pylori: a binary adhesion model in which surface heat-shock (stress) proteins mediate sulfatide recognition in gastric colonization. Infect Immun. 1996;64:2643–2648. doi: 10.1128/iai.64.7.2643-2648.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Fang S, Xiao Y, O’Connor S, Nadler S, Lee D, Jefferson D, Kaplan J, Smith A, Cheng S. Partial restoration of cAMP-stimulated CFTR chloride channel activity in DeltaF508 cells by deoxyspergualin. Am J Physiol. 1998;275:C171–C178. doi: 10.1152/ajpcell.1998.275.1.C171. [DOI] [PubMed] [Google Scholar]
- Lee L, Abe A, Shayman JA. Improved inhibitors of glucosylceramide synthase. J Biol Chem. 1999;274:14662–14669. doi: 10.1074/jbc.274.21.14662. [DOI] [PubMed] [Google Scholar]
- Lingwood C, Hay G, Schachter H. Tissue distribution of sulfolipids in the rat. Restricted location of sulfogalactosylacylalkylglycerol. Can J Biochem. 1981;59:556–563. doi: 10.1139/o81-077. [DOI] [PubMed] [Google Scholar]
- Lingwood CA, Quinn PA, Wilansky S, Nutikka A, Ruhnke H, Miller RB. Common sulfoglycolipid receptor for mycoplasma involved in infertility. Biol Reprod. 1990;43:694–697. doi: 10.1095/biolreprod43.4.694. [DOI] [PubMed] [Google Scholar]
- Loffing J, Moyer BD, Reynolds D, Stanton BA. PBA increases CFTR expression but at high doses inhibits Cl(−) secretion in Calu-3 airway epithelial cells. Am J Physiol. 1999;277:L700–L708. doi: 10.1152/ajplung.1999.277.4.L700. [DOI] [PubMed] [Google Scholar]
- Lukacs G, Mohamed A, Kartner N, Chang X, Riordan J. Conformational maturation of CFTR but not it’s mutant counterpart (Δ508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994;13:6076–6086. doi: 10.1002/j.1460-2075.1994.tb06954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamelak D, Lingwood C. The ATPase domain of Hsp70 possesses a unique binding specificity for 3′sulfogalactolipids. J Biol Chem. 2001;276:449–456. doi: 10.1074/jbc.M006732200. [DOI] [PubMed] [Google Scholar]
- Mamelak D, Mylvaganam M, Whetstone H, Hartmann E, Lennarz W, Wyrick P, Raulston J, Han H, Hoffman P, Lingwood C. Hsp70s contain a specific sulfogalactolipid binding site. Differential aglycone influence on sulfogalactosyl ceramide binding by prokaryotic and eukaryotic hsp70 family members. Biochemistry. 2001a;40:3572–3582. doi: 10.1021/bi001643u. [DOI] [PubMed] [Google Scholar]
- Mamelak D, Mylvaganam M, Tanahashi E, Ito H, Ishida H, Kiso M, Lingwood C. The aglycone of sulfogalactolipids can alter the sulfate ester substitution position required for hsc70 recognition. Carbohydr Res. 2001b;335:91–100. doi: 10.1016/s0008-6215(01)00209-9. [DOI] [PubMed] [Google Scholar]
- McClellan AJ, Brodsky JL. Mutation of the ATP-binding pocket of SSA1 indicates that a functional interaction between Ssa1p and Ydj1p is required for post-translational translocation into the yeast endoplasmic reticulum. Genetics. 2000;156:501–512. doi: 10.1093/genetics/156.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan AJ, Endres JB, Vogel JP, Palazzi D, Rose MD, Brodsky JL. Specific molecular chaperone interactions and an ATP-dependent conformational change are required during posttranslational protein translocation into the yeast ER. Mol Biol Cell. 1998;9:3533–3545. doi: 10.1091/mbc.9.12.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham GC, Patterson P, Zhang W, Younger JM, Cyr DM. The hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol. 2001;3:100–105. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- Mohamed A, Ferguson D, Seibert FS, Cai HM, Kartner N, Grinstein S, Riordan JR, Lukacs GL. Functional expression and apical localization of the cystic fibrosis transmembrane conductance regulator in MDCK I cells. Biochem J. 1997;322:259–265. doi: 10.1042/bj3220259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler SG, Tepper MA, Schacter B, Mazzucco CE. Interaction of the immunosuppressant deoxyspergualin with a member of the Hsp70 family of heat shock proteins. Science. 1992;258:484–486. doi: 10.1126/science.1411548. [DOI] [PubMed] [Google Scholar]
- Nguyen TD, Kim US, Perrine SP. Novel short chain fatty acids restore chloride secretion in cystic fibrosis. Biochem Biophys Res Commun. 2006;342:245–252. doi: 10.1016/j.bbrc.2006.01.127. [DOI] [PubMed] [Google Scholar]
- Nutikka A, Binnington-Boyd B, Lingwood C. Methods for the identification of host receptors for Shiga toxin. In: Philpot D, Ebel F, editors. Methods in Molecular Medicine, Volume 73, E.coli Shiga Toxin Methods and Protocols. Totowa, NY: Humana Press; 2003. pp. 197–208. [DOI] [PubMed] [Google Scholar]
- Pagano RE. Endocytic trafficking of glycosphingolipids in sphingolipid storage diseases. Philos Trans R Soc Lond B Biol Sci. 2003;358:885–891. doi: 10.1098/rstb.2003.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano RE, Puri V, Dominguez M, Marks D. Membrane traffic in sphingolipid storage diseases. Traffic. 2000;1:807–815. doi: 10.1034/j.1600-0854.2000.011101.x. [DOI] [PubMed] [Google Scholar]
- Pasyk EA, Foskett JK. Cystic fibrosis transmembrane conductance regulator-associated ATP and adenosine 3′-phosphate 5′-phosphosulfate channels in endoplasmic reticulum and plasma membranes. J Biol Chem. 1997;272:7746–7751. doi: 10.1074/jbc.272.12.7746. [DOI] [PubMed] [Google Scholar]
- Poschet JF, Boucher JC, Tatterson L, Skidmore J, Van Dyke RW, Deretic V. Molecular basis for defective glycosylation and Pseudomonas pathogenesis in cystic fibrosis lung. Proc Natl Acad Sci USA. 2001;98:13972–13977. doi: 10.1073/pnas.241182598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poschet J, Perkett E, Deretic V. Hyperacidification in cystic fibrosis: links with lung disease and new prospects for treatment. Trends Mol Med. 2002;8:512–519. doi: 10.1016/s1471-4914(02)02414-0. [DOI] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Lyons BM. Sodium 4-phenylbutyrate downregulates Hsc70 expression by facilitating mRNA degradation. Am J Physiol Lung Cell Mol Physiol. 2001;281:L43–L51. doi: 10.1152/ajplung.2001.281.1.L43. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J Respir Crit Care Med. 1998;157:484–490. doi: 10.1164/ajrccm.157.2.9706088. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of DeltaF508-CFTR. Am J Physiol Cell Physiol. 2000;278:C259–C267. doi: 10.1152/ajpcell.2000.278.2.C259. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest. 1997;100:2457–2465. doi: 10.1172/JCI119788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakac D, Zachos M, Lingwood CA. Purification of the testicular galactolipid 3′ phosphoadenosine 5′ phosphosulfate sulfotransferase. J Biol Chem. 1992;267:1655–1659. [PubMed] [Google Scholar]
- Sharma M, Benharouga M, Hu W, Lukacs GL. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem. 2001;276:8942–8950. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- Sharma M, Pampinella F, Nemes C, Benharouga M, So J, Du K, Bache KG, Papsin B, Zerangue N, Stenmark H, Lukacs GL. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164:923–933. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh OV, Vij N, Mogayzel PJ, Jr, Jozwik C, Pollard HB, Zeitlin PL. Pharmacoproteomics of 4-phenylbutyrate-treated IB3-1 cystic fibrosis bronchial epithelial cells. J Proteome Res. 2006;5:562–571. doi: 10.1021/pr050319o. [DOI] [PubMed] [Google Scholar]
- Soriano JM, Gonzalez L, Catala AI. Mechanism of action of sphingolipids and their metabolites in the toxicity of fumonisin B1. Prog Lipid Res. 2005;44:345–356. doi: 10.1016/j.plipres.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, McClellan AJ, Vartikar J, Marks I, Cantalupo P, Li Y, Whyte P, Rundell K, Brodsky JL, Pipas JM. The aminoterminal transforming region of simian virus 40 large T and small t antigens functions as a J domain. Mol Cell Biol. 1997;17:4761–4773. doi: 10.1128/mcb.17.8.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, De Oliveira-Munding CC, van Heeckeren AM, Barr ML, von Kurthy G, Schmid KW, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. 2008;14:382–391. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- Trick D, Decker J, Groene HJ, Schulze M, Wiegandt H. Regional expression of sulfatides in rat kidney: immunohistochemical staining by use of monospecific polyclonal antibodies. Histochem Cell Biol. 1999;111:143–151. doi: 10.1007/s004180050344. [DOI] [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Whetstone D, Lingwood C. 3′Sulfogalactolipid binding specifically inhibits Hsp70 ATPase activity in vitro. Biochemistry. 2003;42:1611–1617. doi: 10.1021/bi026735t. [DOI] [PubMed] [Google Scholar]
- White NM, Corey DA, Kelley TJ. Mechanistic similarities between cultured cell models of cystic fibrosis and niemann-pick type C. Am J Respir Cell Mol Biol. 2004;31:538–543. doi: 10.1165/rcmb.2004-0117OC. [DOI] [PubMed] [Google Scholar]
- White NM, Jiang D, Burgess JD, Bederman IR, Previs SF, Kelley TJ. Altered cholesterol homeostasis in cultured and in vivo models of cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2007;292:L476–L486. doi: 10.1152/ajplung.00262.2006. [DOI] [PubMed] [Google Scholar]
- Wright JM, Zeitlin PL, Cebotaru L, Guggino SE, Guggino WB. Gene expression profile analysis of 4-phenylbutyrate treatment of IB3-1 bronchial epithelial cell line demonstrates a major influence on heatshock proteins. Physiol Genomics. 2004;16:204–211. doi: 10.1152/physiolgenomics.00160.2003. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu C, Clark JC, Whitsett JA. Functional genomic responses to cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR(delta508) in the lung. J Biol Chem. 2006;281:11279–11291. doi: 10.1074/jbc.M512072200. [DOI] [PubMed] [Google Scholar]
- Zeitlin PL, Diener-West M, Rubenstein RC, Boyle MP, Lee CK, Brass-Ernst L. Evidence of CFTR function in cystic fibrosis after systemic administration of 4-phenylbutyrate. Mol Ther. 2002;6:119–126. doi: 10.1006/mthe.2002.0639. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Nijbroek G, Sullivan ML, McCracken AA, Watkins SC, Michaelis S, Brodsky JL. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol Biol Cell. 2001;12:1303–1314. doi: 10.1091/mbc.12.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]