

Abstract
Phenylketonuria (PKU) is caused by a deficiency or inactivity of the enzyme phenylalanine hydroxylase that converts phenylalanine (Phe) to tyrosine (Tyr). It has been proposed that a reduction of brain Tyr levels, as well as reduced activity of the key regulatory enzyme of dopamine (DA) synthesis tyrosine hydroxylase, leads to a depletion in DA activity in patients with PKU. We report a case of a 56-year-old woman with an intellectual disability due to late diagnosis of PKU and parkinsonism, with a modest clinical response to levodopa therapy.
We hypothesize that the signs of parkinsonism might be caused by the depletion of DA activity in the brain. Clinicians should be alert on parkinsonian symptoms in patients with PKU, particularly in those treated with agents that negatively influence DA transmission.
Introduction
Phenylketonuria (PKU) is an autosomal recessive disorder of phenylalanine metabolism caused by a deficiency or inactivity of phenylalanine hydroxylase. This enzyme is responsible for conversion of the amino acid phenylalanine (Phe) to the amino acid tyrosine (Tyr). If left untreated, high levels of Phe cause intellectual disability, microcephalia, behavioral disturbances, dermopathy, and epilepsy (Blau et al. 2010; Williams et al. 2008).
The biochemical consequences of PKU include accumulation of Phe and a deficiency of Tyr in the plasma and brain (Paans et al. 1996; Hanley et al. 2000). Tyr depletion in the brain is aggravated by a competitive mechanism through which Phe and Tyr cross the blood–brain barrier. The movement of large neutral amino acids (LNAA) across this barrier is mediated by a high-affinity, low-capacity transport system: if high levels of one of these amino acids are present in the plasma, the transport system becomes saturated, and the migration of other LNAA into the brain will be inhibited. Elevated levels of Phe in plasma thereby reduce cerebral Tyr brain influx (de Groot et al. 2013; Pietz et al. 1998).
Tyr is a precursor for dopamine (DA), a neurotransmitter that is involved in several functions in the brain, including specific cognitive functions, mood and movement. The biochemical pathway of DA is summarized in Fig. 1. The rate of synthesis of DA may be modified by the brain levels of Tyr. Furthermore, Phe is a competitive inhibitor of the enzyme tyrosine hydroxylase, which catalyzes hydroxylation of Tyr to l-3,4-dihydroxyphenylalanine (l-DOPA).
Fig. 1.
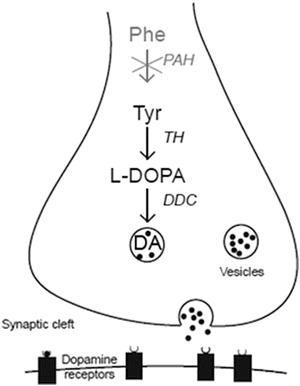
Schematic model of a central dopaminergic nerve terminal illustrating dopamine (DA) synthesis, storage, release, and postsynaptic receptor binding. DA synthesis originates from the amino acid tyrosine (Tyr), and its rate-limiting step is the conversion of Tyr to levodopa (l-DOPA) by the enzyme tyrosine hydroxylase (TH). Consequently, l-DOPA is converted to DA by the enzyme dopa decarboxylase (DDC)
The relationship between these biochemical disturbances and the clinical outcome in patients with PKU remains to be clearly established.
We present a case of an adult patient suffering from PKU and parkinsonism (a neurological syndrome characterized by tremor, hypokinesia, rigidity, and postural instability), with a modest clinical response on levodopa therapy. We hypothesize that signs of parkinsonism may be caused by the depletion of DA activity in the brain of PKU patients.
Patients and Methods
A 56-year-old woman, treated at our outpatient clinic for adults with inherited metabolic diseases, was diagnosed with PKU at the age of one year, after psychomotor developmental delay had occurred. A Phe-restricted diet was immediately started, which partially improved speech and language skills. During childhood and adolescence until she was 30 years of age, her Phe levels were usually high. It was difficult for her to adhere to the diet. Her cognitive abilities appeared to be severely impaired.
The Phe-restricted diet was discontinued between the age of 30 and 46 for unknown reasons. At the age of 46, the diet was reintroduced because of mood swings. She was first seen at our adult clinic at the age of 52. Her main complaints were tremor and mood swings. The diet appeared to be only mildly Phe restricted. An attempt to reintroduce a stricter diet, including adequate supplementation with amino acids, caused severe distress, resulting in an inability to comply fully with protein restriction. Plasma Phe levels remained typically around 1,500 μmol/L, and the diet was discontinued. At 53 years of age, valproic acid was started for aggressive behavior, with limited improvement. On examination there was an action and resting tremor of the head, hands, and arms. This tremor was more pronounced on the right side and impaired drinking and eating. There was hypomimia (masked facies). There was no rigidity, shuffling gait, or decreased arm swing. Reintroduction of a Phe-restricted diet was not considered attainable, and because of the parkinsonian features (resting tremor and hypomimia), the patient was referred to the neurologist at 55 years of age. The signs of parkinsonism (most notably hypomimia and tremor) were confirmed. Imaging of the central dopaminergic system to confirm or exclude loss of nigrostriatal DA neurons could not be performed because of the lack of cooperation from the patient. She was started on levodopa/carbidopa 50/12.5 mg three times daily. The caretakers initially reported a good response, with decreased tremor. Most notably, eating and drinking became much easier with less spill. On follow-up after about 6 months, it appeared that this response was only temporary, and the dose of the medication was gradually increased to 250/25 mg three times daily over a period of about 3 months, with again a temporary effect. Eventually she used levodopa/carbidopa 250/25 mg six times daily and the DA agonist pramipexole 0.125 mg three times daily. At this dose there was a suspicion the patient had visual hallucinations that improved after pramipexole was discontinued. An “On-Off scoring” by a specialized Parkinson nurse was performed while the patient was still using levodopa/carbidopa. The scoring was done using the Unified Parkinson’s Disease Rating Scale part III (UPDRS part III, Ramaker et al. 2002), a systematic motor observation, where a lower score corresponds to better motor skills. This was done before (“off”) and after medication (“on”). The scores were 32 and 28, respectively. The on score was better because there seemed to be a slight decrease in tremor after administration of levodopa/carbidopa 250/25 mg, and drinking was easier. After this test the medication was discontinued because of the small clinical effect. On follow-up caregivers did complain that tremor was worse without treatment, and levodopa was restarted in a low dose (125/12.5 mg thrice daily).
Currently, she lives in an assisted care facility and continuous supervision is required. She is completely ADL (activities of daily living) dependent. She is able to understand some simple verbal commands, and her expressive communicative capacities are limited to brief monosyllabic answers.
Discussion
We described a case of a 56-year-old female PKU patient with severe intellectual disability and inadequate restriction of dietary Phe, who presented with symptoms of parkinsonism at age 52. A modest clinical response to levodopa treatment was observed. Although a chance association between PKU and parkinsonism, and a diagnosis of “true Parkinson’s disease” is possible, the identification of this and previous cases, in combination with a well-defined disorder of brain catecholamine biosynthesis, suggests a possible etiologic association.
The prevalence of neurological symptoms in PKU patients is high: an action or postural tremor is estimated to occur in 5–32% of early-treated adult PKU patients (Cleary et al. 1994; McDonnell et al. 1998; Pietz et al. 1996; Weglage et al. 1995). Hand tremor is present in a third of those afflicted with late diagnosis, and generalized dystonia has also been reported (Brenton and Pietz 2000; Paine 1957). Late motor and cognitive decline may occur in adults who have relaxed their diet (Brenton and Pietz 2000; Thompson et al. 1990). The clinical deterioration is often reversible on resumption of dietary therapy (Thompson et al. 1990).
Several previous studies suggest DA deficiency in PKU patients. For example, using a genetic murine model of PKU, reduced levels of l-DOPA were demonstrated in the medial prefrontal cortex (Pascucci et al. 2012). Furthermore, a positron emission tomography study was performed to measure the utilization of 6-[18F]fluoro-l-dopamine (FDOPA) in the brain of seven adults suffering from PKU with elevated Phe levels, but lacking neurological deficits, compared to healthy subjects (Landvogt et al. 2008). A reduction of the rate of utilization of FDOPA in the striatum of adult PKU patients was found, suggesting reduced DA synthesis. Another study reported a reduction of homovanillic acid (HVA, the main metabolite of DA) levels in cerebrospinal fluid (CSF) in 8 early-treated on-diet PKU adolescents, with Phe levels between 708 and 1,161 μmol/L, in comparison with a control group (Burlina et al. 2000). Six of these patients had brisk deep tendon reflexes, intentional tremor, and/or ankle clonus. McKean found low levels of DA in brain tissue at autopsy. An inverse relation between plasma levels of Phe and HVA in the CSF was also found (McKean 1972). However, a DA deficiency in PKU, as shown above, does not lead to overt parkinsonism in all cases, and reports of clear parkinsonism in PKU are rare. We are aware of one case of levodopa-responsive parkinsonism reported in an adult with PKU (Evans et al. 2004).
Considering the biochemical changes in PKU and the knowledge from abovementioned studies, PKU patients might be more susceptible for neurological symptoms caused by cerebral DA deficiency, especially those with high Phe–Tyr ratios (who relaxed their diet). DA plays a crucial role in voluntary movement. For example, Parkinson’s disease, the most common neurodegenerative cause of parkinsonism, is associated with degeneration of dopaminergic neurons. Therefore clinicians should be alert on possible parkinsonian symptoms in patients with PKU. Notably, a substantial part of the PKU patients with an intellectual disability is prescribed DA antagonists that are associated with extrapyramidal symptoms such as parkinsonism. Other agents that negatively influence DA transmission include the calcium channel blockers flunarizine and cinnarizine (Brücke et al. 1995) and the antiemetic drug metoclopramide. Although no systematic studies exist, and motor symptoms emerge only after pronounced cerebral DA dysfunction (Cummings et al. 2014), prescription of medication that negatively influences DA transmission might carry a risk to cause or aggravate parkinsonian symptoms in PKU patients. Also diet should be optimized whenever possible. If parkinsonism persists, levodopa therapy should be considered. We suggest that this issue should be further explored.
Synopsis
Our case illustrates a slight decrease of symptoms of parkinsonism after introduction of levodopa in a patient suffering from PKU. Signs of parkinsonism might be caused by DA depletion in the brain. Symptoms might be aggravated or provoked by agents that negatively influence DA transmission.
Compliance with Ethics Guidelines
Conflict of Interest
Marieke Velema, Erik Boot, Marc Engelen, and Carla Hollak declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from the legal representative of the patient for being included in the study.
Contribution of the Authors
Marieke Velema: involved in the conception and design and drafting the article
Erik Boot: involved in the conception and design and revising the article critically
Marc Engelen: involved in the conception and design and revising the article critically
Carla Hollak: involved in the conception and design and revising the article critically. Guarantor
Footnotes
Competing interests: None declared
Contributor Information
Carla Hollak, Email: c.e.hollak@amc.uva.nl.
Collaborators: Johannes Zschocke
References
- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376:1417–1427. doi: 10.1016/S0140-6736(10)60961-0. [DOI] [PubMed] [Google Scholar]
- Brenton DP, Pietz J. Adult care in phenylketonuria and hyperphenylalaninaemia: the relevance of neurological abnormalities. Eur J Pediatr. 2000;159(Suppl 2):S114–S120. doi: 10.1007/PL00014373. [DOI] [PubMed] [Google Scholar]
- Brücke T, Wöber C, Podreka I, et al. D2 receptor blockade by flunarizine and cinnarizine explains extrapyramidal side effects. A SPECT study. J Cereb Blood Flow Metab. 1995;15:513–518. doi: 10.1038/jcbfm.1995.63. [DOI] [PubMed] [Google Scholar]
- Burlina AB, Bonafé L, Ferrari V, Suppiej A, Zacchello F, Burlina AP. Measurement of neurotransmitter metabolites in the cerebrospinal fluid of phenylketonuric patients under dietary treatment. J Inherit Metab Dis. 2000;23:313–316. doi: 10.1023/A:1005694122277. [DOI] [PubMed] [Google Scholar]
- Cleary MA, Walter JH, Wraith JE, et al. Magnetic resonance imaging of the brain in phenylketonuria. Lancet. 1994;344:87–90. doi: 10.1016/S0140-6736(94)91281-5. [DOI] [PubMed] [Google Scholar]
- Cummings JL, Fine MJ, Grachev ID, et al. Effective and efficient diagnosis of parkinsonism: the role of dopamine transporter SPECT imaging with ioflupane I-123 injection (DaTscan™) Am J Manag Care. 2014;20:S97–S109. [PubMed] [Google Scholar]
- De Groot MJ, Hoeksma M, Reijngoud DJ, et al. Phenylketonuria: reduced tyrosine brain influx relates to reduced cerebral protein synthesis. Orphanet J Rare Dis. 2013;8:133. doi: 10.1186/1750-1172-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AH, Costa DC, Gacinovic S, et al. L-DOPA-Responsive Parkinson’s syndrome in association with phenylketonuria: in vivo dopamine transporter and D2 receptor findings. Mov Disord. 2004;19:1232–1236. doi: 10.1002/mds.20146. [DOI] [PubMed] [Google Scholar]
- Hanley WB, Lee AW, Hanley AJ, et al. “Hypotyrosinemia” in phenylketonuria. Mol Genet Metab. 2000;69:286–294. doi: 10.1006/mgme.2000.2985. [DOI] [PubMed] [Google Scholar]
- Landvogt C, Mengel E, Bartenstein P, et al. Reduced cerebral fluoro-L-dopamine uptake in adult patients suffering from phenylketonuria. J Cereb Blood Flow Metab. 2008;28:824–831. doi: 10.1038/sj.jcbfm.9600571. [DOI] [PubMed] [Google Scholar]
- McDonnell GV, Esmonde TF, Hadden DR, Morrow JI. A neurological evaluation of adult phenylketonuria in Northern Ireland. Eur Neurol. 1998;39:38–43. doi: 10.1159/000007895. [DOI] [PubMed] [Google Scholar]
- McKean CM. The effects of high phenylalanine concentrations on serotonin and catecholamine metabolism in the human brain. Brain Res. 1972;47:469–476. doi: 10.1016/0006-8993(72)90653-1. [DOI] [PubMed] [Google Scholar]
- Paans AM, Pruim J, Smit GP, Visser G, Willemsen AT, Ullrich K. Neurotransmitter positron emission tomographic-studies in adults with phenylketonuria, a pilot study. Eur J Pediatr. 1996;155:S78–S81. doi: 10.1007/PL00014257. [DOI] [PubMed] [Google Scholar]
- Paine RS. The variability in manifestations of untreated patients with phenylketonuria (phenylpyruvic aciduria) Pediatrics. 1957;20:290–302. [PubMed] [Google Scholar]
- Pascucci T, Giacovazzo G, Andolina D, et al. (2012) In vivo catecholaminergic metabolism in the medial prefrontal cortex of ENU2 mice: an investigation of the cortical dopamine deficit in phenylketonuria. J Inherit Metab Dis. 2012;35:1001–1009. doi: 10.1007/s10545-012-9473-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietz J, Kreis R, Schmidt H, Meyding-Lamade UK, Rupp A, Boesch C. Phenylketonuria: findings at MR imaging and localized in vivo H-1 MR spectroscopy of the brain in patients with early treatment. Radiology. 1996;201:413–420. doi: 10.1148/radiology.201.2.8888233. [DOI] [PubMed] [Google Scholar]
- Pietz J, Dunckelmann R, Rupp A, et al. Neurological outcome in adult patients with early-treated phenylketonuria. Eur J Pediatr. 1998;157:824–830. doi: 10.1007/s004310050945. [DOI] [PubMed] [Google Scholar]
- Ramaker C, Marinus J, Stiggelbout AM, van Hilten BJ. Systematic evaluation of rating scales for impairment and disability in Parkinson’s disease. Mov Disord. 2002;17:867–876. doi: 10.1002/mds.10248. [DOI] [PubMed] [Google Scholar]
- Thompson AJ, Smith I, Brenton D, et al. Neurological deterioration in young adults with phenylketonuria. Lancet. 1990;336:602–605. doi: 10.1016/0140-6736(90)93401-A. [DOI] [PubMed] [Google Scholar]
- Weglage J, Pietsch M, Funders B, Koch HG, Ullrich K. Neurological findings in early treated phenylketonuria. Acta Paediatr. 1995;84:411–415. doi: 10.1111/j.1651-2227.1995.tb13661.x. [DOI] [PubMed] [Google Scholar]
- Williams RA, Mamotte CD, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29:31–41. [PMC free article] [PubMed] [Google Scholar]