

Abstract
Purpose
Mercaptopurine (MP) is the mainstay of curative therapy for acute lymphoblastic leukemia (ALL). We performed a genome-wide association study (GWAS) to identify comprehensively the genetic basis of MP intolerance in children with ALL.
Patients and Methods
The discovery GWAS and replication cohorts included 657 and 371 children from two prospective clinical trials. MP dose intensity was a marker for drug tolerance and toxicities and was defined as prescribed dose divided by the planned protocol dose during maintenance therapy; its association with genotype was evaluated using a linear mixed-effects model.
Results
MP dose intensity varied by race and ethnicity and was negatively correlated with East Asian genetic ancestry (P < .001). The GWAS revealed two genome-wide significant loci associated with dose intensity: rs1142345 in TPMT (Tyr240Cys, present in *3A and *3C variants; P = 8.6 × 10−9) and rs116855232 in NUDT15 (P = 8.8 × 10−9), with independent replication. Patients with TT genotype at rs116855232 were exquisitely sensitive to MP, with an average dose intensity of 8.3%, compared with those with TC and CC genotypes, who tolerated 63% and 83.5% of the planned dose, respectively. The NUDT15 variant was most common in East Asians and Hispanics, rare in Europeans, and not observed in Africans, contributing to ancestry-related differences in MP tolerance. Of children homozygous for either TPMT or NUDT15 variants or heterozygous for both, 100% required ≥ 50% MP dose reduction, compared with only 7.7% of others.
Conclusion
We describe a germline variant in NUDT15 strongly associated with MP intolerance in childhood ALL, which may have implications for treatment individualization in this disease.
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is a prototype of disseminated cancer that can be cured with combination chemotherapy alone.1–3 In particular, prolonged daily exposure to mercaptopurine (MP) during maintenance therapy is the basis of most contemporary ALL treatment regimens and indispensable for the cure of this disease.4–8
ALL is also a model disease for pharmacogenomic research. In fact, genetics-guided thiopurine dose adjustment is one of the first examples of implementation of pharmacogenetics in clinical practice.9–12 Inherited deficiency of thiopurine methyltransferase (TPMT) resulting from nonsynonymous germline polymorphisms leads to higher levels of active metabolites of thiopurines and excess hematologic toxicities during ALL therapy.13 Therefore, pre-emptive genotyping of TPMT enables individualized MP dosing to minimize adverse effects without compromising overall treatment outcomes.14 However, interpatient variability in thiopurine-related myelosuppression is not completely explained by genetic variations in TPMT, and a substantial proportion of patients with wild-type TPMT still experience excessive toxicity that interrupts treatment and may negatively affect survival.13,15 Also, patients of East Asian descent are more intolerant to full dose of thiopurines,16,17 even though TPMT deficiency is less common in this population compared with individuals of European descent,18,19 suggesting the existence of other ancestry-related genetic polymorphisms underlying racial differences in thiopurine response.
To comprehensively identify the genetic basis of interpatient and inter-racial/interethnic variability in MP tolerance, we performed a genome-wide association study (GWAS) in two prospective clinical trials of childhood ALL with common chronic MP treatment regimens. In addition to the previously described risk locus in TPMT, we report a germline variant in nucleoside diphophate–linked moiety X-type motif 15 (NUDT15) that strongly predisposed patients to MP-related toxicities during ALL therapy, contributing to lower drug tolerance in East Asians.
PATIENTS AND METHODS
Patients and MP Therapy
The discovery GWAS cohort consisted of patients prospectively enrolled onto the Children's Oncology Group (COG) clinical trial AALL03N1 (www.clincialtrials.gov identifier NCT00268528), with the specific objectives of characterizing racial differences in MP exposure in children with ALL. Details of enrollment criteria and study design have been described previously.17,20 Briefly, patients were treated on COG frontline protocols for newly diagnosed ALL (or per frontline protocols for small number of patients), were enrolled onto this study for a total of 6 months during maintenance therapy that included a daily dose of oral MP, and were self-identified as white, black, Hispanic, or Asian. The protocol-planned dose of MP during maintenance phase was 75 mg/m2 per day for the COG ALL trials included in this study, with provisions for dose adjustment based on the degree of myelosuppression (WBC count) and/or occurrence of infections. MP dose intensity was defined as the ratio between clinician-prescribed MP dose and protocol dose (%) and was captured on a monthly basis for the 6-month duration of the study. The longitudinal MP dose-intensity data were summarized into a single overall value for each patient, using a linear mixed-effects model with genetic ancestry and the time point of dose-intensity measurement as variables.
The replication cohort consisted of 371 children with ALL treated on the St Jude Children's Research Hospital Total Therapy XV protocol (www.clinicaltrials.gov identifier NCT00137111), with the planned dose of MP at 75 mg/m2 during continuation therapy.21 MP dose intensity for St Jude patients was defined as the ratio of prescribed dose over protocol dose, assessed longitudinally by phase of therapy during continuation therapy, and summarized using a linear mixed-effects model with genetic ancestry and time point of dose-intensity measurement as variables. For patients who stopped therapy early, prescribed and expected protocol doses were calculated only up to that time. This study was approved by respective institutional review boards, and informed consent was obtained from parents, guardians, or patients, as appropriate.
Genotyping and Genetic Ancestry
Germline genomic DNA was extracted from peripheral blood or bone marrow samples obtained during clinical remission and subsequently submitted to genome-wide genotyping using the HumanExome BeadChip (Illumina, San Diego, CA; 244,770 single-nucleotide polymorphisms [SNPs]). Genotype calls were made using Illumina GenomeStudio software, and genotype was coded as 0, 1, or 2 to indicate the number of B alleles (assuming additive genetic model). Samples for which genotypes were ascertained for < 95% of SNPs on the chip were deemed to have failed and were excluded from the analyses. SNPs with poor genotyping quality (call rate < 95%) and/or low allele frequency (minor allele frequency < 1%) were excluded from the GWAS.
Population structure was determined using the principal component analysis in EIGENSTRAT software (http://genetics.med.harvard.edu/reich/Reich_Lab/Software.html).22,23 We also estimated European, African, Native American, and East Asian genetic ancestry using STRUCTURE software (version 2.2.3; http://pritchardlab.stanford.edu/structure.html)24 on the basis of genotypes at 30,000 randomly selected SNPs, with HapMap samples and indigenous Native American references25 as ancestral populations. Europeans, Africans, and East Asians were defined as having ≥ 90% European genetic ancestry, ≥ 70% African ancestry, and ≥ 60% East Asian ancestry, respectively. Hispanics were individuals for whom the proportion of Native American ancestry was ≥ 10% and was greater than the proportion of African ancestry. The rest of the participants were grouped as “other.”
MP Dose-Intensity GWAS and Replication
In the discovery GWAS, we evaluated the correlation between MP dose intensity and genotypes at 40,889 SNPs in the 657 COG patients after quality control, using a linear mixed-effects model. Top-three principal components (PC1, PC2, and PC3, indicating East Asian, African, and Native American genetic ancestry, respectively) were included in the linear mixed-effects regression model to control for population stratification. We also constructed a quantile-quantile plot and observed no evidence of inflation resulting from population stratification (λ = 0.98). SNPs that reached association P < 5 ×10−8 in the discovery GWAS were tested in the replication cohort, following the same statistical procedures.
TPMT Activity
Erythrocyte TPMT activity was measured for the COG and St Jude cohorts at least 90 days after RBC transfusion, using established methods.26,27 The association of SNP genotype with TPMT activity was evaluated using a linear regression model with genetic ancestry as covariates.
Statistical Notes
R statistical software (version 3.0; http://www.r-project.org/) was used for all analyses unless indicated otherwise.
RESULTS
Children with newly diagnosed ALL were prospectively enrolled onto the AALL03N1 study, representing four self-reported races/ethnicities (white, black, Hispanic, and Asian). Genetic ancestry varied significantly among and within each racial/ethnic group (Appendix Fig A1, online only). In particular, self-reported Hispanics had the highest Native American genetic ancestry rate, and self-reported Asians clustered into two groups based on the level of East Asian genetic ancestry. For all subsequent analyses, percent genetic ancestry was treated as a continuous variable, and/or patients were classified as European, African, Hispanic, and East Asian on the basis of respective genetic ancestry (those who did not fall into one of these four categories were grouped as “other”).
The clinical trials stipulated that MP dose was to be adjusted based on toxicities (eg, myelosuppression and/or infections) during therapy. Therefore, MP dose intensity, defined as the ratio of prescribed MP dose over the protocol standard dose of 75 mg/m2 per day, directly reflected host drug sensitivity/tolerance. Dose intensity was measured monthly over the 6-month study period of AALL03N1, and these longitudinal data were summarized into a single overall dose-intensity value for each patient using a mixed linear-effects model. For the four genetic ancestries examined, only East Asian ancestry was significantly associated with MP dose intensity (P < .001), with higher level of East Asian ancestry linked to lower dose intensity (Fig 1). Even when we restricted the analyses to self-reported Asians, those with higher East Asian ancestry (≥ 60%) had lower MP dose intensity compared with other self-identified Asians (P = .04; Appendix Fig A2, online only).
Fig 1.
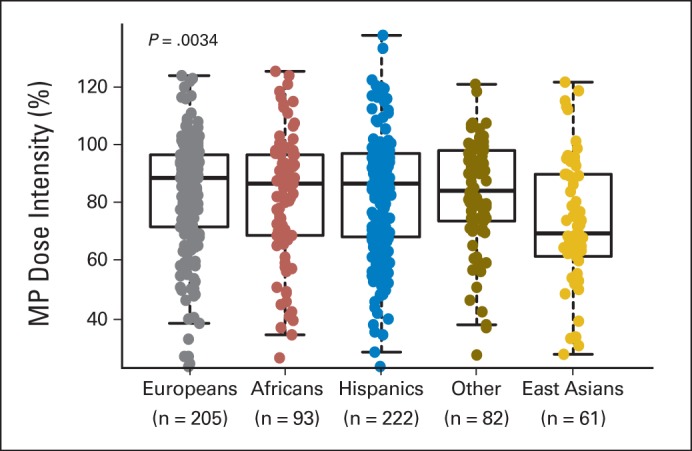
Mercaptopurine (MP) dose intensity and genetic ancestry. For patients enrolled onto AALL03N1 protocol, MP dose was adjusted during maintenance therapy on the basis of host toxicities (myelosuppression and infections), and dose intensity was defined as ratio of prescribed dose over protocol planned dose (75 mg/mg2 per day). Dose intensity was measured longitudinally over 6 months and is shown as single cumulative value for study period. Patients were grouped into five racial/ethnic categories on the basis of genetic ancestry. Genetically defined East Asians had lowest MP dose intensity (ie, most likely to be MP intolerant). P value was estimated by using Kruskal-Wallis test. Each box includes data between 25th and 75th percentiles, with horizontal line indicating median. Whiskers indicate maximal and minimal observations within 1.5× length of box.
In the discovery GWAS of 657 children with ALL in the AALL03N1 cohort, we observed two loci with genome-wide significant associations with MP dose intensity (P < 5 × 10−8): rs1142345 in TPMT (Tyr240Cys, contributing to *3A and *3C variants; P = 8.6 × 10−9) and rs116855232 in NUDT15 (P = 8.8 × 10−9; Fig 2A). Consistent with prior work by us and others, the nonsynonymous rs1142345 variant in TPMT predisposed patients to excessive MP dose reduction (lower dose intensity; Fig 2B), most likely because of increased hematologic toxicities. Only one patient was homozygous for the variant allele (CC genotype) at TPMT SNP rs1142345, and this individual was highly sensitive to MP, with a dose intensity of only 6%, compared with patients with the TC genotype (n = 39) or wild-type TT genotype (n = 617), who tolerated an average dose intensity of 65.7% (standard deviation [SD], ± 27.7%) and 83.5% (SD, ± 22.5%), respectively. The risk allele at rs1142345 was also highly associated with a lower TPMT activity (P = 1.9 × 10−34; Appendix Fig A3A, online only). Combining genotypes at three commonly observed TPMT functional variants (rs1142345, rs1800462, and rs1800460, contributing to *2, *3A, *3B, and *3C variants), TPMT low-activity variants were most prevalent in Africans and least common in East Asians (Appendix Fig A4, online only).
Fig 2.

Genome-wide association study identified TPMT and NUDT15 variants associated with mercaptopurine (MP) dose intensity. (A) Total of 40,889 single-nucleotide polymorphisms (SNPs) were tested for associations with MP dose intensity in 657 children with acute lymphoblastic leukemia in AALL03N1 cohort. Association P value (y-axis) is plotted against respective chromosomal position of each SNP (x-axis). Dashed horizontal line indicates genome-wide significance threshold (P < 5 × 10−8). TPMT and NUDT15 loci are indicated by arrows. MP dose intensity was significantly lower for patients heterozygous or homozygous for risk alleles at (B) TPMT SNP rs1142345 (contributing to *3A and *3C) and (C) NUDT15 SNP rs116855232, compared with those with wild-type genotypes. P value was estimated using linear mixed-effects model, with genetic ancestry as covariate. Each box includes data between 25th and 75th percentiles, with horizontal line indicating median. Whiskers indicate maximal and minimal observations within 1.5× length of box.
The second GWAS hit was the nonsynonymous variant rs116855232 in NUDT15 (Fig 2C). Patients carrying the TT genotype (n = 2) were exquisitely sensitive to MP and tolerated only 8.3% (SD, ± 7%) of the protocol standard dose (ie, dose reduction of 91.7%). The TC genotype group (n = 31) had an average dose intensity of 63% (SD, ± 25.4%), compared with patients with wild-type genotype at this SNP (n = 624), for whom the average dose intensity was 83.5% (SD, ± 22.4%). Longitudinally, the T allele predisposed patients to consistent and prolonged MP dose reduction, especially those homozygous for the risk allele (Appendix Fig A5, online only). This association of rs116855232 with MP dose intensity was replicated in 371 children with ALL from the St Jude Total Therapy XV protocol (P = .024). Remarkably similar to that in the COG group, average dose intensity for the St Jude cohort was 62.5% (SD, ± 14.8%) and 80.1% (SD, ± 18.9%) for patients with CT (n = 9) and CC genotypes (n = 362), respectively (Appendix Fig A6A, online only). No patients in the validation cohort had the TT genotype at this NUDT15 SNP. As expected, the associations of TPMT variant rs1142345 with MP dose intensity (Appendix Fig A6B, online only) and TPMT activity (Appendix Fig A3B, online only) were also highly significant in the St Jude cohort (P = 1.8 × 10−8 and 2.8 × 10−29, respectively).
Importantly, rs116855232 genotype distribution varied substantially by race/ethnicity. In the AALL03N1 cohort, the risk allele was most frequent in East Asians (9.8%), followed by Hispanics (3.9%). It was exceedingly rare in Europeans (0.2%) and not observed in Africans (Fig 3A). This pattern of allele frequency differences was comparable to that observed in general populations from these ancestral backgrounds (eg, 1,000 Genomes Project). When we restricted the analyses to genetically defined East Asians or Hispanics in the AALL03N1 cohort, rs116855232 remained significantly associated with MP dose intensity (P = 8.6 × 10−6 and .0001, respectively; Figs 3B and 3C). We observed a single European patient carrying the risk allele (CT genotype). Although this patient had a lower MP dose intensity relative to those with wild-type genotype (Fig 3D), statistical significance was not achieved because of small numbers.
Fig 3.

NUDT15 variant was more frequent in East Asians but associated with mercaptopurine (MP) dose intensity regardless of ancestry. (A) T (risk) allele at rs116855232 was most common in individuals of East Asian descent followed by Hispanics, rare in Europeans, and not observed in Africans. T allele was consistently linked to lower MP dose intensity in (B) East Asians and (C) Hispanics; (D) similar trend was observed in Europeans. P values were estimated using linear mixed-effects model. Each box includes data between 25th and 75th percentiles, with horizontal line indicating median. Whiskers indicate maximal and minimal observations within 1.5× length of box.
Although effects of this NUDT15 variant were consistent regardless of genetic ancestry, its higher frequency in East Asians suggested possible contribution to the ancestry-related differences in MP tolerance. In fact, in a multivariable analysis, the addition of rs116855232 significantly diminished the association of East Asian ancestry with MP dose intensity (Table 1). In contrast, adding TPMT genotype to the model did not affect the association of either East Asian ancestry or NUDT15 SNP rs116855232 with MP dose intensity.
Table 1.
Multivariable Analyses for Genetic Determinants of MP Dose Intensity
| Variable | Model One |
Model Two |
Model Three |
|||
|---|---|---|---|---|---|---|
| Regression Coefficient Estimate* | P† | Regression Coefficient Estimate* | P† | Regression Coefficient Estimate* | P† | |
| African ancestry | −0.01 | .71 | −0.01 | .66 | −0.009 | .78 |
| East Asian ancestry | −0.14 | .0003 | −0.08 | .019 | −0.102 | .005 |
| Native American ancestry | −0.03 | .43 | 0.0006 | .99 | 0.013 | .75 |
| NUDT15 variant (rs116855232) | −0.22 | 8.8 × 10−9 | −0.22 | 2.7 × 10−8 | ||
| TPMT variants‡ | −0.20 | 4.6 × 10−9 | ||||
NOTE. Three multivariable models illustrate contributions of NUDT15 variants, TPMT variants, and genetic ancestry to interpatient variability in MP dose intensity.
Abbreviations: MP, mercaptopurine; SNP, single-nucleotide polymorphism.
Regression coefficient estimate represents increase (positive value) or decrease (negative value) in percent MP dose intensity for those patients with certain levels of genetic ancestry (model one), those carrying risk allele at NUDT15 SNP (model two), or those carrying risk alleles at TPMT SNPs. For instance, mean MP dose intensity for patients with 100% East Asian genetic ancestry is estimated 14 percentage points lower than that of patients with 100% European genetic ancestry, as in model one. For each additional T allele at rs116855232, mean MP dose intensity is estimated to decrease 22 percentage points while holding ancestry factors at any fixed level, as in model two. In model three, for each additional T allele at rs116855232, mean MP dose intensity is estimated to decrease 22 percentage points while holding ancestry factors and TPMT variation at any fixed levels.
P values were estimated using mixed linear-effects model including independent variables as described.
Genetic variation in TPMT represents composite genotype at rs1142345, rs1800462, and rs1800460 (contributing *2, *3A, *3B, and *3C variants).
To evaluate the combined effects of both TPMT and NUDT15 variants, we compared MP dose intensity for patients with different genotype combinations at these two loci. We first assigned each patient a genetic risk score based on the number of risk alleles at both genes. In the AALL03N1 cohort, there was a significant correlation between the burden of risk alleles and dose intensity (P = 3.2 × 10−16), with individuals homozygous for either TPMT or NUDT15 risk variants (n = 3) showing the lowest MP dose intensity (Fig 4A). Interestingly, patients heterozygous for both TPMT and NUDT15 risk variants (n = 4) also had substantially lower dose intensity compared with those with heterozygous genotype for only one of the two genes (n = 67; P = .018). This trend was also confirmed in the St Jude cohort (P = .03; Fig 4B). In the AALL03N1 cohort, we classified patients as MP intolerant if the actual MP dose was ≤ 50% of the protocol-planned dose (ie, MP ≤ 37.5 mg/m2 per day). Thus, the rate of MP intolerance was 100% (seven of seven) for patients with a genetic risk score of 2 (heterozygous for both TPMT and NUDT15 or homozygous for variant allele at either gene), 25.8% (17 of 66) in patients with a risk score of 1 (heterozygous for either TPMT or NUDT15), and 5.7% (33 of 583) in those with a score of 0 (wild-type genotype for both genes). Therefore, a genetic risk score of 2 in this two-gene model identified patients at risk for MP intolerance with a positive prediction value of 100% (Table 2).
Fig 4.

Combined effects of TPMT and NUDT15 variants on mercaptopurine (MP) intolerance. Each patient in (A) AALL03N1 and (B) St Jude cohorts was first assigned genetic risk score based on genotype at NUDT15 variant (rs116855232) and TPMT variants (rs1142345, rs1800462, and rs1800460; contributing to *2, *3A, *3B, and *3C). MP dose intensity decreased as genetic risk score increased in both cohorts. P value was estimated using Spearman correlation. Each box includes data between 25th and 75th percentiles, with horizontal line indicating median. Whiskers indicate maximal and minimal observations within 1.5× length of box. HET, heterozygous; HOM, homozygous; WT, wild type.
Table 2.
Prediction of MP Intolerance by TPMT and NUDT15 Genetic Variants
NOTE. Genotype and MP dose intensity based on AALL03N1 cohort.
Abbreviation: MP, mercaptopurine.
Overall MP dose intensity was summarized for entire 6-month study period.
Genetic risk score was defined by number of TPMT and/or NUDT15 risk alleles (rs1142345, rs1800462, rs1800460, and rs116855232), as described in Patients and Methods.
DISCUSSION
The inclusion of MP in ALL therapy contributed substantially to the remarkable improvement in survival from this malignancy.28,29 However, thiopurines also have a narrow therapeutic index, particularly in children with ALL, with common dose-limiting toxicities in the hematopoietic tissues.30 Constant MP dose titration is challenging in clinical practice and can negatively affect ALL treatment outcome.31,32 The relationship between TPMT genetic variation and thiopurine toxicities has been well established based on the work by our group13,15 and others,33,34 and preemptive TPMT genotyping is used clinically to guide MP dosing during ALL therapy.35 However, there have been no comprehensive analyses of the genetic basis of MP tolerance in ALL population at the genome-wide level. In this multiethnic GWAS, we took an agnostic approach to systematically evaluate germline SNPs for their association with MP dose intensity during maintenance ALL therapy. We confirmed that variants in TPMT were the strongest predictor of MP tolerance but importantly described NUDT15 as another critical locus associated with susceptibility to MP intolerance resulting from excessive toxicity. The association signals at NUDT15 and TPMT variants were independent of each other. There was a significant correlation between the number of copies of the T allele at the NUDT15 SNP rs116855232 and tolerated MP dose, indicating a gene dosage effect. Patients with homozygous TT genotype were extremely sensitive to MP and tolerated only 8.3% of protocol-planned MP dose. NUDT15 genotyping could identify this group of at-risk children and therefore may have importance for clinical practice. Although patients with heterozygous genotype at the NUDT15 SNP also had a greater MP dose reduction compared with wild-type patients, there was a relatively wide variation in the final tolerated dose, and additional studies with larger sample sizes will be required to fully characterize MP tolerance for this group of patients. Interestingly, four children in the AALL03N1 cohort heterozygous for both TPMT and NUDT15 variants required greater dose reductions compared with those heterozygous at only one of the two loci, indicating at least additive effects of these genes on MP sensitivity.
TPMT is a cytosolic enzyme responsible for the methylation (inactivation) of MP, and the SNP (rs1142345) with the lowest P value in our GWAS is present in the most common inherited nonfunctional alleles of TPMT (*3A and *3C variants).12 This genetic variation leads to an unstable protein that undergoes enhanced proteolysis and therefore loss of TPMT function.36 Results from our GWAS confirmed that TPMT genetic variation is one of the most critical determinants for MP tolerance, particularly in non–East Asian populations.
NUDT15 encodes a nucleoside diphosphatase.37 Also known as MTH2, NUDT15 is shown to degrade oxidized purine nucleoside triphosphates (eg, 8-oxo-dGTP) by dephosphorylation to prevent incorporation into DNA.38 By removing the oxidatively damaged guanine nucleotides, NUDT15 is a safeguard mechanism in mammalian cells to minimize DNA damage and avoid subsequent repair and apoptosis.38 It is well established that MP cytotoxicity is enhanced by its enzymatic conversion into thioguanine nucleotides: thioguanosine mono-, di-, and triphosphates (thio-GMP, thio-GDP, and thio-GTP), as well as deoxy thioguanosine phosphates (thio-dGMP, thio-dGDP, and thio-dGTP). In particular, thio-dGTP is directly incorporated into DNA, which triggers futile DNA damage repair and eventually apoptosis.30 Structurally similar to 8-oxo-dGTP, thio-dGTP is a potential substrate for NUDT15 by which this active metabolite may be hydrolyzed to the inactive thio-dGMP or thio-dGDP. In patients with defective NUDT15, we hypothesize that normal doses of MP will lead to excessive accumulation of thio-dGTP (relative to thio-dGMP and thio-dGDP), extensive DNA damage, and cytotoxicity. Therefore, we posit that the T to A substitution at rs116855232 is likely to be a loss-of-function variant, although its effects on enzymatic activity warrant additional experimental studies. Notably, a recent GWAS in Korean patients with inflammatory bowel diseases also reported the same NUDT15 variant associated with the susceptibility to thiopurine-related leukopenia, with each copy of the T allele conferring a remarkable 35.6-fold increase in toxicity risk.39 These findings compellingly point to NUDT15 as a key modulator of thiopurine cytotoxicity, particularly in patients with Asian or Native American ancestry.
Restricting the analyses to genetically defined Europeans in the discovery GWAS cohort, we observed a single patient carrying the T risk allele at rs116855232. Although this patient had a lower MP dose intensity than the average of wild-type Europeans, the small sample size obviously prevented any meaningful statistical estimates of significance. The risk allele was most common in East Asians and those with higher Native American ancestry (Hispanics), suggesting that this variant allele likely arose originally in East Asian populations and was preserved in Native American diaspora after their migration from East Asia 14,000 years ago.40 Although the higher prevalence of NUDT15 variant in East Asians may have contributed to over-representation of MP intolerance in this population, we also acknowledge that our statistical power was limited by the relatively small number of East Asian patients included in this study. In addition, East Asian genetic ancestry remained associated with MP dose intensity after adjusting for rs116855232, suggesting the existence of other Asian-specific factors related to MP sensitivity.41 Future studies including a greater number of East Asians and/or Hispanics will be required to fully evaluate the effects of the NUDT15 variant and identify additional genetic determinants of MP metabolism and toxicities. Finally, 5.7% of patients who required > 50% MP dose reduction were wild type for both TPMT and NUDT15 variants, and therefore, other genetic and nongenetic factors are still to be discovered to further improve MP dose individualization in children with ALL. In conclusion, taking a GWAS approach, we identified a pharmacogenetic variant in NUDT15 associated with MP intolerance in children with ALL.
Supplementary Material
Acknowledgment
We thank all patients and their parents who participated in the clinical studies by Children's Oncology Group and St Jude.
Glossary Terms
- genetic polymorphisms:
a genetic variant seen in at least 1% of the population. Because proteins are gene products, their polymorphisms reflect allelic differences in the gene. The advent of restriction enzymes, which digest DNA to fragments based on sequence specificity, has ushered in an era of restriction fragment length polymorphisms in which changes in DNA sequences manifest as restriction fragments of different sizes when cleaved with a specific restriction enzyme. Polymorphisms are used in tissue typing, in determining disease, in pharmacogenetics, and in assessing genetic diversity.
- genome-wide association study:
hypothesis-free studies that evaluate the association of genetic variations throughout the entire genome with traits, using high throughput genotyping technologies to assay single nucleotide polymorphisms.
- germline polymorphism:
a difference in DNA sequence among individuals in the germ cells. Unlike somatic cell genetic mutations, these polymorphisms can be transmitted to an organism's offspring. Genetic polymorphisms may be the result of a chance process or may have been induced by external agents (such as viruses or radiation). Changes in DNA sequence that have been confirmed to be caused by external agents are generally called “mutations” rather than “polymorphisms”.
- pharmacogenomics:
the study of how a person's genome can affect their reaction to medications.
- thiopurine S-methyltransferase:
an enzyme that catalyzes the S-methylation of aromatic and heterocyclic compounds with sulphydryl groups and is important in the clearance of drugs such as 6-mercaptopurine and azathioprine. Genetic polymorphisms in the enzyme and their clinical relevance to myelosuppression were the first to be studied in the field of pharmacogenetics.
Appendix
Fig A1.

Genetic ancestry and self-reported race/ethnicity. Genetic ancestry was estimated based on genotypes at 30,000 randomly selected single-nucleotide polymorphisms using STRUCTURE software (version 2.2.3; http://pritchardlab.stanford.edu/structure.html). (A) European, (B) East Asian, (C) Native American, and (D) African ancestries are plotted for four self-reported race/ethnic groups in AALL03N1 cohort.
Fig A2.
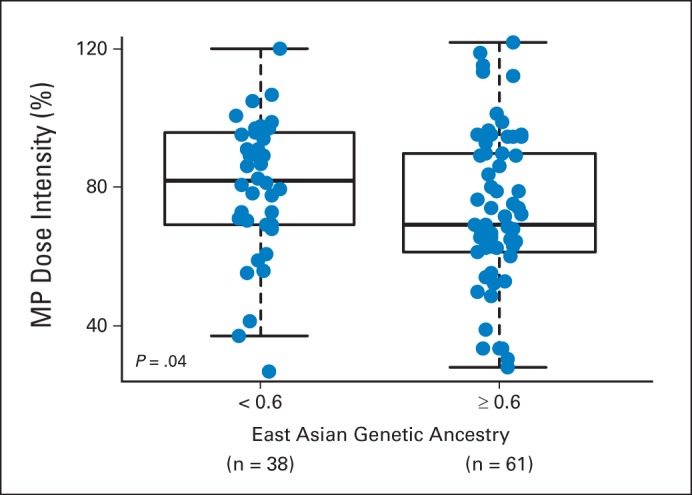
East Asian genetic ancestry was related to lower mercaptopurine (MP) dose intensity within self-reported Asians. In self-identified Asian patients in AALL03N1 cohort, those with greater East Asian genetic ancestry had lower MP dose intensity. P value was estimated using Wilcoxon test. Each box includes data between 25th and 75th percentiles, with horizontal line indicating median. Whiskers indicate maximal and minimal observations within 1.5× length of box.
Fig A3.
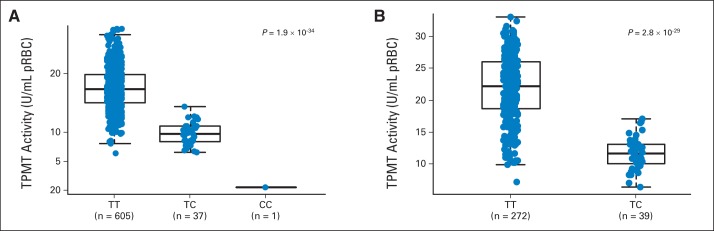
Association of TPMT genotype (rs1142345) and thiopurine methyltransferase (TPMT) enzymatic activity. TPMT activity was measured from erythrocyte extracts for (A) 643 children on AALL03N1 protocol and (B) 311 patients in St Jude cohort. P value was estimated using linear regression with ancestry as covariate. Each box includes data between 25th and 75th percentiles, with horizontal line indicating median. Whiskers indicate maximal and minimal observations within 1.5× length of box.
Fig A4.
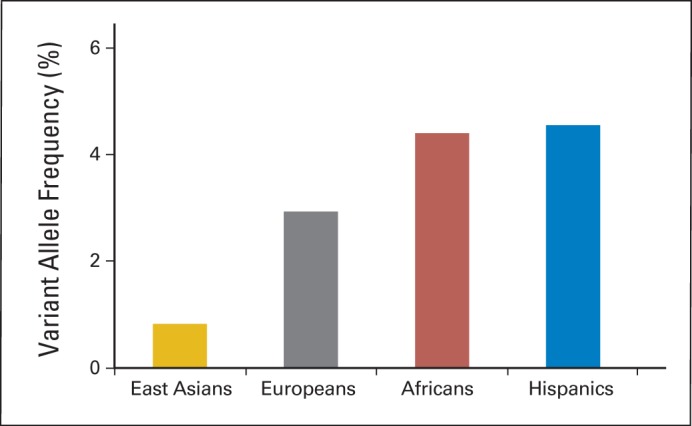
Frequency of TPMT variants in different racial/ethnic groups in AALL03N1 cohort. Composite TPMT genotype was defined by rs1142345, rs1800462, and rs1800460 (contributing to *2, *3A, *3B, and *3C variants). Genetically defined racial/ethnic groups in AALL03N1 cohort were based on genetic ancestry, as described in Patients and Methods.
Fig A5.
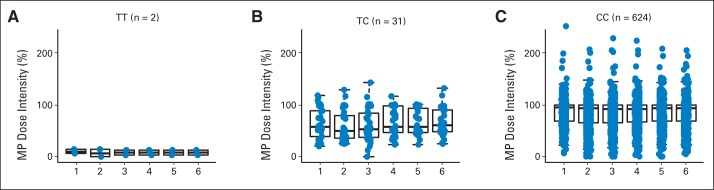
Longitudinal mercaptopurine (MP) dose intensity and genotype at NUDT15 SNP rs116855232. MP dose intensity was determined monthly during 6-month study period of AALL03N1 for patients with (A) TT, (B) TC, and (C) CC genotypes at rs116855232. Numbers on x-axis denote month during which MP dose intensity was measured. Each box includes data between 25th and 75th percentiles, with horizontal line indicating median. Whiskers indicate maximal and minimal observations within 1.5× length of box.
Fig A6.
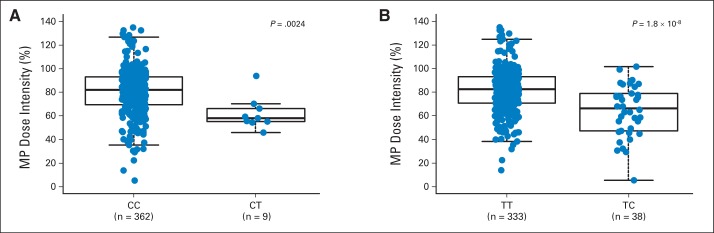
Mercaptopurine (MP) dose intensity and genotype at NUDT15 SNP rs116855232 and TPMT SNP rs1142345 in St Jude cohort. Of 371 children with acute lymphoblastic leukemia treated at St Jude Children's Research Hospital, cumulative MP dose intensity during entire continuation phase (maintenance) was lower in those with CT genotype at (A) rs116855232 (NUDT15) or (B) rs1142345 (TPMT) than in wild-type individuals. P value was estimated using linear mixed-effects regression model with ancestry and number of longitudinal MP dose-intensity measurements as covariates. Each box includes data between 25th and 75th percentiles, with horizontal line indicating median. Whiskers indicate maximal and minimal observations within 1.5× length of box.
Footnotes
See accompanying editorial on page 1230
Processed as a Rapid Communication manuscript.
Supported in part by Grants No. CA096670, CA098543, CA095861, CA36401, CA21765, CA156449, and GM92666 from the National Institutes of Health and by the American Lebanese Syrian Associated Charities. J.J.Y. receives funding as an American Society of Hematology scholar.
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT00268528, NCT00137111.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Jun J. Yang, Smita Bhatia, Mary V. Relling
Financial support: Smita Bhatia
Provision of study materials or patients: Lindsey Hageman, William E. Evans, Ching-Hon Pui, Smita Bhatia
Collection and assembly of data: Jun J. Yang, Wendy Landier, Chengcheng Liu, Lindsey Hageman, Kristine R. Crews, Nancy Kornegay, F. Lennie Wong, Ching-Hon Pui, Smita Bhatia, Mary V. Relling
Data analysis and interpretation: Jun J. Yang, Wenjian Yang, Chengcheng Liu, Cheng Cheng, Deqing Pei, Yanjun Chen, F. Lennie Wong, William E. Evans, Smita Bhatia, Mary V. Relling
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Inherited NUDT15 Variant Is a Genetic Determinant of Mercaptopurine Intolerance in Children With Acute Lymphoblastic Leukemia
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Jun J. Yang
No relationship to disclose
Wendy Landier
Research Funding: Merck Sharpe & Dohme (Inst)
Wenjian Yang
No relationship to disclose
Chengcheng Liu
No relationship to disclose
Lindsey Hageman
No relationship to disclose
Cheng Cheng
No relationship to disclose
Deqing Pei
No relationship to disclose
Yanjun Chen
No relationship to disclose
Kristine R. Crews
No relationship to disclose
Nancy Kornegay
No relationship to disclose
F. Lennie Wong
No relationship to disclose
William E. Evans
Patents, Royalties, Other Intellectual Property: Royalties from licensing TPMT genotyping; royalties from licensing TPMT genotyping (I)
Ching-Hon Pui
No relationship to disclose
Smita Bhatia
No relationship to disclose
Mary V. Relling
Patents, Royalties, Other Intellectual Property: Royalties from licensing TPMT genotyping (Prometheus Labs)
REFERENCES
- 1.Pui CH, Mullighan CG, Evans WE, et al. Pediatric acute lymphoblastic leukemia: Where are we going and how do we get there? Blood. 2012;120:1165–1174. doi: 10.1182/blood-2012-05-378943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pui CH, Carroll WL, Meshinchi S, et al. Biology, risk stratification, and therapy of pediatric acute leukemias: An update. J Clin Oncol. 2011;29:551–565. doi: 10.1200/JCO.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the Children's Oncology Group. J Clin Oncol. 2012;30:1663–1669. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koren G, Ferrazini G, Sulh H, et al. Systemic exposure to mercaptopurine as a prognostic factor in acute lymphocytic leukemia in children. N Engl J Med. 1990;323:17–21. doi: 10.1056/NEJM199007053230104. [DOI] [PubMed] [Google Scholar]
- 5.Relling MV, Hancock ML, Boyett JM, et al. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood. 1999;93:2817–2823. [PubMed] [Google Scholar]
- 6.Childhood ALL Collaborative Group. Duration and intensity of maintenance chemotherapy in acute lymphoblastic leukaemia: Overview of 42 trials involving 12 000 randomised children. Lancet. 1996;347:1783–1788. doi: 10.1016/s0140-6736(96)91615-3. [DOI] [PubMed] [Google Scholar]
- 7.Toyoda Y, Manabe A, Tsuchida M, et al. Six months of maintenance chemotherapy after intensified treatment for acute lymphoblastic leukemia of childhood. J Clin Oncol. 2000;18:1508–1516. doi: 10.1200/JCO.2000.18.7.1508. [DOI] [PubMed] [Google Scholar]
- 8.Schmiegelow K, Schrøder H, Gustafsson G, et al. Risk of relapse in childhood acute lymphoblastic leukemia is related to RBC methotrexate and mercaptopurine metabolites during maintenance chemotherapy: Nordic Society for Pediatric Hematology and Oncology. J Clin Oncol. 1995;13:345–351. doi: 10.1200/JCO.1995.13.2.345. [DOI] [PubMed] [Google Scholar]
- 9.Evans WE, Relling MV. Pharmacogenomics: Translating functional genomics into rational therapeutics. Science. 1999;286:487–491. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 10.Evans WE, Relling MV. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464–468. doi: 10.1038/nature02626. [DOI] [PubMed] [Google Scholar]
- 11.Giacomini KM, Yee SW, Ratain MJ, et al. Pharmacogenomics and patient care: One size does not fit all. Sci Transl Med. 2012;4:153ps18. doi: 10.1126/scitranslmed.3003471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Relling MV, Gardner EE, Sandborn WJ, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011;89:387–391. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91:2001–2008. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 14.Relling MV, Pui CH, Cheng C, et al. Thiopurine methyltransferase in acute lymphoblastic leukemia. Blood. 2006;107:843–844. doi: 10.1182/blood-2005-08-3379. [DOI] [PubMed] [Google Scholar]
- 15.Evans WE, Hon YY, Bomgaars L, et al. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J Clin Oncol. 2001;19:2293–2301. doi: 10.1200/JCO.2001.19.8.2293. [DOI] [PubMed] [Google Scholar]
- 16.Takatsu N, Matsui T, Murakami Y, et al. Adverse reactions to azathioprine cannot be predicted by thiopurine S-methyltransferase genotype in Japanese patients with inflammatory bowel disease. J Gastroenterol Hepatol. 2009;24:1258–1264. doi: 10.1111/j.1440-1746.2009.05917.x. [DOI] [PubMed] [Google Scholar]
- 17.Bhatia S, Landier W, Hageman L, et al. Adherence to oral 6-mercaptopurine in African American and Asian children with acute lymphoblastic leukemia: A Children's Oncology Group study. Blood. 2014;124:2345–2353. doi: 10.1182/blood-2014-01-552166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kham SK, Soh CK, Liu TC, et al. Thiopurine S-methyltransferase activity in three major Asian populations: A population-based study in Singapore. Eur J Clin Pharmacol. 2008;64:373–379. doi: 10.1007/s00228-007-0426-x. [DOI] [PubMed] [Google Scholar]
- 19.Cooper SC, Ford LT, Berg JD, et al. Ethnic variation of thiopurine S-methyltransferase activity: A large, prospective population study. Pharmacogenomics. 2008;9:303–309. doi: 10.2217/14622416.9.3.303. [DOI] [PubMed] [Google Scholar]
- 20.Bhatia S, Landier W, Shangguan M, et al. Nonadherence to oral mercaptopurine and risk of relapse in Hispanic and non-Hispanic white children with acute lymphoblastic leukemia: A report from the Children's Oncology Group. J Clin Oncol. 2012;30:2094–2101. doi: 10.1200/JCO.2011.38.9924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pui CH, Campana D, Pei D, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med. 2009;360:2730–2741. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 23.Yang JJ, Cheng C, Devidas M, et al. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet. 2011;43:237–241. doi: 10.1038/ng.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mao X, Bigham AW, Mei R, et al. A genomewide admixture mapping panel for Hispanic/Latino populations. Am J Hum Genet. 2007;80:1171–1178. doi: 10.1086/518564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yates CR, Krynetski EY, Loennechen T, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: Genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med. 1997;126:608–614. doi: 10.7326/0003-4819-126-8-199704150-00003. [DOI] [PubMed] [Google Scholar]
- 27.McLeod HL, Relling MV, Liu Q, et al. Polymorphic thiopurine methyltransferase in erythrocytes is indicative of activity in leukemic blasts from children with acute lymphoblastic leukemia. Blood. 1995;85:1897–1902. [PubMed] [Google Scholar]
- 28.Burchenal JH, Murphy ML, Ellison RR, et al. Clinical evaluation of a new antimetabolite, 6-mercaptopurine, in the treatment of leukemia and allied diseases. Blood. 1953;8:965–999. [PubMed] [Google Scholar]
- 29.Pinkel D, Hernandez K, Borella L, et al. Drug dosage and remission duration in childhood lymphocytic leukemia. Cancer. 1971;27:247–256. doi: 10.1002/1097-0142(197102)27:2<247::aid-cncr2820270202>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 30.Fotoohi AK, Coulthard SA, Albertioni F. Thiopurines: Factors influencing toxicity and response. Biochem Pharmacol. 2010;79:1211–1220. doi: 10.1016/j.bcp.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 31.Frandsen TL, Abrahamsson J, Lausen B, et al. Individualized toxicity-titrated 6-mercaptopurine increments during high-dose methotrexate consolidation treatment of lower risk childhood acute lymphoblastic leukaemia: A Nordic Society of Paediatric Haematology and Oncology (NOPHO) pilot study. Br J Haematol. 2011;155:244–247. doi: 10.1111/j.1365-2141.2011.08835.x. [DOI] [PubMed] [Google Scholar]
- 32.Schmiegelow K, Björk O, Glomstein A, et al. Intensification of mercaptopurine/methotrexate maintenance chemotherapy may increase the risk of relapse for some children with acute lymphoblastic leukemia. J Clin Oncol. 2003;21:1332–1339. doi: 10.1200/JCO.2003.04.039. [DOI] [PubMed] [Google Scholar]
- 33.Lennard, Lilleyman JS, Van Loon J, et al. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336:225–229. doi: 10.1016/0140-6736(90)91745-v. [DOI] [PubMed] [Google Scholar]
- 34.Black AJ, McLeod HL, Capell HA, et al. Thiopurine methyltransferase genotype predicts therapy-limiting severe toxicity from azathioprine. Ann Intern Med. 1998;129:716–718. doi: 10.7326/0003-4819-129-9-199811010-00007. [DOI] [PubMed] [Google Scholar]
- 35.Hoffman JM, Haidar CE, Wilkinson MR, et al. PG4KDS: A model for the clinical implementation of pre-emptive pharmacogenetics. Am J Med Genet C Semin Med Genet. 2014;166C:45–55. doi: 10.1002/ajmg.c.31391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tai HL, Krynetski EY, Schuetz EG, et al. Enhanced proteolysis of thiopurine S-methyltransferase (TPMT) encoded by mutant alleles in humans (TPMT*3A, TPMT*2): Mechanisms for the genetic polymorphism of TPMT activity. Proc Natl Acad Sci U S A. 1997;94:6444–6449. doi: 10.1073/pnas.94.12.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai JP, Ishibashi T, Takagi Y, et al. Mouse MTH2 protein which prevents mutations caused by 8-oxoguanine nucleotides. Biochem Biophys Res Commun. 2003;305:1073–1077. doi: 10.1016/s0006-291x(03)00864-7. [DOI] [PubMed] [Google Scholar]
- 38.Takagi Y, Setoyama D, Ito R, et al. Human MTH3 (NUDT18) protein hydrolyzes oxidized forms of guanosine and deoxyguanosine diphosphates: Comparison with MTH1 and MTH2. J Biol Chem. 2012;287:21541–21549. doi: 10.1074/jbc.M112.363010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang SK, Hong M, Baek J, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet. 2014;46:1017–1020. doi: 10.1038/ng.3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S, Lewis CM, Jakobsson M, et al. Genetic variation and population structure in native Americans. PLoS Genet. 2007;3:e185. doi: 10.1371/journal.pgen.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krishnamurthy P, Schwab M, Takenaka K, et al. Transporter-mediated protection against thiopurine-induced hematopoietic toxicity. Cancer Res. 2008;68:4983–4989. doi: 10.1158/0008-5472.CAN-07-6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.