

Abstract
Background: Primary carnitine deficiency (PCD) is a disorder of fatty acid oxidation with a high prevalence in the Faroe Islands. Only patients homozygous for the c.95A>G (p.N32S) mutation have displayed severe symptoms in the Faroese patient cohort. In this study, we investigated carnitine levels in skeletal muscle, plasma, and urine as well as renal elimination kinetics before and after intermission with l-carnitine in patients homozygous for c.95A>G.
Methods: Five male patients homozygous for c.95A>G were included. Regular l-carnitine supplementation was stopped and the patients were observed during five days. Blood and urine were collected throughout the study. Skeletal muscle biopsies were obtained at 0, 48, and 96 h.
Results: Mean skeletal muscle free carnitine before discontinuation of l-carnitine was low, 158 nmol/g (SD 47.4) or 5.4% of normal. Mean free carnitine in plasma (fC0) dropped from 38.7 (SD 20.4) to 6.3 (SD 1.7) μmol/L within 96 h (p < 0.05). Mean T1/2 following oral supplementation was approximately 9 h. Renal reabsorption of filtered carnitine following oral supplementation was 23%. The level of mean free carnitine excreted in urine correlated (R2 = 0.78, p < 0.01) with fC0 in plasma.
Conclusion: Patients homozygous for the c.95A>G mutation demonstrated limited skeletal muscle carnitine stores despite long-term high-dosage l-carnitine supplementation. Exacerbated renal excretion resulted in a short T1/2 in plasma carnitine following the last oral dose of l-carnitine. Thus a treatment strategy of minimum three daily separate doses of l-carnitine is recommended, while intermission with l-carnitine treatment might prove detrimental.
Introduction
Primary carnitine deficiency (PCD, OMIM #212140) is an autosomal recessive disorder of fatty acid beta-oxidation caused by a dysfunctional OCTN2 carnitine transporter, coded by the SLC22A5 gene on chromosome five (Shoji et al. 1998; Nezu et al. 1999; Longo et al. 2006). Patients continually lose large amounts of carnitine in urine leading to low blood and tissue levels of carnitine (Scaglia et al. 1998; Tein 2003; Longo et al. 2006). PCD has a relatively high prevalence in the Faroe Islands (1:300) and has been associated with several sudden deaths among young Faroese individuals due to cardiac arrhythmia (Rasmussen et al. 2013, 2014b). Cardiomyopathy has especially been reported in children and patients may develop metabolic disturbances and fatigue – though many remain asymptomatic (Tein 2003; Stanley 2004; Cano et al. 2008; Magoulas and El-Hattab 2012; Rasmussen et al. 2014a). The principal role of carnitine is to transport long-chain fatty acids into the mitochondria for beta-oxidation and to preserve the intracellular CoA homeostasis (Engel et al. 1981; Rebouche 2004). Patients suffering from PCD are treated with daily oral l-carnitine supplements (Lund et al. 2007; Rasmussen et al. 2014a). Four different PCD-related mutations and a risk haplotype have been uncovered in the Faroese population, with the c.95A>G mutation being the most prevalent and severe (Rasmussen et al. 2014b, c). Patients homozygous for the c.95A>G mutation have the lowest mean plasma carnitine levels and mean residual OCTN2 transport activity among the Faroese patients (Rasmussen et al. 2014c). Furthermore all patients found previously to suffer from severe complications and sudden death were homozygous for the c.95A>G mutation indicating a phenotype–genotype correlation (Rasmussen et al. 2013, 2014c). Although it is documented that continued daily l-carnitine supplementation increases blood carnitine levels in PCD patients, the effect on carnitine levels in skeletal muscle tissue, which is the main store of carnitine in the human body, is currently unknown (Rasmussen et al. 2014a). Additionally the kinetics of renal carnitine elimination in PCD patients following interruption of regular l-carnitine supplementation is to our knowledge not fully described.
The objectives of the present study were to investigate and monitor the effects of stopping l-carnitine supplementation in a small cohort of patients homozygous for the c.95A>G mutation during a 5-day period with regard to levels of carnitine in skeletal muscle, blood, and urine prior to and following the intervention.
Materials and Methods
Five male patients with a mean age of 32.6 years (range 19–73) known to be homozygous for the c.95A>G mutation gave informed consent to participate (Table 1). They had all taken l-carnitine supplements continually for at least 3 years. All patients were requested to ingest the same amount of l-carnitine relative to their weight (75 mg/kg/day) in three daily doses a week prior to the study. The patients had no symptoms of PCD and had previously only had symptoms of fatigue, which had been treated effectively by l-carnitine supplementation. One patient was treated with warfarin because of chronic atrial fibrillation; apart from that, the patients did not receive any medication other than l-carnitine.
Table 1.
Baseline values
| Patient #a | Age (years) | Weight (kg) | Height (cm) | BMI (kg/m2) | BSA (m2) | LV mass index (g/m2) | LVEF (%) | B-hemoglobin (mmol/L) | P-glucose (mmol/L) | P-potassium (mmol/L) | P-sodium (mmol/L) | P-ALAT (U/)L | P-creatinine (μmol/L) | Creatinine clearance (mL/min)b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 23 | 90 | 183 | 26.9 | 2.12 | 104 | 55 | 9.1 | 4.9 | 3.6 | 138 | 31 | 83 | 167 |
| 2 | 73 | 120 | 193 | 32.2 | 2.49 | 90 | 56 | 9.1 | 6.7 | 3.9 | 139 | 43 | 106c | 60c |
| 3 | 19 | 70 | 180 | 21.6 | 1.89 | 84 | 61 | 8.0 | 4.6 | 3.6 | 137 | 24 | 64 | 212 |
| 4 | 28 | 83 | 185 | 24.3 | 2.07 | 105 | 64 | 8.0 | 5.0 | 3.8 | 143 | 40 | 75 | 78 |
| 5 | 20 | 85 | 187 | 24.3 | 2.11 | 134c | 60 | 8.5 | 4.5 | 4.0 | 140 | 16 | 75 | 107 |
BMI body mass index, BSA body surface area, LV left ventricle, LVEF left ventricular ejection fraction
aAll homozygous for the c.95A>G mutation
bCorrected for body surface area
cOut of range
The patients were admitted to the National Hospital of the Faroe Islands during the study period under close observation, including continuous heart monitoring. Informed written consent was obtained from all participants and the Faroese Ethics Committee approved the study. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000.
Oral l-carnitine supplements were discontinued when admitted and the patients were without l-carnitine supplementation for initially 97 h (≈4 days). l-carnitine was then infused with a rate of 3.75 mg/kg/h for 4 h and then stopped. The patients were monitored for further 24 h before starting their regular oral supplements again. Blood, drawn with venipuncture from an antecubital vein into regular blood sample tubes containing EDTA, and urine samples were collected with predetermined intervals during the study period. The blood was centrifuged and the resulting plasma collected and frozen at −40°C until analysis in Copenhagen. Some blood was though analyzed in the local hospital laboratory to obtain routine blood parameters, e.g., glucose, electrolytes, creatinine, hemoglobin, etc. Urine was stored at −70°C until analysis. Skeletal muscle biopsies were obtained from the medial part of m. vastus lateralis using the Bergstrom technique before discontinuation of l-carnitine (t = 0 h), midway through the study (t = 48 h), and just before infusion of l-carnitine (t = 96 h) (Bergstrom 1975). Muscle tissue was immediately frozen in liquid nitrogen and stored at −80°C until analysis.
Standard echocardiography was performed before the discontinuation and restart of l-carnitine supplementation using a GE Medical Vivid s6 ultrasound system. Measurements were obtained according to accepted standards and techniques (Lang et al. 2005). All reported measures were the average of two separate measurements.
Plasma, urine, and muscle tissue were analyzed at the Centre for Inherited Metabolic Disease in Rigshospitalet, Copenhagen.
Analyses of acylcarnitines and carnitine in plasma, muscle, and urine were performed using stable-isotope dilution combined with ultra-performance liquid chromatography–tandem mass spectrometry, using a Quattro Micro triple quadrupole mass spectrometer (Waters, Milford, Massachusetts).
d3-Carnitine, d3-acetylcarnitine, d3-propionylcarnitine, d3-butyrylcarnitine, d9-isovalerylcarnitine, d3-octanoylcarnitine, d3-tetradecanoylcarnitine, and d3-hexadecanoylcarnitine (Herman ten Brink, Vrije Universiteit, Amsterdam, The Netherlands) were added to samples before extraction/homogenization. Carnitine and acylcarnitines in all three matrices were quantified using external spiked plasma calibration curves.
Pharmacokinetic Methods
Pharmacokinetic parameters were determined using Phoenix WinNonlin 6.2 (Pharsight Certara) and Microsoft Excel. For each urine collection, interval (the amount of excreted carnitine (Aeti)) was determined (ti represents a particular time interval). The area under the plasma concentration curves (AUCti) for carnitine was determined by non-compartmental analysis in WinNonlin using the same time intervals as the urine collection intervals. Renal clearance (CLR) was estimated as amount excreted divided by the area under the plasma concentration curve Aeti/AUCti. Fraction reabsorbed (Freabs) was calculated as 1-(CLR/CLCrea). Half-lives were calculated as ln(2)/λz, where λz (the terminal rate constant) was determined by linear regression of the semilogarithmic plot of the plasma concentration–time profile.
Statistical Analysis
Data analysis was performed using IBM® SPSS® Statistics Version 19 (SPSS Inc., Chicago, IL, USA). All continuous variables were expressed as mean (standard deviation). Paired student’s T-test was used to test for a significant difference in carnitine levels. Level of significance was set at p < 0.05 (two-tailed test).
Results
The mean level of free carnitine in skeletal muscle was severely depressed before discontinuation of l-carnitine, 158 nmol/g (SD 47.4, range 87.6–217) compared to a mean of 2,914 nmol/g (SD 249) found in healthy individuals (Madsen et al. 2013). There was a tendency for the mean level of free carnitine in muscle to decrease slightly (13.5 nmol/g) during the l-carnitine intermission (p = 0.35) (Fig. 1).
Fig. 1.
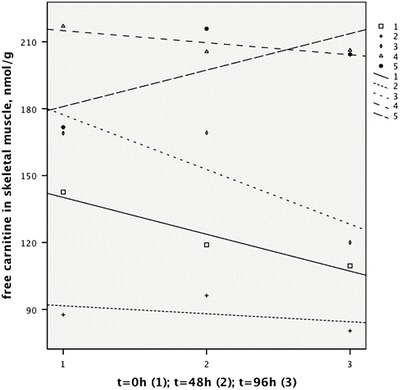
Free carnitine in muscle measured three times during the study
Mean free carnitine in plasma (fC0) was 38.7 (SD 20.4) μmol/L measured 1 h after the last dose of l-carnitine. The mean T1/2 following oral supplementation was approximately 9 h (Table 2). fC0 decreased most rapidly during the first 24 h before leveling off and dropping further to a mean 6.3 (SD 1.7) μmol/L before infusion of l-carnitine at 96 h (p = 0.02) (Fig. 2). During the 4-h infusion of l-carnitine, the mean level of fC0 rose quickly to 174 (SD 51) μmol/L but then fell rapidly to 12.3 (SD 6.8) μmol/L within 26 h (Fig. 2). There was practically no difference in mean renal reabsorption of filtered carnitine following oral supplementation (23%) and following infusion with l-carnitine (25%), combined mean 24% (Table 2), when measured in the lower plasma free carnitine concentration range (5.9–54.3 μmol/L) where reabsorption would be expected to be more than 90% in healthy subjects.
Table 2.
Pharmacokinetics
| Patient # | Means | Oral versus infusion | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CLR (mL/min) | CLcrea (mL/min) | F reabsorption | T 1/2-oral (h) | T 1/2-inf (h) | F reabsorption-oral | F reabsorption-inf | CLR-oral (mL/min) | CLR-inf (mL/min) | |
| 1 | 121.1 | 166.8 | 0.27 | 9.2 | 21.2 | 0.23 | 0.31 | 127.6 | 114.6 |
| 2 | 54.2 | 60.0 | 0.10 | 16.1 | 11.1 | 0.28 | −0.08 | 43.4 | 65.1 |
| 3 | 103.1 | 211.9 | 0.51 | 5.6 | 14.2 | 0.45 | 0.58 | 117.0 | 89.3 |
| 4 | 77.2 | 78.2 | 0.01 | 9.5 | 38.6 | 0.00 | 0.02 | 78.1 | 76.4 |
| 5 | 77.0 | 107.3 | 0.28 | 5.7 | 17.8 | 0.17 | 0.40 | 89.1 | 64.9 |
| Mean (CI 95%) | 86.5 (54.3–118.7) | 124.9 (46.2–203.4) | 0.24 (0.08–0.39) | 9.22 (3.9–14.5) | 20.6 (7.2–33.9) | 0.23 (0.0–0.47) | 0.25 (−0.09–0.58) | 91.0 (49.6–132.5) | 82.1 (56.3–107.8) |
Fig. 2.
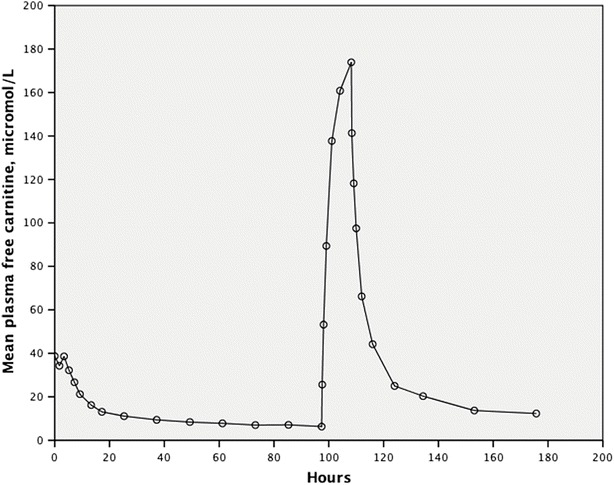
Mean free carnitine in plasma following discontinuation of oral l-carnitine and then during and after l-carnitine infusion
The level of mean free carnitine excreted in urine correlated (R2 = 0.78, p < 0.01) with fC0 in plasma, with a decreased excretion with decreasing plasma levels of fC0 and vice versa (Fig. 3).
Fig. 3.
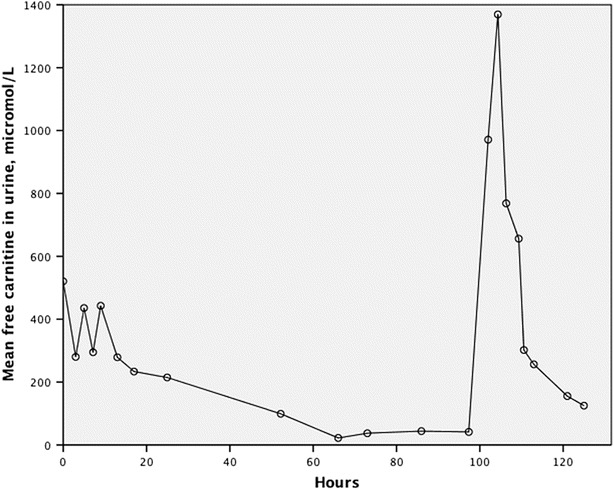
Mean free carnitine in urine following discontinuation of oral l-carnitine and then during and after l-carnitine infusion
The level of mean plasma acetylcarnitine fell significantly within 7 h (p = 0.037) from 23.0 (SD 9.2) μmol/L to 10.7 (SD 5.0) μmol/L and then dropped to 0.7 (SD 0.6) μmol/L (p < 0.001) before infusion of l-carnitine. Following the start of l-carnitine infusion, we measured a nonsignificant increase in the level of mean plasma acetylcarnitine to 3.5 (2.9) μmol/L (p = 0.13), before decreasing again following the infusion. As with fC0, the acetylcarnitine in plasma was rapidly excreted in urine following the intermission of supplementation and then following the l-carnitine infusion (Fig. 4). The elderly patient (#2 in Table 1) who had a decreased renal function exhibited a slower decrease in plasma fC0 than the other younger patients and reached a higher level of plasma acetylcarnitine (from 0.4 to 8.6 μmol/L) following l-carnitine infusion.
Fig. 4.
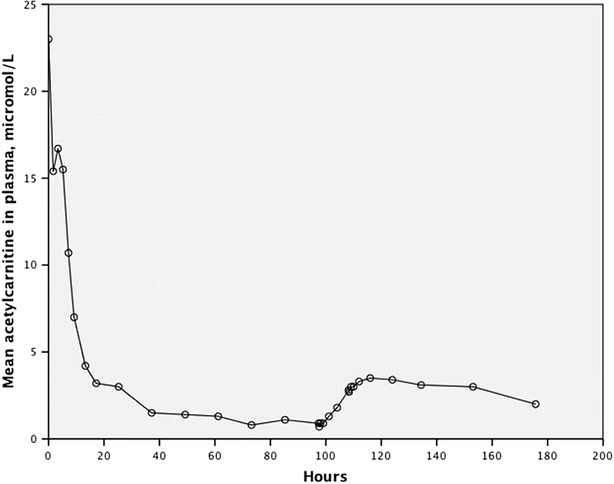
Mean acetylcarnitine in plasma following discontinuation of oral l-carnitine and then during and after infusion of l-carnitine
Baseline values including routine safety blood samples and echocardiography were normal – apart from a mild renal insufficiency in the elderly patient (Table 1). There was no change in echocardiographic parameters following 4 days without l-carnitine supplementation. Except for the changes in carnitine levels, there were no changes in other measured blood parameters, including hemoglobin, glucose, electrolytes, ALAT, and creatinine.
Discussion
Our primary findings were that l-carnitine supplemented adult PCD patients homozygous for the c.95A>G mutation had very limited skeletal muscle carnitine stores and that the level of mean free carnitine in plasma dropped rapidly with a mean T1/2 of 9 h following intermission with oral l-carnitine.
The mean concentration of free carnitine in skeletal muscle was only 158 nmol/g in the participants corresponding to approximately 5.4% of the reported normal mean level of 2,914 nmol/g (Madsen et al. 2013). Previous data from children with PCD, showing only a slight increase in skeletal muscle carnitine following supplementation, supports our findings (Stanley et al. 1991). In normal healthy individuals, skeletal muscle carnitine stores account for 97% of all carnitine in the body with a slow estimated turnover of 105 h (Reuter and Evans 2012). There was a tendency toward a decrease in mean muscle carnitine following intermission with l-carnitine supplementation, which was not significant, possibly due to the low number of subjects. It has been hypothesized that if the intramuscular concentration of carnitine is above 4% of normal then fatty acid oxidation in muscle is not compromised (Stanley et al. 1991). Four of our patients reported prior to the study improved physical ability and endurance following long-term l-carnitine supplementation, compared to before being diagnosed with PCD when they did not receive l-carnitine, indicating that even though the obtained skeletal muscle stores were only just above 5% of normal during supplementation, the patients may have benefited with improved physical fitness.
Mean free carnitine in the participants following long-term l-carnitine supplementation was in the lower normal range (Reuter et al. 2008). Carnitine in plasma accounts for only 0.1% of total body carnitine in normal subjects (Reuter and Evans 2012). Plasma carnitine levels are normally maintained by renal tubular reabsorption, a rapidly equilibrating compartment (liver and kidneys) and a slowly equilibrating compartment (muscle) (Evans et al. 2000). It is likely that a compromised renal reabsorption, and a lack of sufficient carnitine stores to maintain plasma levels in the PCD patients, led to rapidly decreasing carnitine levels toward pretreatment plasma levels and an fC0 half-life of only 9 h following the last oral dose of l-carnitine (Fig. 2).
The patients experienced continued excessive loss of carnitine in urine because the mean renal tubular reabsorption of filtered l-carnitine was only 24% compared to more than 90% in normal subjects under normal conditions (Engel et al. 1981; Rebouche et al. 1993; Rebouche 2004; Steiber et al. 2004). We have previously shown that the mean residual OCTN2 transport activity measured in fibroblasts from patients homozygous for the c.95A>G mutation was only 4% compared to normal – the dysfunctional transporter is thus not capable of adequately reabsorbing the filtered carnitine, which is lost in urine instead (Rasmussen et al. 2014c). When l-carnitine was infused, the plasma levels rose quickly, but the infused l-carnitine was excreted in urine within a short period of time in all participants. One exception was the elderly individual with a decreased glomerular filtration, who may not have been able to clear the l-carnitine as effectively as his younger counterparts. Acetylcarnitine is formed intracellularly during normal metabolic activity when l-carnitine is present (Flanagan et al. 2010). The level of plasma acetylcarnitine in the participants fell when they stopped oral supplementation, which may also indicate decreasing intracellular levels of l-carnitine as plasma levels were decreasing (Fig. 4). Plasma acetylcarnitine levels only increased marginally in the young participants following l-carnitine infusion, which might indicate that the amount of infused l-carnitine reaching the intracellular compartment was negligible (Fig. 4). The rise in the level of plasma acetylcarnitine in the elderly man was more pronounced – which might indicate that a greater fraction of infused l-carnitine reached the intracellular space due to reduced glomerular filtration and a slower excretion of the infused carnitine. This may support a strategy of maintaining plasma carnitine levels at a reasonable level with several daily doses of l-carnitine in order to ensure sufficient intracellular carnitine levels. This was partially achieved clinically as shown by a mean plasma acetylcarnitine level of 23 μmol/L after long-term l-carnitine supplementation. The mean plasma half-life of 9 h in the patients following oral intermission supports a daily regime of at least three daily dosages of l-carnitine, which is also recommended in all Faroese PCD patients.
This study demonstrated that adhesion to continued oral l-carnitine supplementation is necessary to ensure and maintain plasma carnitine levels at close to normal levels. It should not be expected that the patients develop sufficient carnitine stores during continued supplementation to compensate for an intermission in their daily supplementation regime. Even though a short-term pause in oral supplementation might not be harmful, our study shows that the plasma levels fall rapidly, which could lead to decreased intracellular carnitine levels, which again might affect cellular metabolism and prove detrimental.
Conclusion
Patients homozygous for the severe c.95A>G mutation had only limited skeletal muscle carnitine stores following long-term high-dosage l-carnitine supplementation. Mean renal reabsorption of l-carnitine was only 24% and the mean plasma half-life of free carnitine following the last oral dose of l-carnitine was 9 h. A treatment strategy of at least three daily separate doses of l-carnitine is recommended from a pharmacokinetic perspective, and while the biochemical consequences of intermission with supplementation in PCD patients are clear, the associated clinical risk is unknown from this study.
Acknowledgments
The authors would like to thank the participants for their committed effort, the Faroese Biobank for its support, and the technical staff in the National Hospital of the Faroe Islands for their skillful assistance.
Compliance with Ethics Guidelines
Conflict of Interest
Jan Rasmussen, Jákup A. Thomsen, Jess H. Olesen, Trine M. Lund, Magni Mohr, Jón Clementsen, Olav W. Nielsen, and Allan M. Lund have no conflicts of interest to report.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study. Proof that informed consent was obtained is available upon request.
Contributions of Individual Authors
Jan Rasmussen: First author and involved in all aspects of the work including conception, design, recruiting patients, analysis, and drafting of the manuscript. Guarantor of the article.
Jákup A. Thomsen: Involved in recruiting patients, collecting data, and critical revision of the manuscript.
Jess H. Olesen: Analyzed blood, skeletal muscle, and urine for carnitine and was also involved in critically revising the manuscript.
Trine M. Lund: Involved in design, pharmacokinetic analyses, and critical revision of the manuscript.
Magni Mohr: Involved in collecting data, performed the muscle biopsies, and revised the manuscript critically.
Jón Clementsen: Involved in collecting data and biomaterial and critically revising the manuscript.
Olav W. Nielsen: Involved in design, analysis, and critical revision of the manuscript.
Allan M. Lund: Involved in conception, design, analysis of data, and critically revising the manuscript.
Footnotes
Competing interests: None declared
Contributor Information
J. Rasmussen, Email: lsjanra@ls.fo, Email: ras_jan@yahoo.com
Collaborators: Johannes Zschocke
References
- Bergstrom J. Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest. 1975;35(7):609–616. doi: 10.3109/00365517509095787. [DOI] [PubMed] [Google Scholar]
- Cano A, Ovaert C, Vianey-Saban C, et al. Carnitine membrane transporter deficiency: a rare treatable cause of cardiomyopathy and anemia. Pediatr Cardiol. 2008;29(1):163–165. doi: 10.1007/s00246-007-9051-9. [DOI] [PubMed] [Google Scholar]
- Engel AG, Rebouche CJ, Wilson DM, et al. Primary systemic carnitine deficiency. II. Renal handling of carnitine. Neurology. 1981;31(7):819–825. doi: 10.1212/WNL.31.7.819. [DOI] [PubMed] [Google Scholar]
- Evans AM, Faull R, Fornasini G, et al. Pharmacokinetics of L-carnitine in patients with end-stage renal disease undergoing long-term hemodialysis. Clin Pharm Ther. 2000;68(3):238–249. doi: 10.1067/mcp.2000.108850. [DOI] [PubMed] [Google Scholar]
- Flanagan JL, Simmons PA, Vehige J, et al. Role of carnitine in disease. Nutr Metab. 2010;7:30. doi: 10.1186/1743-7075-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang RM, Bierig M, Devereux RB, et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18(12):1440–1463. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Longo N, di San A, Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C(2):77–85. doi: 10.1002/ajmg.c.30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund AM, Joensen F, Hougaard DM, et al. Carnitine transporter and holocarboxylase synthetase deficiencies in The Faroe Islands. J Inherit Metab Dis. 2007;30(3):341–349. doi: 10.1007/s10545-007-0527-9. [DOI] [PubMed] [Google Scholar]
- Madsen KL, Preisler N, Orngreen MC, et al. Patients with medium-chain acyl-coenzyme a dehydrogenase deficiency have impaired oxidation of fat during exercise but no effect of L-carnitine supplementation. J Clin Endocrinol Metab. 2013;98(4):1667–1675. doi: 10.1210/jc.2012-3791. [DOI] [PubMed] [Google Scholar]
- Magoulas PL, El-Hattab AW. Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J Rare Dis. 2012;7:68. doi: 10.1186/1750-1172-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezu J, Tamai I, Oku A, et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet. 1999;21(1):91–94. doi: 10.1038/5030. [DOI] [PubMed] [Google Scholar]
- Rasmussen J, Nielsen OW, Lund AM, et al. Primary carnitine deficiency and pivalic acid exposure causing encephalopathy and fatal cardiac events. J Inherit Metab Dis. 2013;36(1):35–41. doi: 10.1007/s10545-012-9488-8. [DOI] [PubMed] [Google Scholar]
- Rasmussen J, Kober L, Lund AM, et al. Primary Carnitine deficiency in the Faroe Islands: health and cardiac status in 76 adult patients diagnosed by screening. J Inherit Metab Dis. 2014;37(2):223–230. doi: 10.1007/s10545-013-9640-0. [DOI] [PubMed] [Google Scholar]
- Rasmussen J, Nielsen OW, Janzen N, et al. Carnitine levels in 26,462 individuals from the nationwide screening program for primary carnitine deficiency in the Faroe Islands. J Inherit Metab Dis. 2014;37(2):215–222. doi: 10.1007/s10545-013-9606-2. [DOI] [PubMed] [Google Scholar]
- Rasmussen J, Lund AM, Risom L, et al. Residual OCTN2 transporter activity, carnitine levels and symptoms correlate in patients with primary carnitine deficiency. Mol Genet Metab Reports. 2014;1:241–248. doi: 10.1016/j.ymgmr.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebouche CJ. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann N Y Acad Sci. 2004;1033:30–41. doi: 10.1196/annals.1320.003. [DOI] [PubMed] [Google Scholar]
- Rebouche CJ, Lombard KA, Chenard CA. Renal adaptation to dietary carnitine in humans. Am J Clin Nutr. 1993;58(5):660–665. doi: 10.1093/ajcn/58.5.660. [DOI] [PubMed] [Google Scholar]
- Reuter SE, Evans AM. Carnitine and acylcarnitines: pharmacokinetic, pharmacological and clinical aspects. Clin Pharmacokinet. 2012;51(9):553–572. doi: 10.1007/BF03261931. [DOI] [PubMed] [Google Scholar]
- Reuter SE, Evans AM, Chace DH, et al. Determination of the reference range of endogenous plasma carnitines in healthy adults. Ann Clin Biochem. 2008;45(Pt 6):585–592. doi: 10.1258/acb.2008.008045. [DOI] [PubMed] [Google Scholar]
- Scaglia F, Wang Y, Singh RH, et al. Defective urinary carnitine transport in heterozygotes for primary carnitine deficiency. Genet Med. 1998;1(1):34–39. doi: 10.1097/00125817-199811000-00008. [DOI] [PubMed] [Google Scholar]
- Shoji Y, Koizumi A, Kayo T, et al. Evidence for linkage of human primary systemic carnitine deficiency with D5S436: a novel gene locus on chromosome 5q. Am J Hum Genet. 1998;63(1):101–108. doi: 10.1086/301911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley CA. Carnitine deficiency disorders in children. Ann N Y Acad Sci. 2004;1033:42–51. doi: 10.1196/annals.1320.004. [DOI] [PubMed] [Google Scholar]
- Stanley CA, DeLeeuw S, Coates PM, et al. Chronic cardiomyopathy and weakness or acute coma in children with a defect in carnitine uptake. Ann Neurol. 1991;30(5):709–716. doi: 10.1002/ana.410300512. [DOI] [PubMed] [Google Scholar]
- Steiber A, Kerner J, Hoppel CL. Carnitine: a nutritional, biosynthetic, and functional perspective. Mol Aspects Med. 2004;25(5–6):455–473. doi: 10.1016/j.mam.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Tein I. Carnitine transport: pathophysiology and metabolism of known molecular defects. J Inherit Metab Dis. 2003;26(2–3):147–169. doi: 10.1023/A:1024481016187. [DOI] [PubMed] [Google Scholar]