

Abstract
Purpose: Enzyme replacement therapy (ERT) has been shown to improve outcome in classical infantile Pompe disease. The purpose of this study was to assess mortality, morbidity, and shortcomings of ERT in a larger cohort of patients treated outside clinical trials. To accomplish this, we retrospectively analyzed the data of all 23 subjects with classical infantile Pompe disease having started ERT in Germany between January 2003 and December 2010.
Results: Ten patients (43%) deceased and four others (17%) became ventilator dependent. Seven infants (30.5%) made no motor progress at all, while seven (30.5%) achieved free sitting, and nine (39%) gained free walking. Besides all the seven patients (100%) attaining no improvement of motor functions, four out of the seven (57%) achieving to sit without support, and three out of the nine (33%) being able to walk independently, secondarily deteriorated, and died or became ventilator dependent. Sustained reduction of systolic function despite reversal of cardiac hypertrophy (n = 3), gastroesophageal reflux (n = 5), swallowing difficulties or failure to thrive (n = 11), recurrent pneumonias (n = 14), port system complications (n = 4), anesthesia-related incidents (n = 2), severe allergic reactions (n = 6), hearing loss (n = 3), and orthopedic deformities (n = 4) were problems frequently encountered.
Conclusion: Although this study has important shortcomings due to its retrospective nature and because important variables potentially influencing outcome were not available for a substantial amount of patients, these data suggest that classical infantile Pompe disease still remains a life-threatening condition associated with high morbidity and often dismal prognosis. Currently, a relevant number of patients do not benefit definitely from ERT.
Keywords: Enzyme replacement therapy, Glycogen storage disease, Lysosomal storage disorder, Metabolic myopathy, Pompe disease
Introduction
Pompe disease is a rare autosomal recessive disorder caused by deficiency of lysosomal acid α-glucosidase (GAA) (Hirschhorn and Reuser 2001). In classical infantile Pompe disease, virtual absence of enzyme activity leads to marked accumulation of glycogen in the heart, skeletal muscle, and other tissues. Affected patients present with hypertrophic cardiomyopathy (HCM), failure to thrive, and muscular hypotonia and weakness during the first months of life (Hirschhorn and Reuser 2001). The disease is rapidly progressive, and the majority of untreated subjects die within the first year of life without achieving any motor milestone such as turning, sitting, or standing. Survival beyond the age of 18 months is exceptional (van den Hout et al. 2003).
First enzyme replacement therapy (ERT) trials with recombinant human GAA (rhGAA) including small numbers of patients with infantile Pompe disease were conducted at the end of the 1990s and yielded promising results (van den Hout et al. 2000, 2001; Amalfitano et al. 2001). In 2006, a multinational multicenter open-label study was published that had enrolled eight infants ranging in age from 3 to 15 months with HCM and a residual enzyme activity less than 1% (Kishnani et al. 2006). All patients showed a reduction of left ventricular muscle mass reflecting reversal of HCM; five gained motor functions, and three achieved free walking. After 12 months of follow-up, two out of eight patients had died, one had become ventilator dependent, and four patients had died during the extended follow-up phase. Median age of death in the whole group was 21.7 months, being significantly later than in untreated patients (Kishnani et al. 2006).
Since this trial suggested that an early start of ERT may yield better results, a further pivotal study was performed that included 18 infants younger than age 6 months (Kishnani et al. 2007), receiving 20 or 40 mg/kg rhGAA biweekly. After 12 months of ERT, there was a significant reduction of left ventricular muscle mass and all patients were still alive. Thirteen subjects made motor progress, whereas five did not gain motor functions. Seven out of 18 achieved free walking, three were able to stand, and three were sitting without support, while six patients became ventilator dependent. Follow-up of these 18 patients for up to 3 years demonstrated that all of them had survived until age 18 months but that five patients (28%) had died thereafter and that further four subjects (22%) had become ventilator dependent. No significant differences were observed between the groups treated with 20 or 40 mg/kg rhGAA, respectively (Kishnani et al. 2007, 2009). Based on the positive results of these pivotal studies, ERT with rhGAA at a recommended dosage of 20 mg/kg biweekly has been approved by the EMA in 2006 for the treatment of Pompe disease in Europe.
Patients with classical infantile Pompe disease synthesize a nonfunctional form of GAA or are completely unable to form any kind of native enzyme (Kishnani et al. 2010a). Western blot analysis of fibroblasts derived from patients using antibodies directed against GAA can distinguish subjects with a nonfunctional protein and those synthesizing no enzyme at all. The former patients are designated as cross-reactive immunologic material (CRIM) positive, whereas the latter are classified as CRIM negative (Kishnani et al. 2007, 2010a). Alternatively, the CRIM status can be deduced from the results of genotyping, if the effects of a specific mutation on protein synthesis have been characterized (Kishnani et al. 2010a; Banugaria et al. 2011). Recently, it has been demonstrated that CRIM-negative subjects are much more likely to develop high-titer antibodies directed against rhGAA than CRIM-positive individuals and that irrespective of CRIM status, patients with high antibody titers have an attenuated therapeutic response to ERT (Kishnani et al. 2010a; Banugaria et al. 2011).
Since information about mortality and morbidity of larger patient cohorts treated outside clinical trials is yet rare (Chakrapani et al. 2010; van Gelder et al. 2014) and because data about the long-term results of ERT is still limited (Nicolino et al. 2009; Chakrapani et al. 2010; Rohrbach et al. 2010; van Gelder et al. 2014), we retrospectively analyzed outcome as well as problems encountered and mode of treatment in clinical practice of patients with classical infantile Pompe disease, who were born and had started rhGAA (Myozyme®) treatment in Germany between 2003 and 2010.
Patients and Methods
Physicians with special expertise potentially treating patients with classical infantile Pompe disease in Germany were contacted and asked to participate in the study. Informed consent to analyze anonymized data was obtained from all parents of patients alive, and the study was approved by the local ethics committee of the medical faculty of the University of Giessen, Germany.
Criteria for inclusion were a definite diagnosis of classical infantile Pompe disease and treatment with rhGAA (Myozyme®) in children born in Germany between January 2003 and December 2010. Data acquisition took place in August 2013.
Definite diagnosis of classical infantile Pompe disease was accepted when patients had onset of clinical symptoms within the first 6 months of life, HCM diagnosed by echocardiography in combination with muscular hypotonia and proximal muscle weakness, and significantly reduced GAA activity in lymphocytes confirmed by determination in fibroblasts and/or by mutational analysis of the GAA gene. Twenty-three patients met these criteria. Ten subjects (43%) were female. Twelve children were of German, ten of Turkish, and one of Arabian descent. All subjects were treated with ERT.
The medical records of all subjects were retrospectively analyzed and the following data were extracted by the local physicians: age at onset of symptoms, age at diagnosis, age at start of ERT, age at ventilator dependency, age at death, or current age. We also collected data concerning the mode of initial diagnostics and their results, enzyme dosage applied and other treatment modalities, and problems, complications, surgical interventions, or procedures related to disease or to ERT. Furthermore, we analyzed the results of CRIM-status testing, genotype studies, and antibody titer determination if available (Table 1).
Table 1.
Outcome measures and variables potentially related to morbidity and mortality in 23 German patients with classical infantile Pompe disease
| P | Sex | Start ERT (months) | Genotype | CRIM status | Antibody titer | Survival/death (months) | Ventilated | Motor status | ERT dosage (mg/kg biweekly) | Tube feeding | Complications during ERT | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Maternal allele | Paternal allele | Best | Last/current | ||||||||||
| 1 | M | 3.9 | c.2078insA | c.2078insA | n.k. | 1: 800 | Alive, 43.3 | − | Walking | Walking | 20 | − | Recurrent pneumonias |
| 2 | F | 4.7 | c.1799G>A | c.1859G>A | + | 1:1.200 | Alive, 113.7 | − | Sitting | Sitting | 40 | + | Aspiration pneumonia |
| 3 | M | 4.9 | c.IVS17 + 102_IVS18 + 31 | c.1465G>T | + | 1:200 | Alive, 122.1 | − | Sitting | Sitting | 20 | − | Chronic diarrhea |
| 4 | M | 3.3 | c.IVS18-1G>C | c.IVS18-1G>C | + | –a | Died, 8.0 | + | None | None | 20 | + | Aspiration pneumonia, AR IV |
| 5 | M | 0.9 | c.2662G>T | c.2662G>T | n.k. | 1:12,000 | Alive, 76.9 | + | None | None | 40 | + | Recurrent pneumonias, AR III |
| 6 | M | 2.8 | c.915G>A | c.915G>A | n.k. | 1:84,000 | Died, 18.2 | + | Sitting | None | 30 | − | Pneumonia + deterioration, progressive HCM, AR IV |
| 7 | F | 2.3 | c.1933G>A | c.1933G>A | + | 1:800 | Alive, 90.6 | − | Walking | Sitting | 20 | − | Femur fracture, port thrombosis |
| 8 | F | 3.7 | c.266G>A | c.104T>C | + | 1:4.800 | Died, 36.8 | + | Sitting | Sitting | 40 | + | GER, recurrent pneumonias |
| 9 | M | 6.8 | c.1157insA | c.1157insA | n.k. | –d | Died, 16.1 | + | None | None | 40 | + | Port infection, sepsis |
| 10 | M | 7.0 | c.2560C>T | c.2560C>T | n.k. | –d | Died, 21.1 | − | None | None | 40 | + | Recurrent pneumonias |
| 11 | M | 2.6 | c.1548G>A | c.1548G>A | − | 1:51.200 | Died, 14.1 | + | None | None | 40 | + | Aspiration pneumonia, AR IV |
| 12 | F | 9.3 | c.2431insC | c.2431insC | + | –e | Alive, 115.0 | + | Sitting | None | 80 | + | Pneumonia + deterioration |
| 13 | M | 0 | c.IVS11-2A>G | c.IVS11-2A>G | n.k. | n.k. | Alive, 69.2 | - | Walking | Walking | 20 | − | Hearing impairment |
| 14 | M | 1.5 | c.1978C>T | c.1978C>T | n.k. | n.k. | Died, 41.8 | + | Walking | None | 30 | − | Pneumonia + deterioration |
| 15 | F | 4.7 | c.1687C>T | c.1687C>T | − | n.k. | Died, 8.1 | - | None | None | 20 | + | Sepsis, asystolia, port infection, AR IV |
| 16 | F | 3.4 | c.IVS14 + 20A>G | c.1548G>A | + | 1:5,600 | Died, 27.0 | + | Sitting | None | 40 | + | Pneumonia + deterioration, AR III |
| 17 | F | 5.5 | c.1978C>T | c.1978C>T | n.k. | 1:800 | Alive, 62.8 | − | Walking | Walking | 20 | − | Recurrent pneumonias |
| 18 | M | 1.4 | c.896T>C | c.896T>C | + | n.k. | Died, 36.4 | − | Walking | None | 20 | − | Recurrent pneumonias |
| 19 | M | 2.6 | c.12126G>A | c.12126G>A | n.k. | –b | Alive, 50.4 | − | Walking | Sitting | 20 | − | Pneumonia + deterioration |
| 20 | F | 2.9 | c.2740-2742dup | c.2740-2742dup | + | 1:6,400 | Alive, 50.6 | − | Sitting | Sitting | 40 | − | Recurrent pneumonias |
| 21 | M | 0.5 | c.IVS07A>G | c.IVS07A>G | n.k. | –c | Alive, 49.4 | − | Walking | Walking | 20 | + | Swallowing difficulties |
| 22 | F | 0.2 | c.2004 C>A | c.541545delTTAC | n.k. | 1:1.800 | Alive, 47.0 | − | Walking | Walking | 20 | − | Contractures |
| 23 | F | 4.7 | c.1456G>C | c.1456G>C | n.k. | n.k. | Alive, 45.8 | + | None | None | 40 | + | Scoliosis, recurrent pneumonias |
aAt age 4 months
bAt age 18 months
cAt age 32 months
dAt age 12 months
eAt age 36 months
n.k. not known, HCM hypertrophic cardiomyopathy, AR allergic reaction
To assess the effects of ERT on motor function, we asked for the best motor milestone ever achieved and for the current or last motor status (i.e., no sitting, sitting without support, free walking), since standardized tests such as the Bailey scales or the Alberta Infant Motor Scale were not applied in a substantial number of patients.
Cardiac function was analyzed in patients for whom reliable data were available at least at start, after 6 months, and after 12 months of ERT. Echocardiographic findings were used for analysis in case examinations were performed by an experienced pediatric cardiologist with a 5-MHz transducer. Standard apical two- and four-chamber views were analyzed for the determination of interventricular septum (IVS) thickness during diastole, left ventricular posterior wall (LVPW) thickness during diastole, and shortening fraction (SF). IVS and LVPW thickness were assumed to reflect cardiac hypertrophy, and SF was supposed to indicate systolic function. The results were related to normal values (Kampmann et al. 2000).
All results are given as median values and range, the Mann–Whitney-rank sum test was used to compare median values between single groups, and p-values <0.05 were accepted as significant.
Results
At first presentation, all patients displayed elevated CK values (median 814 U/L, range 512–1,387 U/L; normal range <295 U/L) and signs of HCM, while muscular hypotonia and weakness were documented in 20 subjects (87%). Only one patient was able to turn around with help at age 3 months, whereas no other child had reached a motor milestone appropriate for the age at first presentation (i.e., turning around or sitting with or without support). No patient was ventilator dependent. In all subjects, Pompe disease was assumed after measuring a reduced GAA activity in lymphocytes. Diagnosis was confirmed by the determination of reduced enzyme activity in fibroblasts in 6 (26%) and/or by molecular genetic analysis in 22 patients (96%). Muscle biopsies performed in nine subjects (39%) revealed a vacuolar myopathy with increased glycogen content and reduced GAA activity in all.
Patient demographics and baseline characteristics are summarized in Table 2. Median age at first symptoms was 1.4 months (range 0–5.0), which at diagnosis 2.8 months (range 0–8.4), and which at start of ERT 3.3 months (range 0–9.3) (Table 2). The median time interval between diagnosis and start of ERT was 0.5 months (range 0–2.8). ERT was begun within the first month in four (17%), within the second in two (9%), within the third in five (22%), within the fourth in four (17%), and within the fifth month in four children (17%). One subject each started ERT within the sixth, seventh, eighth, and tenth month of age.
Table 2.
Patient demographics and baseline characteristics of 23 German patients with classical infantile Pompe disease
| Sex | |
| Female | 10 (43%) |
| Male | 13 (57%) |
| Age at first symptoms | 1.4 (0–5.0) months |
| Age at diagnosis | 2.8 (0–8.4) months |
| Age at start of ERT | 3.3 (0–9.3) months |
| Outcome | |
| Deceased | 10 (43%) |
| Alive with ventilatory support | 4 (17%) |
| Alive without ventilatory support | 9 (40%) |
| Age at death | 21.1 (8.0–41.8) months |
| Current age of surviving patients | 62.8 (43.3–122.1) months |
| Best motor milestone achieved | |
| None | 7 (30%) |
| Sitting without support | 7 (30%) |
| Walking without support | 9 (40%) |
| Secondary loss of motor milestones | 5 (22%) |
| Cardiac function ( n = 15) | |
| Reversal of cardiac hypertrophy | 14 (96%) |
| No reversal of cardiac hypertrophy | 1 (7%) |
| Sustained impairment of contractility | 3 (13%) |
| CRIM status ( n = 11) | |
| Positive | 9 (39%) |
| Negative | 2 (9%) |
| RhGAA dosage | |
| Standard | 11 (48%) |
| Nonstandard | 12 (52%) |
Data is presented as median value and range or as absolute and percentage numbers
Survival, ventilator-free survival, and motor function represent important outcome measures of ERT in classical infantile Pompe disease. The overall mortality rate in this cohort was 43% (ten subjects). Additionally, four children (17%) became ventilator dependent, and two of them are completely paralyzed. Cause of death was cardiac arrhythmia related to continuously progressive HCM in one subject, whereas lethal tachyarrhythmia was suspected in another. One child died due to severe sepsis in conjunction with respiratory problems. In the remaining seven cases, parents declined start or continuation of invasive ventilation, necessary to treat respiratory insufficiency. Median age at death was 21.1 months (range 8.0–41.8). The current age of the 13 surviving patients at time of data acquisition was 62.8 months (range 43.3–122.1).
To assess whether the effects of ERT on motor development allow estimating survival or ventilator-free survival, we related these two parameters to the best motor milestone achieved (Fig. 1). All seven patients who did not achieve to sit without support (100%) died or became ventilator dependent, while this was the case for four out of seven sitting free (57%) and for three out of nine achieving free walking (33%). To determine whether beginning ERT at young age is associated with a better outcome, we depicted the best motor status achieved and the number of patients becoming ventilator dependent or having deceased in relation to start of therapy within the first 3 months of life (Fig. 2). This showed that approximately two thirds of patients starting ERT at young age achieved walking independently and that a further quarter reached the ability to sit without support.
Fig. 1.

Outcome in relation to the best motor milestone achieved in 23 German patients with classical infantile Pompe disease
Fig. 2.
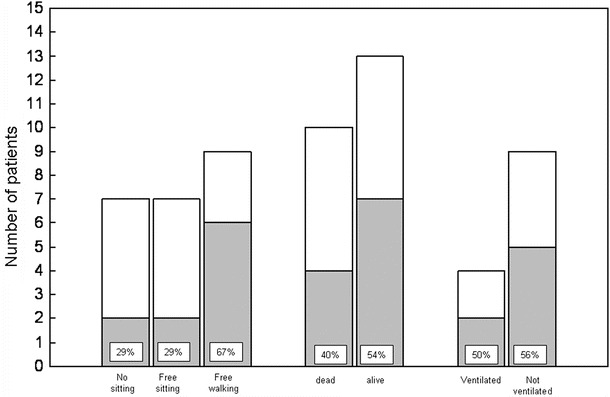
Best motor milestone achieved, survival and ventilator dependency, in relation to young age at start of ERT in 23 German patients with classical infantile Pompe disease. Gray areas correspond to the number of subjects starting ERT within the first 3 months of life
The CRIM status, assumed to present an important predictor of outcome, was known for 11 subjects only. Two of these patients were CRIM negative and died at age 8 and 14 months, respectively (100%), without making any motor progress. Three CRIM-positive patients (37.5%) deceased at age 27, 31, and 36 months, respectively. Two of these subjects were able to sit without support and one individual achieved free walking. Determination of GAA antibody titers, also known to be related to outcome, was not performed on a regular basis in this cohort, but maximum antibody titers determined in 18 patients are presented in Table 1. Seroconversion occurred in 12 of these subjects (67%). Two individuals with very high titers (1:84,000 + 1:51,000) deceased at age 14 and 18 months, respectively. One of them made no motor progress at all, whereas the other subject deceased due to relentlessly progressive HCM. Three out of five subjects with negative antibody titers also died. Two of these patients started ERT beyond the age of 6 months, while in the third one, the antibody titer had been determined only once, shortly after start of ERT.
Problems related to the underlying disease, intercurrent illnesses, and complications of ERT may also change outcome and contribute to morbidity in individual patients. Table 3 summarizes operative procedures, problems, and complications occurring in the patient group. Port implantation was the most frequent operative procedure performed. Problems related to ERT occurred in eight patients (35%). Port complications encompassed infections in two subjects and thrombosis of the catheter in one child. Allergic reactions grade III or IV necessitating immediate interruption of intravenous application of rhGAA were documented in six patients (26%). Orthopedic deformities emerged in four subjects (17%) and included scoliosis in one and talipes in three patients. Beneath swallowing difficulties and gastroesophageal reflux necessitating gastric tube placement and sometimes fundoplication, recurrent pneumonias were a major factor contributing to morbidity in this cohort. Five subjects (22%) experienced a distinct deterioration of their functional status during pneumonias. Three of these patients were able to sit without support but lost this ability and became ventilator dependent, while two subjects, who had walked independently, showed an abrupt and unexpected worsening of their functional status resulting in ventilatory failure at age 3 and 3½ years, respectively.
Table 3.
Synopsis of surgical interventions performed and problems and complications encountered in 23 German patients with classical infantile Pompe disease
| Surgical interventions | N | (%) |
|---|---|---|
| Fundoplication | 5 | 22 |
| Tracheostomy | 5 | 22 |
| Muscle biopsy | 9 | 39 |
| Gastric tube placement | 11 | 48 |
| Port implantation | 15 | 65 |
| Problems/complications | ||
| Gastroesophageal reflux | 7 | 30 |
| Failure to thrive/swallowing difficulties | 15 | 65 |
| Recurrent pneumonias | 14 | 61 |
| Sudden deterioration during infection | 5 | 22 |
| Anesthetic complication | 2 | 9 |
| Complication related to port system | 3 | 13 |
| Allergic reaction grade III/IV | 6 | 26 |
| Orthopedic deformities | 4 | 17 |
| Hearing impairment | 3 | 13 |
Augmentation of the enzyme dosage is a further factor that may influence the course of disease. In 12 patients (52%), the recommended dosage of rhGAA (20 mg/kg biweekly) was intermittently or permanently increased. It was doubled in nine and raised to 30 mg/kg weekly in two, while the maximum dosage administered in one child was 40 mg/kg weekly. Reasons to increase enzyme dosage were secondary worsening of motor or pulmonary function in nine, profound muscle weakness at start of therapy in one, and cardiac insufficiency or protracted improvement of cardiac function in two subjects. Figure 3 shows the outcome of patients treated with the standard regimen and of those receiving higher rhGAA dosages. While only less than 10% of individuals with increased dosages achieved free walking, nearly three quarter of subjects treated with the standard dosage reached this motor milestone. In addition, more than 50% of patients getting higher enzyme dosages had deceased during follow-up, while nearly 75% of patients treated with 20 mg/kg biweekly were still alive. Moreover, 80% of the surviving patients receiving higher rhGAA dosages had to be ventilated, whereas this was the case for about 10% of subjects treated with the standard regimen only.
Fig. 3.
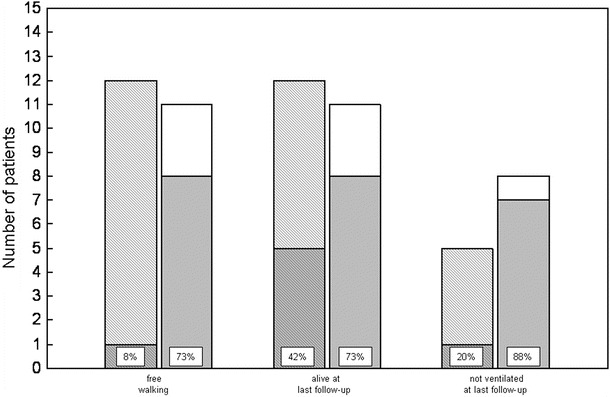
Achievement of free walking, survival, and ventilator-free survival in relation to the enzyme dosage applied in 23 German patients with classical infantile Pompe disease. Hatched bars correspond to subjects treated with nonstandard (higher) dosages, and white bars reflect individuals receiving the standard regimen (20 mg/kg biweekly). Gray areas correspond to patients achieving free walking, being alive, and being alive and not ventilated, respectively
Clinical, laboratory, or echocardiographic symptoms of cardiac insufficiency prompting treatment with anticongestive medication were present in 17 patients (74%). Medications used included beta-blockers in 11, diuretics in 10, angiotensin-converting enzyme inhibitors in 4, calcium channel blocker in 1, and digoxins in 2 children.
Reliable echocardiographic data were available for 17 subjects (74%) at start, for 15 (65%) after 6, and for 12 (52%) after 12 months of ERT. By definition, IVS and LVPW were thickened in all subjects at the start of ERT, while SF was decreased in 6 out of 17 (35%) patients. IVS thickness had decreased significantly after 6 months and LVPW thickness after 12 months of ERT (Fig. 4). IVS thickness was normalized after 6 months of ERT in seven, had markedly decreased in seven, but had further increased in one subject despite an increase of enzyme dosage. SF remained reduced in 4 out of 15 subjects after 6 (2× <30% and 2× <25%) and in 3 out of 12 after 12 months of ERT (Fig. 4).
Fig. 4.
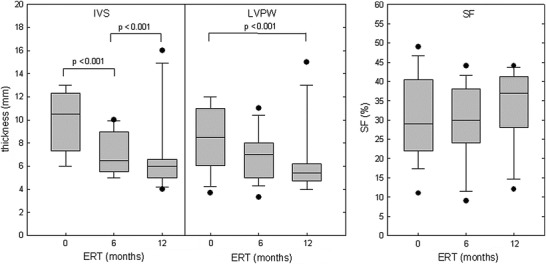
Interventricular septum (IVS) thickness during diastole, left ventricular posterior wall (LVPW) thickness during diastole, and shortening fraction (SF) in 17 subjects with classical infantile Pompe disease at start, in 15 after 6 months, and in 12 after 12 months of ERT
Discussion
This study retrospectively assessed the outcome of patients with classical infantile Pompe disease who were born and had started treatment with ERT in Germany between January 2003 and December 2010. Follow-up of the cohort was performed until death or for a period of at least 30 months of ERT. Major findings of this study with longer follow-up were that nearly 40% of patients achieved free walking but that about 60% of the patients died or became ventilator dependent and that approximately 50% of the patients with an initially positive response to ERT showed a secondary loss of acquired motor milestones during the further course of disease. These results are similar to those obtained in the pivotal study analyzing safety and efficacy of ERT in classical infantile Pompe disease (Kishnani et al. 2009), even though these children were selected for the pivotal trial and forming a more homogenous group of patients that were all treated in the same way and received utmost care. Our results are also in-line with the published UK experience on 20 patients treated from 2000 to 2009, which suggested a poorer outcome than initially hoped for (Chakrapani et al. 2010). In the British cohort, 35% of patients (7 out of 20) died, and another 30% (6/20) were alive but ventilator dependent. Moreover, our findings are also consistent with the recently reported outcome of 11 Dutch infants followed up for 0.3–13.7 years. In this cohort that included nine subjects starting ERT within the first 4 months of life, three patients died and another two became ventilator dependent (van Gelder et al. 2014).
The exact number of subjects with classical infantile Pompe disease born and treated with ERT in Germany is not known since an official registry does not exist. Based on annual birth numbers of 665,000 to 707,000 in Germany during the years 2003 to 2010 (Poetsch 2012), the calculated incidence of classical infantile Pompe disease in our study was ~1:208,000, which is lower than the frequency of 1:140,000 per year expected according to the literature (Hirschhorn and Reuser 2001). Thus, it cannot be excluded with certainty that some individuals with classical infantile Pompe disease receiving or not receiving ERT have been missed. The high level of cooperation between specialized institutions potentially being in charge of such children, however, makes a substantial bias of data unlikely.
Patients with limited benefit from ERT included infants that did not make any substantial motor progress from the very beginning as well as children with an initial positive response but a secondary loss of functions that untreated patients would not have attained. A decline of motor and respiratory capabilities after initial improvement has also been observed in other studies with longer follow-up periods (Kishnani et al. 2009; Nicolino et al. 2009).
Factors assumed to influence outcome of patients with classical infantile Pompe disease are age at onset of symptoms, age at start of ERT, negative CRIM status due to mutations of the GAA gene resulting in complete enzyme absence, and high antibody titers against rhGAA (Kishnani et al. 2010a; van Gelder et al. 2014).
In this study, ERT started earlier than in the UK cohort (Chakrapani et al. 2010) and also as compared to the pivotal study (Kishnani et al. 2009) (3.3 vs. 6.5 vs. 5.3 months). This was caused by a younger age at diagnosis (2.8 vs. 4.3 vs. 5.8 months) and by short intervals between diagnosis and start of ERT (0–2.8 months), suggesting that a late diagnosis and a delayed start of therapy were no factors negatively influencing the outcome in our cohort. The observation that two children who began ERT within the first 6 weeks of life gained no motor functions at all underscores that an early start of treatment does not guarantee a positive response (Kishnani et al. 2009; Chakrapani et al. 2010). Nevertheless, subjects starting ERT within the first 3 months of life were more likely to achieve free walking than those beginning later (Fig. 2). This supports the idea that early diagnosis and timely treatment allow a better motor development (Kishnani et al. 2009; van Gelder et al. 2014). These results are in contrast to the unexpected finding of the UK study that children starting ERT later had a better outcome compared to those beginning early (Chakrapani et al. 2010). This discrepancy can be explained by differences in the study populations. All individuals from the present study manifested clinically within the first 6 months of life as it is characteristic of the classical infantile type (van Gelder et al. 2014). By contrast, the UK cohort included three subjects with onset of clinical symptoms beyond the first year of life, being compatible with a more attenuated phenotype (Hirschhorn and Reuser 2001).
Unfortunately, information about the CRIM status was not available for a substantial amount of our patients, and antibody titers were not assessed systemically, thereby limiting interpretation of our results. This lack of data is mainly explained by difficulties in performing such tests on a routine basis, especially in the first years after the introduction of ERT. However, our findings are also compatible with the assumption that high antibody titers and negative CRIM status are predictors of poorer outcome (Kishnani et al. 2010a; Banugaria et al. 2011).
Notably, the outcome of patients treated with higher enzyme dosages concerning motor function, survival, and ventilator-free survival seemed to be less favorable than that of subjects receiving the standard dosage of 20 mg/kg rhGAA biweekly (Fig. 3). This unsuspected result can be explained by the fact that enzyme dosages were increased mainly in patients showing a deterioration of their functional status or in those responding not well to the standard regimen. These findings may also suggest that starting to treat patients with higher enzyme dosages after deterioration of their functional status is of limited benefit.
In contrast to other European countries such as the Netherlands and the UK, prescription of rhGAA therapy in Germany is not restricted to a single institution or to few designated centers. Experience in treating an extremely rare disorder may also contribute to outcome of affected patients (Kishnani et al. 2010b). In our cohort, we documented a multitude of medical problems and complications related to the underlying disease and to ERT. Therefore, complexity of the disease and high morbidity require a multidisciplinary approach and render supervision right from the start by centers experienced in the care of such patients mandatory. Although international treatment guidelines for Pompe disease are crucial (Kishnani et al. 2010b), national recommendations accommodating the peculiarities of a specific country are also meaningful (Llerena et al. 2009; Hahn et al. 2012).
Severity of HCM is a further factor known to influence the outcome in classical infantile Pompe disease (van den Hout et al. 2003). Several studies have demonstrated normalization of left ventricular muscle mass during ERT even in subjects showing no improvement of motor functions (Klinge et al. 2005; Kishnani et al. 2007, 2009). The information obtained on cardiac function in our retrospective study has to be interpreted cautiously since reliable data were available only for a limited number of patients. Nonetheless and in-line with others, we observed a marked reduction or even normalization of left ventricular muscle wall thickness in the majority of patients (van Gelder et al. 2014). However, progressive and lethal HCM despite ERT in one subject and suspected malignant tachyarrhythmia in another demonstrate that cardiac dysfunction still contributes to mortality in the era of ERT. Moreover, systolic function, as expressed by reduced shortening fractions, remained permanently impaired despite reversal of cardiac hypertrophy in about 20% of patients with serial echocardiographies.
In summary, rhGAA therapy has dramatically improved the outcome of patients with classical infantile Pompe disease but still raises important ethical problems (Kishnani et al. 2010b). While ERT should start as soon as possible, it is not known in many cases whether the infant will respond well or not. ERT may substantially prolong life by reversing HCM, whereas its inferior effects on skeletal muscles may not prevent complete paralysis and long-term artificial ventilation (Chakrapani et al. 2010). Moreover, no recommendations exist on how to proceed if unfavorable outcome predictors (e.g., negative CRIM status) are identified during course.
Our study has several important shortcomings such as its retrospective nature, data ascertainment by independently acting institutions, and lack of data for factors potentially influencing outcome in a substantial amount of subjects. However, the data compiled in this investigation are of avail, since knowledge about potential problems occurring during ERT in clinical practice and about the limitations of this therapy are essential to provide accurate advice to families.
Acknowledgment
The authors would like to thank all patients and their parents for participating in this study. We are also grateful to Stephan Gromer, MD, Genzyme, for his support.
Key Sentence
A retrospective analysis of 23 infants with classical infantile Pompe disease starting ERT in Germany between January 2003 and December 2010 confirms that ERT has substantially improved long-term outcome but also demonstrates that this disorder still remains a life-threatening condition associated with high morbidity and often dismal prognosis.
Compliance with Ethics Guidelines
Conflicts of Interest
Andreas Hahn, Susanne Praetorius, Nesrin Karabul, Martina Baethmann, Julia B. Hennermann, Nicole Muschol, Thorsten Marquardt, Martina Huemer, Marianne Rohrbach, Gökce Seyfullah, and Eugen Mengel have received speaker honoraria and/or research grants from Genzyme Corporation, Germany.
Johanna Dießel, Dorle Schmidt, Reinald Motz, Claudia Haase, René Santer, Claudia Thiels, Martin Smitka, and Ann Meyer declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all parents of patients alive for being included in the study.
Animal Rights
This article does not contain any studies with animal subjects performed by any of the authors.
Details of the Contributions of Individual Authors
Andreas Hahn: planning of the study, analysis and interpretation of data, and drafting of the manuscript.
Julia B. Hennermann, Thorsten Marquardt, and Eugen Mengel: planning of the study, analysis and interpretation of data, and critical reading of the manuscript with significant intellectual contribution.
Susanne Praetorius, Nesrin Karabul, Martina Baethmann, Nicole Muschol, Martina Huemer, Marianne Rohrbach, Gökce Seyfullah, Johanna Dießel, Dorle Schmidt, Reinald Motz, Claudia Haase, René Santer, Claudia Thiels, Martin Smitka, and Ann Meyer: acquisition of data, analysis and interpretation of data, and critical reading of the manuscript with significant intellectual contribution.
Footnotes
Competing interests: None declared
Contributor Information
Andreas Hahn, Email: Andreas.Hahn@paediat.med.uni-giessen.de.
Collaborators: Johannes Zschocke
References
- Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. [PubMed] [Google Scholar]
- Banugaria SG, Prater SN, Ng YK, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med. 2011;13:729–736. doi: 10.1097/GIM.0b013e3182174703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrapani A, Vellodi A, Robinson P, Jones S, Wraith JE. Treatment of infantile Pompe disease with alglucosidase alpha: the UK experience. J Inherit Metab Dis. 2010;33:747–750. doi: 10.1007/s10545-010-9206-3. [DOI] [PubMed] [Google Scholar]
- Hahn A, Hennermann JB, Marquardt T, et al. M. Pompe im Kindesalter: Aktueller Stand der Diagnostik und Therapie. Monatsschr Kinderheilkd. 2012;160:1243–1250. doi: 10.1007/s00112-012-2789-z. [DOI] [Google Scholar]
- Hirschhorn R, Reuser AJJ. Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. In: Scriver C, Beaudet A, Valle D, Sly W, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3389–3420. [Google Scholar]
- Kampmann C, Wiethoff CM, Wenzel A, et al. Normal values of M mode echocardiographic measurements of more than 2000 healthy infants and children in central Europe. Heart. 2000;83:667–672. doi: 10.1136/heart.83.6.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Nicolino M, Voit T, et al. Chinese hamster ovary cell-derived recombinant human α-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid α-glucosidase. Major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:1–11. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Corzo D, Leslie ND, et al. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009;66:329–335. doi: 10.1203/PDR.0b013e3181b24e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Austin SL, Arn P, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12:446–463. doi: 10.1097/GIM.0b013e3181e655b6. [DOI] [PubMed] [Google Scholar]
- Klinge L, Straub V, Neudorf U, Voit T. Safety and efficacy of recombinant alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscul Disord. 2005;15:24–31. doi: 10.1016/j.nmd.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Llerena JC, Jr, Horovitz DM, Marie SK, et al. The Brazilian consensus on the management of Pompe disease. J Pediatr. 2009;155(4 Suppl):S47–S56. doi: 10.1016/j.jpeds.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11:210–219. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- Poetsch O (2012) Geburten in Deutschland. Statistisches Bundesamt, Wiesbaden, Bestellnummer 0120007-12900-1
- Rohrbach M, Klein A, Köhli-Wiesner A, et al. CRIM-negative infantile Pompe disease: 42-month treatment outcome. J Inherit Metab Dis. 2010;33:751–757. doi: 10.1007/s10545-010-9209-0. [DOI] [PubMed] [Google Scholar]
- Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A, Van der Ploeg AT. Recombinant human α-glucosidase from rabbit milk in Pompe patients. Lancet. 2000;356:397–398. doi: 10.1016/S0140-6736(00)02533-2. [DOI] [PubMed] [Google Scholar]
- Van den Hout JM, Reuser AJ, de Klerk JB, Arts WF, Smeitink JA, Van der Ploeg AT. Enzyme therapy for Pompe disease with recombinant human α-glucosidase from rabbit milk. J Inherit Metab Dis. 2001;24:267–275. doi: 10.1023/a:1010383421286. [DOI] [PubMed] [Google Scholar]
- Van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- van Gelder CM, Hoogeveen-Westerveld M, Kroos MA, Plug I, van der Ploeg AT, Reuser AJ (2014) Enzyme therapy and immune response in relation to CRIM status: the Dutch experience in classic infantile Pompe disease. J Inherit Metab Dis [DOI] [PMC free article] [PubMed]