

Abstract
Objectives: Hurler syndrome is characterized by progressive multisystem deterioration leading to early death in childhood. This prospective study evaluated the long-term outcomes of patients with Hurler syndrome who underwent umbilical cord blood transplantation from unrelated donors.
Study design: Only patients with Hurler syndrome who underwent umbilical cord blood transplantation between December 1995 and March 2006 (n = 25) and who were followed for at least 5 years (n = 19) were included in the analysis. The patients were longitudinally evaluated by a multidisciplinary team of specialists following a standardized protocol.
Results: Median age at transplantation was 15.9 months (range 2.1–35), and patients were followed up until a median age of 10.1 years (range 7.2–14.9). Overall survival was 80%. All successfully transplanted patients achieved full donor chimerism and normal enzyme levels, and all children continue to make gains in development. Gross motor function was the most affected area. Vision and hearing were compromised in a minority of the patients, with some requiring corneal transplant or hearing aids. Cardiopulmonary function improved. Some children required orthopedic surgery, but severe complications were prevented in most patients. Although longitudinal growth was lower than that of unaffected children, it was considerably higher than expected from the natural course of the disease. Head circumference normalized. Hydrocephalus was not observed at longer follow-up, and cerebral atrophy decreased over time.
Conclusions: In this descriptive study of children with Hurler syndrome, unrelated umbilical cord blood transplantation was associated with improved somatic disease and neurodevelopment.
Introduction
Hurler syndrome is the most severe form of mucopolysaccharidosis type I (MPS I), an autosomal recessive lysosomal storage disorder caused by a deficiency of α-l-iduronidase (IDUA). The resulting accumulation of glycosaminoglycans leads to generalized cell, tissue, and organ dysfunction. Without treatment, patients with Hurler syndrome experience multisystem manifestations including mental retardation, skeletal deterioration, severe cardiopulmonary disease, hepatosplenomegaly, visual impairment, and deafness, usually leading to death within the first decade of life (Neufeld and Muenzer 2001).
Allogeneic hematopoietic stem cell (HSC) transplantation was first performed in a patient with Hurler syndrome three decades ago and has been extensively performed since that time (Hobbs et al. 1981). The engrafted donor-derived stem cells provide a continuous source of IDUA throughout the body, including the central nervous system. Since enzyme replacement therapy (ERT) is not capable of crossing the blood–brain barrier, HSC transplantation is the treatment of choice for Hurler syndrome. Clinical outcomes include reversal of organomegaly, preservation of neurocognitive development, and improved hearing, vision, and cardiopulmonary function in most transplanted patients (Guffon et al. 1998; Whitley et al. 1993; Shapiro et al. 1995; Peters et al. 1996; Vellodi et al. 1997; Peters et al. 1998; Souillet et al. 2003; Aldenhoven et al. 2008; Malm et al. 2008). However, outcomes are highly variable, and manifestations in certain organ systems continue to progress. These results were often based on small study populations or short follow-up. Additionally, there is significant heterogeneity among the stem cell sources used, conditioning regimens applied, and methods of clinical evaluation. Finally, enzyme levels were suboptimal in a significant percentage of these children because of mixed chimerism or use of a carrier donor.
In addition to bone marrow and peripheral blood, unrelated umbilical cord blood is an effective HSC source. There is evidence of improved neurocognitive outcomes after umbilical cord blood transplantation (UCBT) in children with Hurler syndrome, adrenoleukodystrophy, and Krabbe disease, particularly when performed early in the disease course (Staba et al. 2004; Escolar et al. 2005; Beam et al. 2007; Prasad et al. 2008). Various studies of UCBT in Hurler syndrome patients report high rates of sustained engraftment with full donor chimerism and normal IDUA activity (Staba et al. 2004; Martin et al. 2006; Prasad et al. 2008). Furthermore, umbilical cord blood is readily available, reducing the time between diagnosis and transplantation, which is important for optimal prognosis (Staba et al. 2004; Escolar et al. 2005).
The purpose of this manuscript is to describe the outcomes of 25 patients with Hurler syndrome who underwent UCBT in two institutions and were prospectively followed long-term using a standardized protocol.
Methods
Patient Characteristics
We reviewed the data of patients with Hurler syndrome referred for neurodevelopmental evaluations to the Program for the Study of Neurodevelopment in Rare Disorders (NDRD) at the University of North Carolina at Chapel Hill. The patients underwent UCBT between December 1995 and March 2006 at Duke University Medical Center and the University of Minnesota and were followed by the NDRD for at least 5 years. In all patients, diagnosis was confirmed by clinical phenotype and low IDUA activity in peripheral blood leukocytes. A standardized protocol was used to assess all patients. Somatic disease was evaluated at various departments at Duke University, University of North Carolina, or University of Minnesota. The transplant data of all the patients were previously reported (Prasad et al. 2008; Orchard et al. 2010). This study was reviewed by the institutional review board of the University of North Carolina at Chapel Hill. Parents of all patients provided written informed consent before enrollment.
Mutation Analysis
Mutation analysis was performed on DNA from skin fibroblasts by the Department of Genetic Medicine of the Government of South Australia Children, Youth and Women’s Health Service in North Adelaide, Australia. All patients were initially tested for four common mutations (p.Q70X, p.A327P, p.W4O2X, p.P533R). If the mutations could not be identified by targeted mutation testing, sequencing of the IDUA gene was performed, and mutations were confirmed by restriction fragment length analysis or allele-specific oligonucleotide analysis. The decision to proceed with UCBT was based primarily on clinical phenotype and/or family history and low enzyme activity. The rationale for this was that although some common nonsense mutations are associated with the severe Hurler syndrome phenotype, many MPS I patients have at least one private mutation, which makes phenotype prediction difficult (Beesley et al. 2001; Terlato and Cox 2003).
Transplantation Procedure
All patients underwent conditioning with busulfan, cyclophosphamide, and horse antithymocyte globulin. Prophylaxis against graft-versus-host disease (GvHD) was given using cyclosporine for 9 months and methylprednisolone for 2–3 months. Supportive care was provided as previously described (Staba et al. 2004; Thomas et al. 2006; Prasad et al. 2008). Most patients underwent tonsillectomy, adenoidectomy, and pressure-equalizing tube placement before transplant.
Clinical Follow-Up
Patients were evaluated at baseline and every 6–12 months after transplant by multiple pediatric subspecialists, including a cardiologist, audiologist, ophthalmologist, otolaryngology specialist, orthopedic surgeon, and a neurodevelopmental pediatrician working with speech therapists, psychologists, and physical therapists. Brain magnetic resonance imaging (MRI) and neurophysiology tests (electromyography, brainstem auditory evoked responses) were performed at the specified intervals. Serial neuroimaging and neurophysiologic and neurodevelopmental studies were all performed within 1 week at each clinical time point.
Neurodevelopmental Assessment
All patients underwent comprehensive neurodevelopmental examinations using standardized and validated neurobehavioral tools, and outcomes were compared to norms of typically developing children (Martin et al. 2006). Age equivalents were used to allow comparison across tests and determine the acquisition of new skills. Gross motor, cognitive, language (receptive and expressive), adaptive behavior, and fine motor skills were longitudinally assessed. Adaptive behavior is a standardized measure of independent functioning and self-help skills based on parents’ perceptions of their child’s abilities.
Somatic Assessments
Patients were assessed for somatic outcomes including:
Cardiopulmonary outcome: Echocardiograms and cardiologic and otolaryngology consultations were performed to assess valvular insufficiency, cardiomyopathy, and upper respiratory obstruction.
Orthopedic outcome: Orthopedic assessments were carried out to determine the need for orthopedic surgery, including those related to kyphoscoliosis, atlantoaxial instability, cord compression, hip dysplasia, genu valgum, or carpal tunnel syndrome.
Audiologic outcome: Behavioral audiometry and brainstem auditory evoked responses were performed to detect hearing loss and need for hearing aids. Ophthalmologic outcome: Ophthalmologic examinations were performed at baseline and during follow-up to assess corneal clouding and need for corneal transplantation.
Brain MRI: Brain MRI images were analyzed for abnormalities including hydrocephalus and cerebral atrophy.
Growth: Weight, longitudinal height, and head circumference were compared to gender- and age-specific growth charts from the Centers for Disease Control and Prevention. A subgroup of the patients was tested for growth hormone deficiency and evaluated to determine the need for growth hormone treatment.
All patients were followed at least 5 years; we divided the follow-up before and after 2 years to help identify outcomes that occurred closer to the time of transplantation and those that were at least 2 years from transplantation.
Statistical Analysis
The cumulative incidences of engraftment and GvHD were calculated by standard methods. The probability of event-free survival (survival with durable engraftment of donor cells) was calculated by Kaplan–Meier analysis. The cutoff date for data analysis was December 1, 2011. To evaluate neurodevelopment, a general linear mixed model was fit to the data. Age at evaluation and age-equivalent scores were used to describe and compare development across different domains of function (neurocognitive, language, motor, and adaptive). Descriptive statistics were used to describe somatic outcomes.
Results
Patient Characteristics
Twenty-five children (11 boys, 14 girls) were included in the study, and 19 children were followed up for >5 years after UCBT. Median follow-up time was 13.0 years (range 9.9–18.5) after transplantation, and median age at last follow-up was 10.1 years (range 7.2–14.9). Most (88%) of the children were of European descent (Table 1). Median age at diagnosis was 11.0 months (range 0–28 months), and median age at transplantation was 15.9 months (range 2.1–35). None of the patients received ERT. Only patients transplanted at Duke fit the inclusion criteria and were used for the analysis.
Table 1.
Patient and donor graft characteristics, engraftment, graft-versus-host disease, and survival of Hurler syndrome patients who underwent umbilical cord blood transplantation
| Patient Characteristics | n (%) | ||
|---|---|---|---|
| Total number of patients (N = 25) | |||
| Gender | Male | 11 (44) | |
| Female | 14 (56) | ||
| Race | American Indian/Alaska Native | 1 (4) | |
| Asian | 1 (4) | ||
| African American | 1 (4) | ||
| White | 22 (88) | ||
| Ethnicity | Hispanic | 3 (12) | |
| Median | Range | ||
| Age at diagnosis (months) | 11 | 0–28 | |
| Age at UCBT (months) | 15.9 | 2.1–35 | |
| Age at last follow-up (years) living and engrafted | 10.1 | 7.2–14.9 | |
| Age at last follow-up (years) (n = 6 deceased or graft failure) | 1.8 | 0.9–3.2 | |
Mutation Analysis
Mutation analysis was performed for 19/25 patients. At least two putative disease-causing IDUA mutations were detected in 14 patients, one disease-causing mutation in four patients, and an unknown mutation in one patient (Table 2). Fifteen patients were homozygous or compound heterozygous for mutations associated with the severe phenotype. In four patients an unknown mutation was identified. Targeted mutation analysis for the four common mutations detected two (p.Q70X, p.W4O2X) IDUA mutations in eight patients, and targeted mutation analysis combined with gene sequencing detected both mutations in seven other patients. For four patients targeted mutation analysis detected only one mutation, with no additional mutations identified by gene sequencing, and for two patients gene sequencing identified both mutations. The following six IDUA mutations were identified by sequence analysis: p.M133I, p.Y202X, Q400X, p.R628X, c.1614delG, and IVS9-1G>T.
Table 2.
Mutation analysis for 19 of 25 patients
| Patient | Allele 1 mutation | Allele 2 mutation |
|---|---|---|
| 1 | W402X | W402X |
| 2 | W402X | M133I/Y202X |
| 3 | Q70X | W402X |
| 4 | None detected | None detected |
| 5 | Missing | Missing |
| 6 | Q70X | Q70X |
| 7 | W402X | Unknown |
| 8 | Missing | Missing |
| 9 | W402X | Unknown |
| 10 | Missing | Missing |
| 11 | Missing | Missing |
| 12 | W402X | W402X |
| 13 | W402X | W402X |
| 14 | W402X | E299X |
| 15 | W402X | W402X |
| 16 | W402X | c.1614delG |
| 17 | Q400X | IVS9-1G>T |
| 18 | W402X | Q70X |
| 19 | Q400X | IVS9-1G>T |
| 20 | W402X | W402X |
| 21 | W402X | Unknown |
| 22 | W402X | Unknown |
| 23 | W402X | R628X |
| 24 | Missing | Missing |
| 25 | Not performed | Not performed |
Engraftment and GvHD
Six children had chronic and extensive GvHD, whereas seven patients had mild cases with only skin involvement.
Survival
As of December 1, 2011, five of the 25 children died post-transplant because of infection (n = 2), hepatic/gastrointestinal bleeding (n = 1), respiratory failure (n = 1), or chronic GvHD (n = 1). Graft failure occurred in two patients, both of whom underwent a second transplant. One of these patients died, and the second failed to engraft. Overall survival was 80%. All patients who underwent successful transplantation achieved full donor chimerism (>90%), and all showed IDUA activity within the reference range.
Neurodevelopmental Function
Post-transplantation neurodevelopmental function of children with Hurler syndrome was compared to the neurodevelopmental function of unaffected children. All six areas of development (gross/fine motor, cognitive, receptive/expressive language, adaptive behavior) contribute to overall functioning. Gross and fine motor functions were the most affected, progressing at significantly lower rates compared to those of unaffected children of the same age. On the other hand, after successful UCBT, patients gained cognitive skills at a normal rate and maintained the gains over time (Fig. 1). Because of the degree of motor involvement, many activities of daily living were affected, leading to lower adaptive behavior compared to age-matched unaffected children.
Fig. 1.
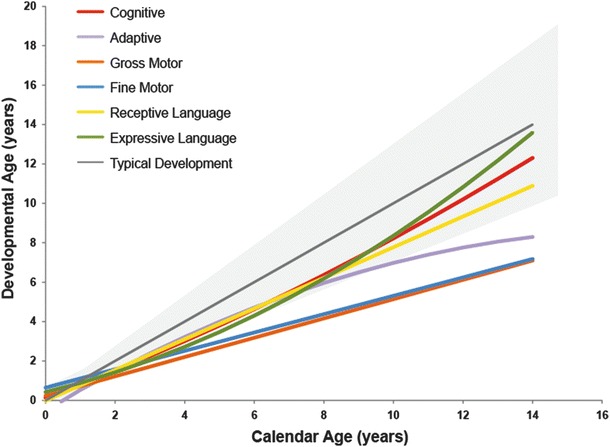
Neurodevelopmental function of children with Hurler syndrome after umbilical cord blood transplantation compared to that of unaffected children. Age-equivalent scores were used to compare and monitor developmental progress. The colored lines depict the mean developmental curves (i.e., cognitive, adaptive, gross motor, fine motor, receptive language, and expressive language) of the surviving patients. These lines were plotted against the mean typical cognitive growth curve (gray continuous line) and approximate variability (95%; gray area) observed in typically developing children
Somatic Function
Table 3 outlines somatic manifestations present before treatment, during the initial 2 years after transplantation, and after the subsequent 2-years to understand which outcomes are expected closer and further away from the time of transplant.
Table 3.
Somatic manifestations in Hurler syndrome patients who were evaluated more than 5 years after receiving umbilical cord blood transplantation
| Organ system | Manifestation | At baseline (pre-UCBT) n (%) | At 0–2 years FU (post-UCBT) n (%) | At >2 years FU (post-UBCT) n (%) | |
|---|---|---|---|---|---|
| Cardiopulmonary | Valvulopathy: insufficiency | 4/14 (29) | 4/16 (25) | 13/19 (68) | |
| Cardiomyopathy | 2/13 (15) | 4/15 (27) | 3/19 (16) | ||
| Upper respiratory obstruction | 4/13 (31) | 0/15 (0) | 13/19 (68) | ||
| Orthopedic | Thoracolumbar kyphosis: spinal surgery | 0/13 (0) | 0/15 (0) | 6/19 (32) | |
| Hip dysplasia: surgery | 0/13 (0) | 0/15 (0) | 4/19 (21) | ||
| Genu valgum: surgery | 0/13 (0) | 0/15 (0) | 12/19 (63) | ||
| Carpal tunnel syndrome: surgery | 0/13 (0) | 0/15 (0) | 9/19 (47) | ||
| Cord compression: surgery | 0/13 (0) | 0/15 (0) | 0/19 (0) | ||
| Atlantoaxial instability: surgery | 0/13 (0) | 0/15 (0) | 0/19 (0) | ||
| Other surgery* | 0/12 (0) | 0/15 (0) | 4/19 (21) | ||
| Audiologic | Hearing loss: BAER abnormalities | 2/13 (15) | 0/15 (0) | 1/19 (5) | |
| Hearing loss: amplification | 0/13 (0) | 4/15 (27) | 7/19 (37) | ||
| Ophthalmologic | Corneal clouding | 8/14 (57) | 8/16 (50) | 18/19 (95) | |
| Degree | None | 6/14 (43) | 8/16 (50) | 1/19 (5) | |
| Mild | 5/14 (36) | 6/16 (38) | 8/19 (42) | ||
| Moderate | 3/14 (21) | 2/16 (13) | 7/19 (37) | ||
| Severe | 0/14 (0) | 0/16 (0) | 3/19 (16) | ||
| Corneal transplant | 0/13 (0) | 0/15 (0) | 4/19 (21) | ||
| Cerebral MRI | Hydrocephalus: surgery (VP shunt)** | 3/13 (16) | 5/15 (33) | 6/19 (32) | |
| Cerebral atrophy | 8/16 (50) | 5/15 (33) | 6/19 (32) | ||
| Growth | GH deficiency | 0/13 (0) | 0/15 (0) | 7/19 (37) | |
| GH treatment | 0/13 (0) | 0/15 (0) | 11/19 (58) | ||
*Other operations: trigger finger release 2/24 (8%), ankle surgery 2/24 (8%), Achilles tendon lengthening 1/24 (4%)
**New cases of VP shunt insertion
Cardiopulmonary Outcome
Cardiomyopathy was observed at baseline (15%), during the first 2 years post-transplant (27%), and after the 2-year follow-up (16%). However, cardiomyopathy resolved in all but one patient after longer follow-up. In contrast, cardiac valve insufficiency appeared to progress over time, detected in 29% of patients before UCBT, 25% during the first 2 years post-transplant, and 68% after the 2-year follow-up.
Orthopedic Outcome
Orthopedic manifestations requiring surgical intervention included kyphoscoliosis (32%), hip dysplasia (21%), genu valgum (63%), and carpal tunnel syndrome (47%). Other operations performed were trigger finger surgery, surgery for valgus deformity of the ankles, and Achilles tendon lengthening. All orthopedic interventions were performed more than 2 years after transplantation. No interventions were required for atlantoaxial instability, and none of the patients had cord compression.
Audiologic Outcome
Amplification for hearing loss was required for 0% of patients before UCBT, 27% of patients during the first 2 years post-transplant, and 37% of patients after the 2-year follow-up. After 2 years only one patient had abnormal brainstem auditory evoked responses.
Ophthalmologic Outcome
After 2 years of follow-up, 95% of the patients had corneal clouding (mild, n = 8; moderate, n = 7; severe, n = 3). Corneal clouding progressed over time in 56% of patients, stabilized in 38% of patients, and improved in 6% of patients. Corneal transplantation was required for severe corneal clouding in 21% of patients >2 years post-transplant.
Neurological Outcome
Neurosurgical intervention with a ventriculoperitoneal shunt (VPS) was performed for hydrocephalus in 16% of patients before transplantation, 33% of the patients required a VPS in the first 2 years post-transplantation, and 32% continued to require a VPS >2 years post-transplant. However, none required additional VPS insertions 2 years after transplantation.
Cerebral MRI
Cerebral atrophy was present in 50% of patients at baseline but was detected in only 32% of patients >2 years post-transplant.
Growth
Longitudinal height, weight, and head circumference are shown in Fig. 2a–f. After successful transplantation, longitudinal height remains most affected, ranging from <5th to 50th percentile in both boys and girls. Eleven patients received growth hormone treatment. Weight was normal in the vast majority of patients, and head circumference normalized during longer follow-up.
Fig. 2.
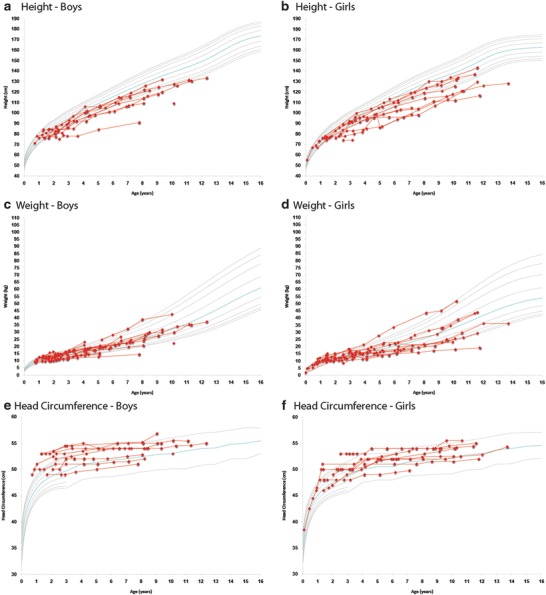
Longitudinal height, weight, and head circumference in boys and girls from umbilical cord blood transplantation until last follow-up visit. The gray curves represent standard growth curves (3rd, 5th, 10th, 25th, 50th, 75th, 90th, 95th, and 97th percentiles) for (a, b) height, (c, d) weight, and (e, f) head circumference. The red lines represent individual patients
Discussion
In this study we evaluated the long-term functional outcomes of 19 children who underwent UCBT for Hurler syndrome. The safety and efficacy of UCBT from unrelated donors was similar to that reported by previous studies (Staba et al. 2004; Martin et al. 2006; Prasad et al. 2008). However, this is the only comprehensive prospective longitudinal study on a large cohort of patients with Hurler syndrome who were evaluated before and after transplantation using a standardized protocol. Transplantation was performed using a single graft source (umbilical cord blood) and the same conditioning regimen for all patients. Full donor chimerism and normal enzyme levels were obtained in all successfully transplanted patients. Clinical follow-up consisted of multidisciplinary evaluations performed within a single week and at regular intervals for a mean of 8.6 years (range 0.1–14.9) after transplantation. Neurodevelopmental data provided information in the six areas of functional skills.
Our results show that UCBT successfully provided a means of enzyme replacement and stabilized the disease in these patients. Motor skills were more severely affected than other functional domains and responded less well to treatment, affecting adaptive behavior, self-help skills, and ability to carry out activities of daily living. On the other hand, cognitive development was preserved, with continued gains in cognitive function.
Long-term somatic outcomes were encouraging. Cardiopulmonary function significantly improved in most patients, with cardiomyopathy or severe respiratory obstruction rarely seen after the 2-year follow-up. Transient cardiomyopathy was observed before or within the first 2 years after transplantation, mainly due to steroid use or as a transplantation-related complication. Valve involvement was still common at longer follow-up but did not appear to significantly interfere with cardiac function. More than 2 years after transplantation, hearing was considerably compromised in some patients, requiring amplification. Although residual corneal clouding was common, progression to an advanced state requiring corneal transplantation was prevented in most patients.
Overall, the skeletal system responded less well to UCBT, as demonstrated by the need for orthopedic interventions. Importantly, severe complications including cord compression and atlantoaxial instability were prevented, and linear growth was significantly higher than would be expected during the natural course of the disease. Without treatment, children with Hurler syndrome show severe growth failure by the age of 2 years, achieving a maximal height of 110 cm (Neufeld and Muenzer 2001). However, this increased height after transplantation may have contributed to the progression of orthopedic complications. Eleven patients received growth hormone treatment; however, whether a defined subgroup of patients will benefit from treatment with growth hormone needs further evaluation. Hydrocephalus did not develop at longer follow-up in most patients, and a VPS was placed mainly before, during, or immediately after transplantation. Furthermore, longitudinal head circumference normalized and subsequently stabilized during long-term follow-up. The presence of cerebral atrophy on MRI also decreased over time.
At present, HSC transplantation is the only treatment able to provide the IDUA enzyme throughout the body, including the central nervous system, making it the treatment of choice for Hurler syndrome patients. For patients who do not undergo transplantation because of delayed diagnosis, lack of a compatible donor, or major concerns of parents, ERT has been used in an attempt to improve quality of life (Thomas et al. 2006; Tokic et al. 2007; Wraith et al. 2007). Although some clinical benefits have been reported for ERT, continued musculoskeletal and central nervous system deterioration have been observed. In a study of patients with severe MPS I younger than 5 years of age, follow-up was too short to draw any conclusions on neurocognitive outcome (Wraith et al. 2007). Another potential limitation of ERT is the induction of an immune response to the therapeutic enzyme. Antibodies produced against IDUA may neutralize the effect of enzyme replacement by reducing the efficiency of enzyme uptake and redirecting the enzyme to other target tissues (Dickson et al. 2008). To circumvent the blood–brain barrier, intrathecal ERT is currently being studied; however, the long-term safety and efficacy remain unclear (Tolar et al. 2009). Transplantation of HSCs genetically modified to express supranormal levels of IDUA showed promising results in MPS I mice (Visigalli et al. 2010). Whether this therapy represents an effective and safe therapeutic option for human patients in the future warrants further clinical studies.
In several other lysosomal storage disorders, early treatment with HSC transplantation has resulted in superior neurodevelopmental outcomes. We have also demonstrated benefits for Hurler syndrome, with transplantation before 9 months of age associated with improvements in cognitive function, receptive and expressive language, and adaptive behavior (Poe et al. 2014). Early treatment may have similar effects for some of the somatic manifestations of Hurler syndrome, but whether orthopedic complications can be prevented remains to be elucidated. Our results regarding neurodevelopmental function and growth suggest that newborn screening for Hurler syndrome is needed to optimize the benefits from transplantation. The challenges with newborn screening, however, will be predicting which children who screen positive will develop neurological disease and therefore require transplantation.
Our results are consistent with previous studies reporting that certain disease manifestations continue to progress despite successful transplantation. Insufficient enzyme delivery to poorly vascularized tissues is thought to account for musculoskeletal deterioration after transplantation, similar to that of untreated children (Weisstein et al. 2004; Aldenhoven 2008). Although HSCT may improve ventricular hypertrophy and ventricular function, cardiac valve disease persists (Braunlin et al. 2010), and hearing impairment, persistent corneal clouding, and noninfectious pulmonary complications (e.g., diffuse alveolar hemorrhage, idiopathic pneumonia syndrome) after transplantation are also common (Souillet et al. 2003; Kharbanda et al. 2006). Therefore, these patients require close follow-up consisting of at least annual assessment by a multidisciplinary team with experience treating children with MPS I (Muenzer et al. 2009).
In our study long-term follow-up of patients with Hurler syndrome who underwent UCBT showed significant improvement of cardiopulmonary function and neurocognitive functioning, prevention of hydrocephalus and severe orthopedic complications, amelioration of vision and hearing impairment, and preservation of cognitive development in the large majority of the patients. These effects accounted for the dramatically increased life expectancy and improved quality of life after transplantation. Further studies are needed to determine whether even earlier transplantation might further improve the prognosis of these patients.
Acknowledgments
We would like to acknowledge the families who travelled for the evaluation, some of which self-referred for follow-up, and all the clinicians at the University of North Carolina, University of Minnesota, and Duke University Medical Center who participated in the care of these patients. This study was funded by the Caterina Marcus Foundation.
Synopsis
Unrelated umbilical cord transplantation is associated with improved cognitive development and cardiopulmonary function, attenuated vision and hearing impairment, and prevention of hydrocephalus and severe orthopedic complications.
Compliance with Ethics Guidelines
Conflict of Interest
Hannah Y. Coletti, Mieke Aldenhoven, Karina Yelin, Michele D. Poe, Joanne Kurtzberg, and Maria L. Escolar declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients included in the study.
Details of the Contributions of Individual Authors
HYC and KY collected and entered the data and drafted the initial manuscript; MLE was the principal investigator of the study and secured funding; MA and MLE finished the manuscript; MDP performed statistical analysis; MLE interpreted the results; and JK and MLE critically revised the manuscript.
Footnotes
Competing interests: None declared
Contributor Information
Maria L. Escolar, Email: maria.escolar@chp.edu
Collaborators: Johannes Zschocke
References
- Aldenhoven M, Boelens JJ, de Koning TJ (2008) The clinical outcome of Hurler syndrome after stem cell transplantation. Biol Blood Marrow Transplant 14:485–498 [DOI] [PubMed]
- Beam D, Poe MD, Provenzale JM, et al. Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy. Biol Blood Marrow Transplant. 2007;13:665–674. doi: 10.1016/j.bbmt.2007.01.082. [DOI] [PubMed] [Google Scholar]
- Beesley CE, Meaney CA, Greenland G, et al. Mutational analysis of 85 mucopolysaccharidosis type I families: frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum Genet. 2001;109:503–511. doi: 10.1007/s004390100606. [DOI] [PubMed] [Google Scholar]
- Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2010;34:1183–1197. doi: 10.1007/s10545-011-9359-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson P, Peinovich M, McEntee M, et al. Immune tolerance improves the efficacy of enzyme replacement therapy in canine mucopolysaccharidosis I. J Clin Invest. 2008;118:2868–2876. doi: 10.1172/JCI34676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- Guffon N, Souillet G, Maire I, Straczek J, Guibaud P. Follow-up of nine patients with Hurler syndrome after bone marrow transplantation. J Pediatr. 1998;133:119–125. doi: 10.1016/S0022-3476(98)70201-X. [DOI] [PubMed] [Google Scholar]
- Hobbs JR, Hugh-Jones K, Barrett AJ, et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet. 1981;2:709–712. doi: 10.1016/S0140-6736(81)91046-1. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Panoskaltsis-Mortari A, Haddad IY. Inflammatory cytokines and the development of pulmonary complications after allogeneic hematopoietic cell transplantation in patients with inherited metabolic storage disorders. Biol Blood Marrow Transplant. 2006;12:430–437. doi: 10.1016/j.bbmt.2005.12.026. [DOI] [PubMed] [Google Scholar]
- Malm G, Gustafsson B, Berglund G, et al. Outcome in six children with mucopolysaccharidosis type IH, Hurler syndrome, after haematopoietic stem cell transplantation (HSCT) Acta Paediatr. 2008;97:1108–1112. doi: 10.1111/j.1651-2227.2008.00811.x. [DOI] [PubMed] [Google Scholar]
- Martin PL, Carter SL, Kernan NA, et al. Results of the cord blood transplantation study (COBLT): outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with lysosomal and peroxisomal storage diseases. Biol Blood Marrow Transplant. 2006;12:184–194. doi: 10.1016/j.bbmt.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Muenzer J, Wraith JE, Clarke LA. International consensus panel on management and treatment of mucopolysaccharidosis I. Pediatrics. 2009;123:19–29. doi: 10.1542/peds.2008-0416. [DOI] [PubMed] [Google Scholar]
- Neufeld EF, Muenzer J. The mucopolysaccharidosis. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3421–3452. [Google Scholar]
- Orchard PJ, Milla C, Braunlin E, DeFor T, Bjoraker K, Blazar BR. Pre-transplant risk factors affecting outcome in Hurler syndrome. Bone Marrow Transplant. 2010;45:1239–1246. doi: 10.1038/bmt.2009.319. [DOI] [PubMed] [Google Scholar]
- Peters C, Balthazor M, Shapiro EG, et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood. 1996;87:4894–4902. [PubMed] [Google Scholar]
- Peters C, Shapiro EG, Anderson J, et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. The storage disease collaborative study group. Blood. 1998;91:2601–2608. [PubMed] [Google Scholar]
- Poe MD, Chagnon SL, Escolar M. Early treatment is associated with improved cognition in Hurler syndrome. Ann Neurol. 2014;76(5):747–753. doi: 10.1002/ana.24246. [DOI] [PubMed] [Google Scholar]
- Prasad VK, Mendizabal A, Parikh SH, et al. Unrelated donor umbilical cord blood transplantation for inherited metabolic disorders in 159 pediatric patients from a single center: influence of cellular composition of the graft on transplantation outcomes. Blood. 2008;112:2979–2989. doi: 10.1182/blood-2008-03-140830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro EG, Lockman LA, Balthazor M, Krivit W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J Inherit Metab Dis. 1995;18:413–429. doi: 10.1007/BF00710053. [DOI] [PubMed] [Google Scholar]
- Souillet G, Guffon N, Maire I, Pujol M, et al. Outcome of 27 patients with Hurler’s syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 2003;31:1105–1117. doi: 10.1038/sj.bmt.1704105. [DOI] [PubMed] [Google Scholar]
- Staba SL, Escolar ML, Poe M, Kim Y, et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N Engl J Med. 2004;350:1960–1969. doi: 10.1056/NEJMoa032613. [DOI] [PubMed] [Google Scholar]
- Terlato NJ, Cox GF. Can mucopolysaccharidosis type I disease severity be predicted based on a patient’s genotype? A comprehensive review of the literature. Genet Med. 2003;5:286–294. doi: 10.1097/01.GIM.0000078027.83236.49. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Jacobs S, Kierstein J, Van HJ. Outcome after three years of laronidase enzyme replacement therapy in a patient with Hurler syndrome. J Inherit Metab Dis. 2006;29:762. doi: 10.1007/s10545-006-0457-y. [DOI] [PubMed] [Google Scholar]
- Tokic V, Barisic I, Huzjak N, Petkovic G, Fumic K, Paschke E. Enzyme replacement therapy in two patients with an advanced severe (Hurler) phenotype of mucopolysaccharidosis I. Eur J Pediatr. 2007;166:727–732. doi: 10.1007/s00431-006-0316-8. [DOI] [PubMed] [Google Scholar]
- Tolar J, Dickson P, Orchard PJ. Intravenous and intrathecal enzyme replacement before and after hematopoietic cell transplantation for Hurler syndrome. Biol Blood Marrow Transplant. 2009;15(Suppl 2):74. doi: 10.1016/j.bbmt.2008.12.230. [DOI] [Google Scholar]
- Vellodi A, Young EP, Cooper A, et al. Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Child. 1997;76:92–99. doi: 10.1136/adc.76.2.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visigalli I, Delai S, Politi LS, et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood. 2010;9(116):5130–5139. doi: 10.1182/blood-2010-04-278234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisstein JS, Delgado E, Steinbach LS, Hart K, Packman S. Musculoskeletal manifestations of Hurler syndrome long-term follow-Up after bone marrow transplantation. J Pediatr Orthop. 2004;24:97–101. doi: 10.1097/01241398-200401000-00019. [DOI] [PubMed] [Google Scholar]
- Whitley CB, Belani KG, Chang PN, et al. Long-term outcome of Hurler syndrome following bone marrow transplantation. Am J Med Genet. 1993;46:209–218. doi: 10.1002/ajmg.1320460222. [DOI] [PubMed] [Google Scholar]
- Wraith JE, Beck M, Lane R, et al. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: results of a multinational study of recombinant human alpha-L-iduronidase (laronidase) Pediatrics. 2007;120:e37–e46. doi: 10.1542/peds.2006-2156. [DOI] [PubMed] [Google Scholar]