

Abstract
Background: Hypoglycaemic drugs that close the KATP channel have been tested in patients with permanent neonatal diabetes due to glucokinase mutations (PNDM-GCK). From the results obtained, it has been suggested that this treatment may be beneficial in patients carrying GCK mutations with mild kinetic defects. The aim of this study was to evaluate the kinetic analysis of glucokinase activity as a predictive factor for response to sulphonylureas in PNDM-GCK.
Methods: The clinical characteristics of two siblings with PNDM born to non-consanguineous parents are described. Mutation analysis of KCNJ11, INS and GCK genes was done by sequencing. A comprehensive functional characterisation of GCK mutation was undertaken. Glibenclamide treatment was assayed for 16 weeks in one child. Response to treatment was evaluated by means of fasting glycaemia, C-peptide and HbA1c levels.
Results: Compound heterozygous GCK mutations (p.Ile19Asn and p.Ser441Trp) were identified. Functional analysis of GCK(p.Ile19Asn) indicated that this mutant retained more than 70% of wild-type catalytic activity in vitro, with a slight increase of thermolability. This mutation did not impair the interaction with the glucokinase regulatory protein, and the enzymatic activity of the GCK(p.Ile19Asn) mutant is restored to wild-type levels in the presence of GCK allosteric activator LY2121260. However, glibenclamide treatment of the patient on a reduced dose of insulin did not reduce HbA1c levels, and C-peptide increased only very slightly.
Conclusion: Hypoglycaemic drugs acting on the KATP channel might not be useful in the treatment of PNDM-GCK, even in patients carrying GCK mutations with mild kinetic defects.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2014_383) contains supplementary material, which is available to authorized users.
Keywords: Glucokinase, Glucose sensor, Hypoglycaemic drugs, KATP channel, Neonatal diabetes, PNDM-GCK, Sulphonylurea receptor
Introduction
Inactivating mutations in both alleles of the glucokinase gene (GCK) are a cause of permanent neonatal diabetes mellitus (PNDM-GCK), which is generally characterised by onset within the first months of life, low birth weight and lifelong insulin therapy (Rubio-Cabezas et al. 2011). So far, 38 PNDM-GCK cases have been reported in 28 families (Njølstad et al. 2001; Njølstad et al. 2003; Porter et al. 2005; Turkkahraman et al. 2008; Rubio-Cabezas et al. 2008; Bennett et al. 2011; Wajda-Cuszlag et al. 2012; Durmaz et al. 2012; Raimondo et al. 2014). Most of them are homozygous due to consanguinity. Only two cases show compound heterozygous mutations (Njølstad et al. 2003; Wajda-Cuszlag et al. 2012). PNDM-GCK patients lack the glucose sensor that integrates blood glucose levels and insulin secretion, since glucokinase activity is drastically reduced or completely absent. The glycolytic enzyme glucokinase has a major role in the body glucose homeostasis. GCK activity controls the glycolytic flux necessary for insulin secretion in the pancreatic β-cells and also regulates glucose metabolism in the liver. The specific role of GCK results from the particular kinetic characteristics of this enzyme (i.e., low affinity and cooperativity for glucose) and its unique regulation through protein interactions with other protein partners (mainly the inhibitory glucokinase regulatory protein – GCKRP – in hepatocytes) (Matschinsky 2009).
Although insulin is the treatment of choice in patients with PNDM-GCK, hypoglycaemic drugs have been tried in some cases with varied results (Bakri et al. 2004; Turkkahraman et al. 2008; Bennett et al. 2011; Durmaz et al. 2012). It has been suggested that the relative success of these treatments depends on the functional severity of the GCK mutations carried out by the patients (Bakri et al. 2004; Turkkahraman et al. 2008). In this study, we aimed to test the hypothesis that functional in vitro GCK analysis can be used as a predictive factor for responsiveness of PNDM-GCK patients to sulphonylurea treatment. We describe two siblings affected by PNDM that bear compound heterozygous mutations in the GCK gene (p.Ile19Asn and p.Ser441Trp). We report the functional characterisation of mutation p.Ile19Asn and the clinical response of one of these patients to glibenclamide therapy. We found that while mutation p.Ile19Asn has mild effects on GCK kinetics, this patient did not respond to treatment. Thus, we provide evidence that sulphonylurea might not be useful in the treatment of PNDM-GCK even in patients with GCK mutations resulting in mild kinetic defects.
Case History and Glibenclamide Treatment
A male baby of Caucasian ancestry was born at 39 weeks’ gestation from non-consanguineous parents. Birth weight was 2,260 g. During pregnancy, his mother showed altered oral glucose tolerance test (OGTT) and was treated with diet. His father was not known to have diabetes. Within the first day of life, the patient presented with nausea, vomiting and hyperglycaemia (13.4 mmol/L). Treatment with insulin was initiated (0.25 U/kg/day). Serum C-peptide was 0.44 μg/L, and diabetes-associated autoantibodies were negative. One month later, the baby was referred to the Hospital Universitario La Fe. Baby’s weight was 3,100 g, glycaemia 19.2 mmol/L and plasma insulin 0.8 mg/L, with no acidosis or ketosis. After 3 months, a genetic test identified compound heterozygous GCK mutations. Biochemical analysis of parents revealed impaired fasting glucose. Mother’s fasting plasma glucose (FPG) was 7.5 mmol/L, HbA1c 6.4% and OGTT at 120 min 5.8 mmol/L. Father’s FPG was 7.9 mmol/L, HbA1c 6.2% and OGTT 7.1 mmol/L. Two years later, his sister was born at 39 weeks’ gestation by caesarean section due to poor foetal growth and oligohydramnios. Her birth weight was 2,350 g and plasma glucose 14.4 mmol/L. Requirements of insulin were 0.02–0.04 U/kg/h, but with large glucose level fluctuations.
At age of 4 years, the boy was admitted for a trial of glibenclamide therapy. Before the treatment, OGTT values were 7.8 and 19.4 mmol/L at 0 and 120 min, respectively, and C-peptide <0.1 μg/L. At that time he was treated with subcutaneous insulin (1U/kg/day) and HbA1c was 8.8%. Oral glibenclamide was initiated at 0.1 mg/kg/day split into two equal doses and increased gradually up to 0.75 mg/kg/day over a week. This value is within the same range as that used in a similar case (Turkkahraman et al. 2008) and above the median dose administered in patients with PNDM due to KATP channel mutations (http://www.diabetesgenes.org). Plasma biochemical analysis and liver function tests showed no side effects or hypoglycaemia. Insulin doses were gradually reduced until reaching a dose of 0.6 U/kg/day. C-peptide was measured at 1 and 2 months after initiating the treatment. Fasting capillary blood glucose levels were measured daily during the trial.
Methods
Genetic Studies
Genomic DNA was extracted by standard procedures. Exons and flanking introns of KCNJ11 (NM_000525.3), INS (NM_000207.2) and GCK (NM_000162.3) genes were amplified and sequenced (BigDye Terminator v3.1 Cycle Sequencing Kit; Applied Biosystems, Foster City, CA). PCR products were purified and run on ABI Prism Genetic Analyzer 3130.xl (Applied Biosystems).
Enzymatic Analysis of Wild-Type and Mutant GCK
Recombinant human wild-type β-cell GCK fused to glutathionyl-S-transferase (GST-GCK) was prepared as described previously (Galán et al. 2006). Mutation p.Ile19Asn was produced using the oligonucleotides 5′-gtagagcagaacctggcagagttccaactgcaggag-3′ and 5′-ctcctgcagttggaactctgccaggttctgctctac-3′ and checked by sequencing and digestion with PvuII. Determination of kinetic parameters and thermal inactivation were done as previously described (Galán et al. 2006). The relative activity index was normalised to 5 mmol/L basal blood glucose (Christesen et al. 2002). GCK activator LY2121260 (Elli Lilly and Co) was prepared as described (García-Herrero et al. 2012).
Statistical Analysis
Normality of distributions was tested by Shapiro–Wilk test. Normally distributed parameters were compared by two-tailed Student t-test in combination with Levene’s test for equality of variances. Mann–Whitney U test was used for nonnormally distributed variables. P < 0.05 was considered statistically significant.
Results
Identification of GCK Mutations
Sequence analysis of the proband did not reveal any mutation in KCNJ11 and INS genes. Two mutations, both in heterozygosis, were identified in the GCK gene: c.56T>A (p.Ile19Asn) in exon 2 and c.1322C>G (p.Ser441Trp) in exon 10. Both were previously described as cause of familiar hyperglycaemia (MODY-GCK) in the heterozygous state (Massa et al. 2001; Estalella et al. 2007). Mutation analysis confirmed that the father carried the MODY-GCK(p.Ser441Trp) mutation, the mother carried the MODY-GCK(p.Ile19Asn) mutation and the sister was also a compound heterozygous for both mutations.
Functional Characterisation of p.Ile19Asn Mutation
Kinetic analysis of the GCK(p.Ser441Trp) mutant, reported by Barbetti et al. (2009) and reflected in Table 1, showed low enzymatic activity compared to the wild-type GCK. However, no functional data was available for the p.Ile19Asn mutation. Kinetic parameters of recombinant wild-type GST-GCK and mutant GST-GCK(p.Ile19Asn) are shown in Table 1. Mutation p.Ile19Asn produced a slight but significant decrease in the catalytic constant, cooperativity and affinity for glucose (as shown by reduced Kcat and nH and increased S0.5 values, respectively) and a small increase in ATP affinity (reduced ATP Km value) that results in a slight reduction of the calculated relative activity index (Iar = 0.72; p = 0.073). We also observed that the GCK synthetic allosteric activator LY2121260 activates the mutant protein as much as the wild type (Table 1) and that this mutation does not impair the interaction of GCK with GCKRP in the two hybrid assay (supplementary Fig. 1). To ascertain whether protein instability could contribute to reduce activity of GCK(p.Ile19Asn), we tested the activity of the purified fusion proteins at different temperatures. Our results indicate that mutation p.Ile19Asn confers a mild thermal instability to the GCK protein (Fig. 1).
Table 1.
Kinetic parameters of wild-type GST-GCK and mutant GST-GCK(p.Ile19Asn) and effect of GCK activator LY2121260
| Protein | Yield (mg/L) (n = 3) | Kinetic parameters | Effect of LY2121260 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| K cat (s−1) | Glucose S0.5 (mmol/L) | n H | ATP Km (mmol/L) | I ar | Fold increased K cat | Fold decreased Glucose S0.5 | Fold decreased n H | Fold increased ATP Km | Fold increased I a | ||
| GST-GCK (n = 6) | 4.79 ± 0.73 | 51.29 ± 2.40 | 7.85 ± 0.68 | 1.53 ± 0.03 | 0.49 ± 0.04 | 1.00 ± 0.19 | 1.41 ± 0.09 | 5.09 ± 0.86 | 1.5 ± 0.14 | 1.54 ± 0.27 | 50 ± 16 |
| GST-GCK (p.Ile19Asn) (n = 6) | 3.79 ± 0.95 | 37.22 ± 3.1** | 10.68 ± 0.8* | 1.30 ± 0.08* | 0.33 ± 0.04* | 0.72 ± 0.28 | 1.34 ± 0.16 | 8.05 ± 1.44* | 1.4 ± 0.15 | 1.44 ± 0.50 | 78 ± 19 |
| WT/p.Ser441Trp (Barbetti et al. 2009) | 65.57 ± 7.19/20.72 ± 4.56 | 7.97 ± 0.31/15.89 ± 2.49 | 1.71 ± 0.07/1.38 ± 0.07 | 0.45 ± 0.05/1.12 ± 0.14 | 1/0.11 | ||||||
Activity data are shown as mean ± SD for six independent experiments (n = 6) using three separate enzyme preparations, for wild-type and mutant GST-GCK. The Km for ATP was measured at a glucose concentration of 7.5 and 11 mmol/L for wild type and mutant, respectively. The Hill coefficient (n H) and the relative activity index (I ar) are unit less. For comparison, means of GST-GSK wild-type and p.Ser441Trp mutant kinetic parameter data from Barbetti et al. (2009) are included in the table. To assay the effect of LY2121260, GCK activity was measured in the absence and presence of 10 μmol/L activator. Since LY2121260 was dissolved in a buffer containing 0.8% DMSO, all assays contained the same final concentration of DMSO. Fold variation has been calculated dividing values in the presence of LY2121260 by their corresponding values in its absence. (*) p < 0.005, Student’s t-test with Levene’s test for equality of variances. (**) p < 0.005, non-parametric Mann–Whitney U test
Fig. 1.
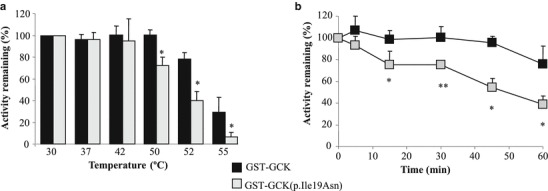
Assessment of thermostability for wild-type GST-GCK and mutant GST-GCK(p.Ile19Asn) proteins. Enzyme solutions were diluted to 250 mg/L as described in Galán et al. (2006). (a) The enzyme solution was incubated for 30 min at different temperatures ranging from 30 to 55°C and then assayed at 30°C as described in Methods. (b) The enzyme solution was incubated for different periods of time from 5 to 60 min at 50°C. After incubation, GCK activity was assayed at a glucose concentration of 100 mmol/L. Means and SD of six independent experiments (n = 6), from three enzyme preparations, are shown. (*) p < 0.005, Student’s t-test with Levene’s test for equality of variances. (**) p < 0.005, non-parametric Mann–Whitney U test
Glibenclamide Treatment
Insulin doses were reduced to 0.6 U/Kg/day but were increased back because HbA1c levels were rising (Table 2). After 1 and 2 months of treatment, C-peptide levels were of 0.33 and 0.20 μg/L, respectively. Insulin injections could not be stopped, and after 1 month, because of poor response and in order to avoid secondary effects, glibenclamide dose was reduced to 0.5 mg/kg/day and maintained 3 months more. After 16 weeks, the boy needed insulin doses similar to those dispensed previously. Then the treatment was discontinued, and patient was kept on continuous subcutaneous insulin infusion (1 U/kg/day), obtaining a better metabolic control, although glycaemia was not optimally controlled (HbA1c: 7.7%).
Table 2.
Glibenclamide treatment and blood biochemical levels
| Time of treatment | |||||
|---|---|---|---|---|---|
| Before treatment | 1 month | 2 months | 3 months | 4 months | |
| Insulin dose (U/kg/day) | 1 | 0.6 | 0.6 | 0.6 | 0.9 |
| glibenclamide dose (mg/kg/day) | – | 0.75 | 0.5 | 0.5 | 0.5 |
| C-peptide (μg/L) | < 0.1 | 0.33 | 0.2 | ND | ND |
| HbA1c (%) | 8.8 | 10.2 | 10 | 10 | 8.7 |
| FPG (mmol/L) | 7.8 | 12.3 | 14.8 | ND | ND |
| Fasting capillary blood glucose (mmol/L) | −4 w: 8.5 ± 4.9 | 1 w: 13.9 ± 2.6 | 5 w: 8.0 ± 3.2 | 9 w: 8.3 ± 4.8 | 13 w: 6.6 ± 2.5 |
| −3 w: 12.4 ± 6.7 | 2 w: 8.5 ± 2.7 | 6 w: 9.5 ± 2.7 | 10 w: 8.8 ± 3.6 | 14 w: 7.8 ± 3.0 | |
| −2 w: 9.4 ± 4.1 | 3 w: 7.1 ± 3.7 | 7 w: 10.7 ± 2.2 | 11 w: 9.8 ± 3.5 | 15 w: 9.3 ± 4.5 | |
| −1 w: 10.6 ± 2.8 | 4 w: 9.1 ± 3.2 | 8 w: 12.8 ± 2.8 | 12 w: 9.4 ± 3.8 | 16 w: 11.9 ± 4.2 | |
| Mean: 10.2 ± 4.8 | Mean: 9.7 ± 3.9 | Mean: 10.2 ± 3.1 | Mean: 8.9 ± 4.0 | Mean: 8.9 ± 4.0 | |
FPG fasting plasma glucose, ND not determined
Fasting capillary blood glucose data are means ± SD of one measurement per day, 7 days per week (w) of treatment
Discussion
Most neonatal diabetic patients with undetectable insulin secretion need lifelong insulin therapy. Insulin secretion by the pancreatic β-cell occurs in response to high blood glucose concentration, which is detected by the glucose sensor glucokinase. Increased glucokinase activity stimulates the glycolytic flux rate and increases ATP levels, which in turn induces the closure of the KATP channel, thus resulting in membrane depolarization, calcium influx and insulin release.
Hypoglycaemic drugs closing the KATP channel have been tested in PNDM-GCK patients. Insulin treatment was replaced by repaglinide in two patients, a homozygous (IV8+2/IV8+2) and a compound heterozygous (IV8+2/p.Gly264Ser), with a good response only in the latter (Bakri et al. 2004). Interestingly, mutant GCK(p.Gly264Ser) showed near normal kinetics (Njølstad et al. 2003). Glibenclamide was administered to homozygous patients for mutations p.Thr168Ala (Turkkahraman et al. 2008; Durmaz et al. 2012) and p.Gln98X (Bennett et al. 2011). Although sulphonylurea treatment resulted in HbA1c reduction and increased C-peptide, insulin administration could not be stopped. For such severe GCK mutations, it was proposed that the total absence of GCK activity could limit the ATP production to restore insulin secretion, even in conditions in which sulphonylureas would close the KATP channel (Turkkahraman et al. 2008). From these studies it was inferred that sulphonylurea treatment could be tried in PNDM patients with less severe GCK mutations.
Here we described two PNDM-GCK siblings carrying compound heterozygous mutations. Mutation p.Ser441Trp, inherited from the father, retains about 11% of wild-type GCK activity (Barbetti et al. 2009). Mutation p.Ile19Asn, inherited from the mother, has a much lighter effect on glucokinase kinetics. Although this mutation produces small but significant defects in the kinetic parameters, the mutant enzyme retains more than 70% of wild-type catalytic activity in vitro. Thus, our kinetic results suggest that these PNDM patients might keep a reduced but significant level of GCK activity in vivo. Moreover, the relative mild intrauterine growth retardation, shown by a birth weight above 2,200 g, suggests that insulin secretion during that period was not as severely impaired as in the majority of the PNDM-GCK, which is also consistent with the remaining GCK activity detected in vitro.
However, despite the apparent mild effect of mutation p.Ile19Asn on GCK kinetics, the patient did not respond to the glibenclamide treatment. It is known that GCK mutations may cause other defects that cannot be detected by kinetic assays in vitro and result in a stronger reduction of activity in vivo, such as protein instability or impairment of interaction with other cellular partners (Osbak et al. 2009). Actually, it has been recently reported that protein instability is a major determinant of the clinical severity of PNDM-GCK (Raimondo et al. 2014). However, it might not be a general rule since the homozygous p.Arg275Cys mutation, which confers protein instability but has no significant effect on GCK kinetics, causes a MODY-like clinical phenotype, but not PNDM (Negahdar et al. 2014). We found a very mild effect of mutation p.Ile19Asn on the thermal stability of GCK if compared to those caused by some other mutations previously described (Raimondo et al. 2014). Furthermore, mutation p.Ile19Asn does not appear to impair other regulatory mechanisms of GCK activity, such as allosteric regulation, as shown by the normal response of the mutant enzyme to allosteric activator LY2121260 or the interaction of GCK with GCKRP. From our results, we conclude that hypoglycaemic drugs acting on the KATP channel might not be useful in the treatment of PNDM-GCK even in patients carrying mutations that result in mild kinetic defects. Although sulphonylurea treatment enables KATP channel closure, the lack of glucose sensor integrity may still prevent normal insulin secretion and improvement of glycaemic control. Synthetic glucokinase activators are also being developed as potential antidiabetic drugs in the treatment of type 2 diabetes (Matschinsky 2013). We have found that the enzymatic activity of the GCK(p.Ile19Asn) mutant is restored to wild-type levels in the presence of the allosteric activator LY2121260 (Table 1). Therefore, this opens the possibility that GCK allosteric activators could provide a more physiological approach in the treatment of such PNDM-GCK cases.
Electronic Supplementary Material
Supplementary Fig. 1. Two hybrid interaction between GCK(p.Ile19Asn) and the human GCKRP. Two hybrid interactions of Gal4-binding domain–GCKRP with Gal4-activating domain–GCK were tested in S. cerevisiae strain Y187 as described previously (Galán et al. 2006). The plasmid encoding the human GCKRP fusion protein to the Gal4 binding domain (GBD) was constructed as follows: the human GCKRP cDNA was amplified by PCR from plasmid p-FLAG CTC-hGCKRP (Brocklehurst et al. 2004), using primers 5′-ggaattcatgccaggcacaaaacggtttc-3′ and 5′-gggatcctactgaacgtcaggctctaggatttc-3′ and inserted into EcoR1 and BamH1 sites of pGBKT7 (Clontech). The plasmid encoding the fusion of mutant GCK to the Gal4 activating domain (GAD) was derived from pACTII (Clontech) by inserting a BamHI-XhoI fragment from the GST-GCK(p.Ile19Asn) into the same sites of the polylinker. Filter lift assays were developed for 1 h. In each case, independent transformants were tested with similar results
Acknowledgements
This work was partially supported by the Instituto de Salud Carlos III grant PI10/00424.
Synopsis
This is the first report providing evidence that sulphonylureas might not be useful in the treatment of PNDM-GCK even in patients carrying glucokinase mutations that result in mild kinetic defects.
Compliance with Ethics Guidelines
Conflict of Interest
Josep Oriola, Francisca Moreno, Angel Gutiérrez-Nogués, Sara León, Carmen-María García-Herrero, Olivier Vincent and María-Angeles Navas declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible institutional committees on human experimentation and with Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients, or their parents, for being included in the study.
Animal Rights
This article does not contain any studies with animals performed by any of the authors.
Contributions of Individual Authors
Josep Oriola, Francisca Moreno and Maria-Angeles Navas conceived and designed the experiments. Francisca Moreno and Sara Leon collected the clinical information and conducted the clinical treatment. Josep Oriola, Angel Gutierrez-Nogués, Carmen-Maria Garcia-Herrero and Olivier Vincent performed genetics and biochemical experiments. Josep Oriola, Francisca Moreno, Angel Gutierrez-Nogués, Olivier Vincent and Maria-Angeles Navas analysed the data. Josep Oriola, Francisca Moreno, Olivier Vincent and Maria-Angeles Navas wrote the manuscript.
All authors contributed to and have approved the final manuscript.
Footnotes
Competing interests: None declared
Francisca Moreno and Angel Gutiérrez-Nogués have contributed equally to this work.
Contributor Information
María-Angeles Navas, Email: manavas@med.ucm.es.
Collaborators: Johannes Zschocke
References
- Bakri D, Gershoni-Baruch R, Shehadeh N. Permanent neonatal diabetes. Isr Med Assoc J. 2004;6:290–291. [PubMed] [Google Scholar]
- Barbetti F, Cobo-Vuilleumier N, Dionisi-Vici C, et al. Opposite clinical phenotypes of glucokinase disease: description of a novel activating mutation and contiguous inactivating mutations in human glucokinase (GCK) gene. Mol Endocrinol. 2009;23:1983–1989. doi: 10.1210/me.2009-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett K, James C, Mutair A, Al-Shaikh H, Sinani A, Hussain K. Four novel cases of permanent neonatal diabetes mellitus caused by homozygous mutations in the glucokinase gene. Pediatr Diabetes. 2011;12:192–196. doi: 10.1111/j.1399-5448.2010.00683.x. [DOI] [PubMed] [Google Scholar]
- Brocklehurst KJ, Payne VA, Davies RA, et al. Stimulation of hepatocyte glucose metabolism by novel small molecule glucokinase activators. Diabetes. 2004;53:535–541. doi: 10.2337/diabetes.53.3.535. [DOI] [PubMed] [Google Scholar]
- Christesen HB, Jacobsen BB, Odili S, et al. The second activating glucokinase mutation (A456V): implications for glucose homeostasis and diabetes therapy. Diabetes. 2002;51:1240–1246. doi: 10.2337/diabetes.51.4.1240. [DOI] [PubMed] [Google Scholar]
- Durmaz E, Flanagan S, Berdeli A, et al. Variability in the age at diagnosis of diabetes in two unrelated patients with a homozygous glucokinase gene mutation. J Pediatr Endocrinol Metab. 2012;25:805–808. doi: 10.1515/jpem-2012-0077. [DOI] [PubMed] [Google Scholar]
- Estalella I, Rica I, Perez de Nanclares G, et al. Mutations in GCK and HNF-1alpha explain the majority of cases with clinical diagnosis of MODY in Spain. Clin Endocrinol. 2007;67:538–546. doi: 10.1111/j.1365-2265.2007.02921.x. [DOI] [PubMed] [Google Scholar]
- Galán M, Vincent O, Roncero I, et al. Effects of novel maturity-onset diabetes of the young (MODY)-associated mutations on glucokinase activity and protein stability. Biochem J. 2006;393:389–396. doi: 10.1042/BJ20051137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Herrero CM, Rubio-Cabezas O, Azriel S, et al. Functional characterization of MODY2 mutations highlights the importance of the fine-tuning of glucokinase and its role in glucose sensing. PLoS One. 2012;7:e30518. doi: 10.1371/journal.pone.0030518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa O, Meschi F, Cuesta-Munoz A, et al. High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetologia. 2001;44:898–905. doi: 10.1007/s001250100530. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM. Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov. 2009;8:399–416. doi: 10.1038/nrd2850. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM. GKAs for diabetes therapy: why no clinically useful drug after two decades of trying? Trends Pharmacol Sci. 2013;34:90–99. doi: 10.1016/j.tips.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Negahdar M, Aukrust I, Molnes J, et al. GCK-MODY diabetes as a protein misfolding disease: the mutation R275C promotes protein misfolding, self-association and cellular degradation. Mol Cell Endocrinol. 2014;382:55–65. doi: 10.1016/j.mce.2013.08.020. [DOI] [PubMed] [Google Scholar]
- Njølstad PR, Søvik O, Cuesta-Muñoz A, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344:1588–1592. doi: 10.1056/NEJM200105243442104. [DOI] [PubMed] [Google Scholar]
- Njølstad PR, Sagen JV, Bjørkhaug L, et al. Permanent neonatal diabetes caused by glucokinase deficiency: inborn error of the glucose-insulin signaling pathway. Diabetes. 2003;52:2854–2860. doi: 10.2337/diabetes.52.11.2854. [DOI] [PubMed] [Google Scholar]
- Osbak KK, Colclough K, Saint-Martin C, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30:1512–1526. doi: 10.1002/humu.21110. [DOI] [PubMed] [Google Scholar]
- Porter JR, Shaw NJ, Barrett TG, Hattersley AT, Ellard S, Gloyn AL. Permanent neonatal diabetes in an Asian infant. J Pediatr. 2005;146:131–133. doi: 10.1016/j.jpeds.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Raimondo A, Chakera AJ, Thomsen SK, et al. Phenotypic severity of homozygous GCK mutations causing neonatal or childhood-onset diabetes is primarily mediated through effects on protein stability. Hum Mol Genet. 2014;23:6432–6440. doi: 10.1093/hmg/ddu360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Díaz González F, Aragonés A, Argente J, Campos-Barros A. Permanent neonatal diabetes caused by a homozygous nonsense mutation in the glucokinase gene. Pediatr Diabetes. 2008;9:245–249. doi: 10.1111/j.1399-5448.2007.00361.x. [DOI] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Kupla T, Malecki MT. Permanent neonatal diabetes mellitus-the importance of diabetes differential diagnosis in neonates and infants. Eur J Clin Invest. 2011;41:323–333. doi: 10.1111/j.1365-2362.2010.02409.x. [DOI] [PubMed] [Google Scholar]
- Turkkahraman D, Bircan I, Tribble ND, Akçurin S, Ellard S, Gloyn AL. Permanent neonatal diabetes mellitus caused by a novel homozygous (T168A) glucokinase (GCK) mutation: initial response to oral sulphonylurea therapy. J Pediatr. 2008;153:122–126. doi: 10.1016/j.jpeds.2007.12.037. [DOI] [PubMed] [Google Scholar]
- Wajda-Cuszlag M, Witkowski D, Piontek E, et al. Glucokinase gene mutation as a causative factor of permanent neonatal diabetes mellitus. Pediatr Endocrinol Diabetes Metab. 2012;18:45–47. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. Two hybrid interaction between GCK(p.Ile19Asn) and the human GCKRP. Two hybrid interactions of Gal4-binding domain–GCKRP with Gal4-activating domain–GCK were tested in S. cerevisiae strain Y187 as described previously (Galán et al. 2006). The plasmid encoding the human GCKRP fusion protein to the Gal4 binding domain (GBD) was constructed as follows: the human GCKRP cDNA was amplified by PCR from plasmid p-FLAG CTC-hGCKRP (Brocklehurst et al. 2004), using primers 5′-ggaattcatgccaggcacaaaacggtttc-3′ and 5′-gggatcctactgaacgtcaggctctaggatttc-3′ and inserted into EcoR1 and BamH1 sites of pGBKT7 (Clontech). The plasmid encoding the fusion of mutant GCK to the Gal4 activating domain (GAD) was derived from pACTII (Clontech) by inserting a BamHI-XhoI fragment from the GST-GCK(p.Ile19Asn) into the same sites of the polylinker. Filter lift assays were developed for 1 h. In each case, independent transformants were tested with similar results