

Abstract
Peramine is an insect-feeding deterrent produced by Epichloë species in symbiotic association with C3 grasses. The perA gene responsible for peramine synthesis encodes a two-module nonribosomal peptide synthetase. Alleles of perA are found in most Epichloë species; however, peramine is not produced by many perA-containing Epichloë isolates. The genetic basis of these peramine-negative chemotypes is often unknown. Using PCR and DNA sequencing, we analyzed the perA genes from 72 Epichloë isolates and identified causative mutations of perA null alleles. We found nonfunctional perA-ΔR* alleles, which contain a transposon-associated deletion of the perA region encoding the C-terminal reductase domain, are widespread within the Epichloë genus and represent a prevalent mutation found in nonhybrid species. Disparate phylogenies of adjacent A2 and T2 domains indicated that the deletion of the reductase domain (R*) likely occurred once and early in the evolution of the genus, and subsequently there have been several recombinations between those domains. A number of novel point, deletion, and insertion mutations responsible for abolishing peramine production in full-length perA alleles were also identified. The regions encoding the first and second adenylation domains (A1 and A2, respectively) were common sites for such mutations. Using this information, a method was developed to predict peramine chemotypes by combining PCR product size polymorphism analysis with sequencing of the perA adenylation domains.
INTRODUCTION
Fungal secondary metabolites are a diverse group of important but often nonessential organic compounds with a wide range of properties that are likely to be advantageous for the producing organism or in some cases essential for pathogenicity or developmental stages (1–3). These low-molecular-weight compounds tend to only be produced under certain environmental or growth conditions. The biosynthetic pathways for production of any particular class of secondary metabolites are common to many fungi, but production of a specific secondary metabolite is often unique to a small phylogenetic group of species (4). Epichloë species are fungal endophytes of C3 grasses that are known to produce several bioactive alkaloids that provide bioprotective properties to the host plant (5). These secondary metabolites include the indole-diterpenes, ergot alkaloids, lolines, and peramine (Fig. 1) (6, 7). The indole-diterpene lolitrem B and ergot alkaloid ergovaline have significant detrimental effects on the health and production of stock animals that graze infected pastures (7, 8). The lolines are insecticidal (9), and peramine is a potent deterrent of feeding by insects, including the agriculturally important invertebrate pest Listronotus bonariensis (Argentine stem weevil) (10–12).
FIG 1.
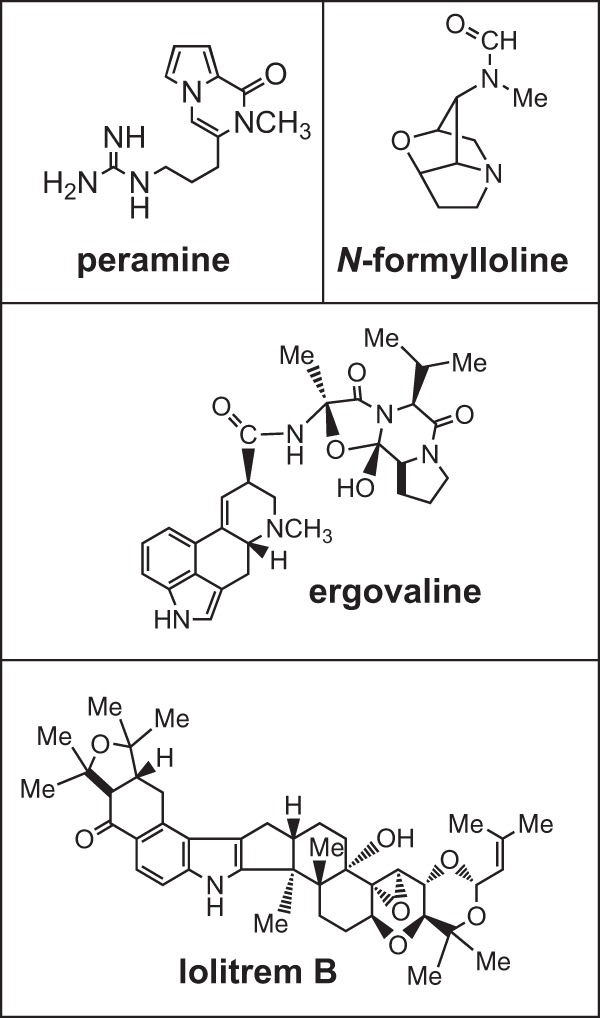
Chemical structures of alkaloid examples produced by Epichloë species.
Peramine synthesis is catalyzed by the two-module nonribosomal peptide synthetase (NRPS), peramine synthetase (PerA), encoded by the 8.3-kb gene perA (12). The first module of PerA contains an adenylation (A1) domain responsible for selection and activation of the proposed substrate amino acid 1-pyrroline-5-carboxylate and a thiolation (T1) domain that bonds this substrate as a thioester via a 4′-phosphopantetheine (4′PPT) linker. The second module contains adenylation (A2) and thiolation (T2) domains for selection, activation, and thiolation of the substrate proposed to be arginine. The second module also contains a methylation (M) domain proposed to N-methylate the alpha-amine of the arginine moiety, a condensation (C) domain that catalyzes peptide bond formation, and a variant reductase domain (R*) (13) at the C terminus, proposed to be responsible for intramolecular cyclization and release of the dipeptide product.
The genus Epichloë (including former Neotyphodium spp.) consists of sexual nonhybrid species and asexual, nonpathogenic endophytes that are derived either directly from the sexual species or by hybridization of two or more Epichloë progenitors (14, 15). Hybrid Epichloë species contain duplicate or even triplicate copies of most genes due to inheritance of an allele from each progenitor. Alleles of perA are found in nearly all Epichloë species, with the notable exceptions of Epichloë glyceriae and Epichloë gansuensis (16), but perA null alleles are common. One such allele, first identified in the genome sequence of Epichloë festucae isolate E2368 (16), has a deletion of the region encoding the C-terminal R* domain of PerA. This deletion is associated with the insertion of the miniature inverted-repeat transposable element (MITE) designated 3m (17). However, there are many other cases of peramine-negative (per−) isolates for which the genetic basis is unknown (18, 19).
Peramine production is an important trait when considering endophyte strains for deployment in forage grasses and likely provides a selective advantage to endophyte-infected wild grasses. As such, diagnostic methods are useful to identify suitable endophyte isolates and associations for use in agriculture around the world. The objective of this study was to identify and characterize the mutations causing perA null alleles in a collection of hybrid and nonhybrid Epichloë species and strains. Using this information, we developed a PCR method to predict the peramine chemotype of endophytes from pure culture and in endophyte-infected plant material.
MATERIALS AND METHODS
Endophyte strains and growth conditions.
Isolates of Epichloë species (see Table S1 in the supplemental material) were grown and maintained as previously described (20, 21). Endophyte-infected plant samples (see Table S2 in the supplemental material) were obtained from plant lines maintained under greenhouse conditions at 23°C during the day and 20°C at night, with 16 h of light starting at 0600; light intensity varied throughout the year, depending on the season.
Peramine analysis.
Peramine was analyzed by AgResearch Grasslands (Palmerston North, New Zealand) from plant material using a modification of the method described by Rasmussen et al. (22). A 50-mg freeze-dried sample taken from endophyte-infected Lolium perenne whole tillers was extracted for 1 h with 1 ml of extraction solvent (50% [vol/vol] methanol) with 2.064 ng/ml homoperamine nitrate (AgResearch Grasslands) as an internal standard. The sample was then centrifuged for 5 min at 8,000 × g, and a 500-μl aliquot of the supernatant was transferred to an amber 12- by 32-mm high-performance liquid chromatography (HPLC) vial via a 0.22-μm-pore polyvinylidene difluoride (PVDF) syringe filter. Separation was achieved on a Synergi Polar-RP 100- by 2.00-mm (2.5-μm) column (Phenomenex, Torrance, CA) using a linear gradient profile (where eluent A is aqueous 0.1% formic acid and eluent B is acetonitrile), with time 0 (T0) at 5% B, T9 at 40% B, T11 at 90% B, and T13 at 90% B, followed by equilibration to initial conditions over the following 8 min. Peramine was quantified by mass spectroscopy (using homoperamine as an internal standard) according to the parameters described by Rasmussen et al. (22). Peramine is expected to have a retention time of 8.6 min with an MS1 ion of 248.1 m/z, and homoperamine is expected to have a retention time of 9.9 min with an MS1 ion of 262.1 m/z. A 5-μl injection volume gave a limit of detection for this technique of 0.1 μg/g for herbage.
Genomic DNA isolation.
Genomic DNA was isolated from freeze-dried mycelium of Epichloë species using the ZR Fungal/Bacterial DNA MiniPrep kit (Zymo Research, Irvine, CA) as per the manufacturer's instructions. Total plant DNA (including endophyte) was extracted using the MagAttract 96 DNA plant core kit (Qiagen, Inc., Valencia, CA) as per the manufacturer's instructions.
Primer design.
Primers for PCR amplification and sequencing (see Table S3 in the supplemental material) were designed using a multiple-sequence alignment of all available perA and flanking gene sequences from the Epichloë genome database (www.endophyte.uky.edu) (16), which included 10 strains from seven species. Primers were designed to maximize conservation of the target binding sequence between species. Primers for sequencing specific alleles from hybrid species were designed to contain at least two single nucleotide polymorphisms (SNPs) specific to each allele, with one of these SNPs located at the 3′ terminus wherever possible.
PCR amplification and product purification.
Genomic DNA templates were amplified using GoTaq DNA polymerase (Promega, Madison, WI) under the conditions described by Takach et al. (18). PCR products for sequencing were purified using the QIAquick PCR purification kit (Qiagen). Where insufficient PCR product was produced for direct sequencing, a second PCR using a 102- or 103-fold dilution of the initial reaction was used as a template to increase PCR product concentration; this was often necessary when amplifying the perA gene directly from endophyte-infected plant material.
Sequencing of perA.
Three overlapping DNA fragments, perA-1, perA-2, and perA-3, or perA-3ΔR*, covering the whole perA or perA-ΔR* gene, was amplified using primer sets defined in Table S3 in the supplemental material and sequenced with BigDye chemistry v3.1 (Applied Biosystems, Foster City, CA) using an Applied Biosystems 3730 DNA analyzer. Sequences from nonhybrid isolates were assembled using MacVector 12.6 with Assembler (MacVector, Inc., Cary, NC), with further sequencing completed using isolate-specific primers where required. Sequences from hybrid isolates were similarly assembled, but this assembly was then used as a reference to design allele-specific primers based on polymorphic regions to sequence each perA allele.
Phylogenetic reconstruction.
A2 domain DNA sequences from 39 perA alleles (1,785 bp in length from positions 3575 to 5358 for perA from E. festucae Fl1) were aligned using ClustalW (23) and manually edited where necessary with MacVector 12.6. DNA sequences spanning from the middle of the T2 domain until the perA-ΔR* truncation location were similarly aligned from 38 perA alleles. These alignments were analyzed using Mega 5.1 (24) via the maximum likelihood method using the gamma-distributed (5 categories) Tamura three-parameter nucleotide substitution model (25) and the subtree-pruning-regrafting (level 3) heuristic method on all sites and codons. The bootstrap method with 1,000 repetitions was used to test the phylogeny.
Nucleotide sequence accession numbers.
The GenBank accession numbers for perA sequences generated by this study are listed as follows: KP347845 to KP347877 and KP719965 to KP719973. Details about these and additional perA accession numbers from other studies (5, 22) are presented in Table S4 in the supplemental material.
RESULTS
Peramine chemotypes of E. festucae isolates.
The distribution of peramine production within E. festucae was evaluated from herbage samples of Lolium perenne plants symbiotic with E. festucae isolate E189, Fg1, Fl1, Frc5, Frc7, Fr1, or Frr1. Of these associations, only the plants infected with Fl1, Frc7, or Frr1 contained peramine (Per+) (Table 1). These data demonstrated that peramine chemotypes can be highly variable and discontinuous, even between isolates of a single Epichloë species.
TABLE 1.
Peramine concentrations of whole tillers from Lolium perenne infected by different Epichloë festucae isolates
Analysis of perA across multiple Epichloë species.
Genomic DNA extracted from mycelium of 34 different isolates spanning nine nonhybrid Epichloë species, including the E. festucae isolates mentioned above, was used in a PCR-based size polymorphism analysis to evaluate the presence and integrity of the PER locus. PCR primers were designed to amplify each perA domain in overlapping DNA fragments, as well as the conserved flanking genes mfsA and qcrA. The genes mfsA and qcrA, but not perA, were detected in E. glyceriae E2772 and Epichloë elymi E184 (Fig. 2). The majority of isolates (30/34) gave either a full complement of perA fragments or all fragments except the R*-domain fragment (no amplification with the primer pair perA3_3/perA3_R), indicating that these alleles likely lacked R* (which we designate perA-ΔR*). Although the regions encoding the A2 and C domains did not amplify from Epichloë baconii As6 and Epichloë bromicola E799, respectively, we know from sequencing and other PCR that these fragments are present (data not shown).
FIG 2.

Analysis of perA integrity within nonhybrid Epichloë species. PCR-based size polymorphism analysis of perA from genomic DNA. The PCR products were produced using primers designed to the conserved sequence in or near each of the major perA domains and the two flanking genes mfsA and qcrA (see Table S3 in the supplemental material). Gray shading indicates the regions amplified. The dashed lines indicate the region that will amplify with primers perA-mid2-F2/perA_17bp_R only from isolates containing perA-ΔR* (Fig. 3). The regions encoding the adenylation (A), thiolation (T), condensation (C), methylation (M), and reductase* (R*) domains are shown within the perA gene map, with numbers indicating whether they are located in the first or second module. Isolates for which perA was subsequently sequenced or for which the perA sequence was already available (16, 33, 42) are shown in boldface. Low-intensity PCR products indicate the presence of SNPs in the primer target binding sequence. A host species labeled “NA” indicates the endophyte strain is the result of a controlled sexual cross. Festuca rubra subsp. commutata is abbreviated Festuca rubra ssp. comm. nt, not tested. Known peramine chemotypes are indicated as Per+ (peramine producer) or per− (peramine nonproducer) (5, 6, 16, 29, 43, 44). Peramine production was predicted for isolates with unknown peramine chemotypes based on the presence of the expected PCR products amplified from all perA domains and is indicated by + or −. Although not apparent in this screen, a perA remnant retaining the R* domain remains in E184 (39).
Alleles of perA-ΔR* were first observed in the genome sequence of E. festucae E2368, E. festucae var. lolii Lp14, and Epichloë typhina subsp. poae E5819 (16, 17). The region encoding the perA R* domain was also missing from both Epichloë sylvatica isolates tested and was discontinuously distributed within E. baconii, E. bromicola, E. festucae, and E. typhina (Fig. 2). An additional deletion was observed within the region encoding the T1 domain of perA-ΔR* from E. sylvatica isolates E354 and E503 (Fig. 2). Also detected was a deletion in the A2 domain fragment from the otherwise full-length E. festucae Fg1 perA allele (Fig. 2). The identification of perA-ΔR* in E. festucae E189, Frc5, and Fr1 and an A2 domain deletion in E. festucae Fg1 explains the observed per− chemotype of these isolates (Table 1).
Repetitive elements associated with perA-ΔR* alleles.
Previous genome sequencing of E. festucae E2368, E. festucae var. lolii Lp14, and E. typhina subsp. poae E5819 indicated that deletion of the reductase domain was associated with repetitive elements, in particular the MITE 3m (16, 17). Additional representative isolates containing the perA-ΔR* alleles were analyzed by sequencing amplification products that span from the perA T2 domain to the adjacent gene, qcrA (Fig. 3A). This region failed to amplify from E. baconii As6, E. baconii E424, and E. bromicola E799. Comparison of the sequence data indicated variation of the repeats between isolates. In particular, different regions of MITE 3m were retained, and some isolates had the addition of MITE 25m (Fig. 3B). Also associated with the perA-ΔR* alleles is a unique 17-bp sequence located immediately downstream of the perA truncation (Fig. 3B). BLAST analysis using the 17-base sequence as a query revealed it is present only in the genome sequence of isolates containing perA-ΔR* and is not part of a repetitive element. The sequence was utilized as a primer (perA-17bp_R) (see Table S3 in the supplemental material) for PCR tests to determine if the 17-bp sequence was common to all perA-ΔR* alleles. DNA from all isolates identified as perA-ΔR* could be amplified with the perA-T2_F/perA-17bp_R primer set, confirming the association of the common 17-bp region with the deletion of the R* domain (Fig. 2 and 4). Thus, the perA-17bp_R primer allowed specific positive identification of the perA-ΔR* allele.
FIG 3.

Analysis of perA-ΔR* downstream repeat sequences. Schematic representation of Epichloë isolates that lack the perA-R*domain. (A) Overview of the functional perA gene required for peramine production and the associated flanking gene qcrA from E. festucae Fl1. The domains of PerA are detailed in Fig. 2. (B) Schematic comparisons of regions from the perA-T2 domain to qcrA. The regions from E. festucae isolates Fl1 and E2368 and E. typhina subsp. poae E5819 were drawn from perA GenBank accession no. AB205145, JN640287, and JN640289, respectively, and E. bromicola AL0426_2 was generated from a genome sequence. The remaining examples were amplified with primer set perA-mid2-F2/Ef104-R using genomic DNA. Maps are arranged to illustrate synteny and do not necessarily suggest an evolutionary history. Syntenic regions (which may include small indels) between sequences are indicated by light gray polygons. Black vertical boxes indicate MITE 3m or 25m. Repeat sequence 16 (a putative retrotransposon) (16) is indicated by a dark gray horizontal box. The primer region for perA-17bp_R is shown in all perA-ΔR* sequences. (C) Schematic representation of the PER loci from a draft genome sequence of the hybrid species E. siegelii e915 demonstrating the inverted orientation of perA-ΔR* allele 2 relative to the flanking gene mfsA. Linkage between perA-ΔR* allele 2 and qcrA allele 2 is likely but cannot be proven due to the position of these genes on the ends of their respective contigs. The progenitor species from which each E. siegelii allele is derived is indicated in parentheses as “Efe” for E. festucae and “Ebm” for E. bromicola. Repeat sequences Ono and 28 (putative retrotransposons) (16) are indicated by dark gray horizontal boxes.
FIG 4.

Analysis of perA integrity from infected plant material. PCR analysis of perA from plants infected with Epichloë species. The PCR products were produced using primers designed to conserved sequence in or near each of the major perA domains and the two flanking genes mfsA and qcrA (see Table S3 in the supplemental material). Gray shading indicates the regions amplified. The dashed lines indicate the region that will amplify with primers perA-mid2-F2/perA_17bp_R only from isolates containing perA-ΔR* (Fig. 3). The PerA domains shown above the perA gene map are listed in Fig. 2. Isolates for which perA was subsequently sequenced are shown in boldface. Endophytes with unknown hybrid status are labeled “unknown.” LAE, Lolium-associated endophyte; Eam, E. amarillans; Ebm, E. bromicola; Eel, E. elymi; Efe, E. festucae; Ety, E. typhina; Etp, E. typhina subsp. poae; nt, not tested. Known peramine chemotypes are indicated as Per+ (peramine producer) or per− (peramine nonproducer) (5, 6, 18, 19, 29, 43, 44). Peramine production was predicted for isolates with unknown peramine chemotypes based on the presence of expected PCR products amplified from all perA domains and are indicated by + or −.
Analysis of perA from endophyte-infected plant material.
Epichloë species for which no mycelium samples were readily available were evaluated directly from endophyte-infected plant samples. Total DNA was extracted from pseudostem or blade samples from 33 different Epichloë-infected plants spanning 13 grass species to evaluate the presence and integrity of perA. Of these plant symbionts, seven contained nonhybrid endophyte strains, 20 contained hybrid endophyte strains, and the hybrid status of endophytes in the remaining six samples was unknown. These DNA samples were used as the templates for PCR amplifications with the same primers described above for the mycelial genomic DNA templates. In most cases, PCR products were of significantly reduced intensity relative to those amplified from the mycelial genomic DNA samples (Fig. 4). This was expected because the endophyte DNA usually accounts for less than 2% of the total plant DNA (26, 27).
The majority of samples tested appeared to contain at least one intact perA gene. As previously shown, the tested Epichloë sp. FaTG-2 hybrid isolates NFe45079 and NFe45115 and the Epichloë sp. FaTG-3 hybrid isolate NFe1100 contained deletions in the regions encoding the A1 and A2 domains of the perA alleles known to be inherited from a Lolium-associated endophyte (LAE) progenitor (18, 28). Epichloë cabralii BlaTG-2 isolate NFe661, Epichloë sp. FaTG-2 G3 isolate NFe45115, and an isolate of Epichloë uncinata E167 have known per− chemotypes (18, 19, 29), yet each of these isolates appeared to contain at least one full-length perA allele (Fig. 4). The presence of full-length copies of perA suggested that small mutations within these gene copies likely generated perA null alleles.
The perA-ΔR* allele was identified in E. typhina OR10, Epichloë siegelii e915, and an undescribed endophyte, Epichloë sp. isolate e4768, from Festuca versuta (Fig. 4). The absence of the R*-domain product in E. siegelii e915, a two-parent hybrid, suggests that this strain contains two perA-ΔR* alleles. For Epichloë sp. strain e4768, the successful amplification of both the R*-domain and ΔR* deletion-specific PCR product indicates this isolate is a hybrid containing both the perA and perA-ΔR* alleles.
A draft genome sequence of E. siegelii e915 was used to explore the region flanking the two perA-ΔR* alleles (Fig. 3C). Annotation of perA-ΔR*, mfsA, qcrA, and repeat sequences that flank these genes revealed that the perA-ΔR* allele 1 was nearly identical to the arrangement found in E. festucae E2368 (Fig. 3B). Interestingly, the e915 perA-ΔR* allele 2, originating from the E. bromicola progenitor, was oriented toward mfsA rather than qcrA, indicating a gene inversion event has occurred. Although the common 17-bp region was still associated with this allele, there were no longer any downstream repetitive sequences. The perA-ΔR* allele of E. bromicola isolate E799 contained a similar orientation (data not shown). Linkage between the contigs from e915 containing perA-ΔR* allele 2 and qcrA allele 2 cannot be determined from this sequence and were not able to be connected by PCR, likely due to the AT-rich repeat sequence that flanks qcrA (Fig. 3C).
Sequencing and characterization of perA variants.
To determine why some isolates appeared to contain an intact perA gene but did not produce peramine, the perA alleles from 27 nonhybrid and seven hybrid isolates were sequenced or evaluated from genome sequences (Fig. 5; see Table S4 in the supplemental material). Of these 34 isolates, 11 were known to be Per+, and 17 were known to be per−; the peramine chemotypes of the remaining 6 isolates were unknown.
FIG 5.

Genetic events disrupting perA. Gene maps of sequenced perA annotated with mutations likely to cause loss of function. (A) Gene maps of full-length perA sequences. The PerA domains shown on the functional perA gene map are listed in Fig. 2. The 13 Per+ perA alleles represented by the first gene map are from E. amarillans E57 and E4668, E. baconii E1031, E. brachyelytri E4804, E. bromicola AL0434, E501, and E502, E. elymi E56, E. festucae Fl1, E. typhina E8, E. typhina subsp. poae BlaTG-1 isolate NFe671 (incomplete allele sequence), Epichloë sp. FaTG-2 G2 isolate NFe45079, and Epichloë sp. FaTG-3 isolate NFe1100. Gene maps of perA per− alleles detail causative mutations of full-length perA null alleles. (B) Partial gene maps of perA null alleles. (C) Gene maps of perA-ΔR* alleles. The 9 alleles represented by the initial gene map originate from E. baconii As6 and E424, E. festucae E189 and E2368 (identical alleles), E. siegelii e915 allele 1, E. typhina E1022 and OR10 (incomplete allele sequence), E. typhina subsp. poae E5819, and Epichloë sp. e4768 (incomplete allele sequence). Gene maps of other perA-ΔR* alleles detail changes that result in frameshift, nonsense, or large deletion mutations upstream of the conserved perA-ΔR* deletion site. Hollow arrows indicate the coding sequence for each allele, with the arrow ending at the first stop codon. Gene maps are annotated as follows. Frameshift-causing insertions or deletions are shown as white or black circles, respectively, with the number indicating how many nucleotides were inserted or deleted. SNPs that generate nonsense-mutation are shown as black hexagons. SNPs that disrupt the ATG start codon are shown as white squares. Large sequence deletions are shown as dashed lines, with the deletion shown relative to perA from E. festucae Fl1, and solid lines indicate conserved sequence located downstream of a premature stop codon. Eam, E. amarillans; Ebm, E. bromicola; Efe, E. festucae; Ety, E. typhina; Etp, E. typhina subsp. poae; LAE, Lolium-associated endophyte.
The identification of perA-ΔR* explained the per− chemotype for 13 per− isolates (Fig. 5C), and the presence of a deletion in the A2 domain in E. festucae Fg1 explained the per− chemotype of this isolate (Fig. 5A). The remaining three per− isolates, E. cabralii BlaTG-2 isolate NFe661, E. uncinata isolate e167, and Epichloë sp. FaTG-2 G3 isolate NFe45115, were hybrids (18, 19, 29). A 1-bp insertion causing a frameshift mutation was identified in E. cabralii BlaTG-2 NFe661 allele 1 (Fig. 5B), and an SNP that resulted in a nonsense mutation was identified in allele 2 (Fig. 5B). Analyses of the allele sequences from the hybrid isolate E. uncinata e167 identified independent frameshift mutations in alleles 1 and 2, generating perA null alleles (Fig. 5A). An SNP identified in the first adenylation domain of Epichloë sp. FaTG-2 G3 isolate NFe45115 allele 2 resulted in a nonsense mutation, while deletions are present in both A domains of allele 1 (Fig. 5A). These mutations explain the per− chemotype of all three isolates.
The peramine chemotype was unknown for 6 of the 34 isolates from which perA was sequenced, so predictions were made by analyzing these sequence data. Of the six isolates, E. baconii E1031 and E. bromicola AL0434 were predicted to be Per+ because they both contained full-length alleles with no nonsense, frameshift deletion, or insertion mutations. Of the four remaining isolates, E. baconii As6 and E. bromicola AL0426_2 were nonhybrid isolates containing perA-ΔR* alleles and were therefore predicted to be per−. The perA allele from Epichloë mollis AL9924 contained multiple small (<10-bp) insertions causing frameshift mutations (Fig. 5A) and was therefore predicted to be per−. Based on phylogenetic analysis, we judged the partial sequence from hybrid Epichloë sp. e4768 allele 2 to be derived from an E. typhina progenitor (Fig. 6B), and it is considered a perA null allele due to a 1-bp deletion that results in a frameshift mutation (Fig. 5B). Isolate e4768 allele 1 was perA-ΔR* and was derived from an E. festucae E2368-like progenitor (Fig. 6B). Given that this isolate is a hybrid and has two null alleles, it is predicted to have a per− chemotype.
FIG 6.

Phylogenetic tree of perA A2-domain DNA sequence. Shown are unrooted maximum likelihood phylogenetic trees generated from all available perA and perA-ΔR* A2-domain and perA-ΔR* sequences spanning from the middle of the T2 domain to the perA-ΔR* truncation location (T2-ΔR*), ending immediately prior to where homology between the perA and perA-ΔR* sequences stops. The regions used to generate each phylogenetic tree are shown relative to a perA gene map at the top of the figure, with PerA domain abbreviations detailed in the legend to Fig. 2. Black triangles next to isolate numbers identify the perA-ΔR* alleles. Numbers next to entries from hybrid species indicate which allele is represented.
Analysis of the 18 sequenced perA-ΔR* alleles identified eight isolates that contained frameshift mutations, large deletions, and/or nonsense mutations in addition to the known 3m MITE-associated R*-domain deletion (Fig. 5C). A large deletion spanning the junction between the regions encoding the A1 and T1 domains and two small insertions were present in the alleles from E. sylvatica E354 and E503 (Fig. 5C), and nonsense mutations that should significantly truncate the translated protein were present in the E. typhina E505 and E. typhina subsp. clarkii Holcus 3 alleles. The E. bromicola AL0426_2 perA-ΔR* allele contained an SNP that disrupted the start codon, but a potential alternate ATG codon was located 189 bp downstream that does not truncate any conserved A-domain motifs. The nonsense mutations identified in the perA-ΔR* alleles from E. bromicola E799 and E. typhina ORE04 and ORE06 were located close to the existing perA-ΔR* truncation, and no other perA domains were affected (Fig. 5C). Both alleles from the hybrid isolate E. siegelii e915 were confirmed to be perA-ΔR*. A nonsense mutation that would result in truncation of the translated protein was identified in e915 allele 2 (Fig. 5C).
Phylogeny of perA A2- and T2-domain DNA sequences.
Unrooted maximum likelihood phylogenetic trees were generated from the A2-domain DNA sequence of 39 perA and perA-ΔR* alleles, and the DNA sequence spanning from the middle of the T2 domain to the location of the perA-ΔR* truncation (T2-ΔR*) from 38 perA and perA-ΔR* alleles (Fig. 6). The A2-domain phylogeny revealed that all E. typhina complex-derived perA-ΔR* alleles grouped in a clade distinct from E. typhina complex perA alleles (69% bootstrap support). The E. bromicola-derived perA-ΔR* alleles also grouped separately from the E. bromicola perA alleles (80% bootstrap support). The perA-ΔR* alleles derived from E. festucae and E. baconii isolates grouped separately from perA alleles from Epichloë amarillans, E. baconii, and E. festucae isolates (97% bootstrap support). In contrast, the phylogeny of the T2-ΔR* region showed all perA-ΔR* alleles grouped together separated from the perA alleles despite originating from multiple different species (Fig. 6). The A2 domain of FaTG-2 G3 NFe45115 perA allele 2 (derived from E. festucae) grouped with the E. baconii- and E. festucae-derived perA-ΔR* alleles, despite this allele still retaining the R* domain, as was previously observed by Takach et al. (18) (Fig. 6). In contrast the T2-ΔR* region from NFe45115 allele 2 grouped with related perA alleles (Fig. 6).
DISCUSSION
Peramine has been reported as the most commonly produced alkaloid by Epichloë species (66%), yet a discontinuous distribution is found within and between species (5, 6). In this study, we identify nonfunctional perA alleles from hybrid and nonhybrid Epichloë species. Although E. glyceriae and E. gansuensis have been previously shown to lack perA (16), we show the discontinuous distribution of peramine producers across Epichloë species is most frequently associated with mutations within perA that abolish peramine production. Analyses of apparently nonfunctional perA alleles show that each inactivating mutation is either isolate specific or is shared between closely related isolates of nonhybrids or closely related genomes in hybrids. Also, the DNA sequences encoding the first and second adenylation (A1 and A2) domains are common sites for such inactivating mutations (Fig. 5). These data indicate that independent mutation events have inactivated perA many times. The only exception to this rule is the identical R*-domain deletion found in all perA-ΔR* alleles. Identification of these inactivating perA mutations provides information to aid in prediction of peramine producers for isolates with unknown peramine chemotypes and genetic diagnosis for isolates known to be per−.
In this study, the PER locus was evaluated using PCR to assess the integrity of each perA domain from 67 isolates representing at least 20 Epichloë species. We were able to distinguish the full-length perA alleles found in peramine producers and the perA-ΔR* alleles found in strains that are unable to produce peramine. Amplification of genomic DNA using a primer pair specific to perA-ΔR* (perA-T2_F and perA-17bp_R) identified 18 (27%) isolates missing the R* domain, of which two isolates, E. siegelli e915 and Epichloë sp. e4768, were hybrid species and the remaining were nonhybrids. However, the PCR analyses used to detect the presence of each domain did not reveal all perA mutants. Sequencing of perA from isolates unable to make peramine revealed frameshift and nonsense mutations predominantly within the regions encoding the A1 and A2 domains that would render perA nonfunctional. From these sequence data, we were able to explain the mutations responsible for the peramine-negative chemotype previously identified in E. uncinata e167, Epichloë sp. FaTG-2 G3 isolate NFe45115, and E. cabralii BlaTG-2 isolate NFe661 (18, 19, 29).
The isolate- or lineage-specific nature of mutations that have resulted in perA null alleles contrasts sharply with the taxonomic distribution of perA-ΔR*. The perA-ΔR* alleles are distributed widely within the Epichloë genus, occurring in a subset of isolates from each of the E. baconii, E. bromicola, E. festucae, and E. typhina complex (ETC) clades (Fig. 6). Given that the R* domain was deleted at identical sites within perA and there is high sequence conservation immediately downstream of the perA-ΔR* alleles, it is unlikely this deletion occurred more than once. In support of the possibility that a single event was responsible is the consistent association of perA-ΔR* with a downstream MITE 3m sequence and a unique 17-bp sequence containing an in-frame stop codon. Both of these features were absent from all perA alleles, so the MITE 3m insertion seems likely to have been involved in the deletion of the region encoding the R* domain.
The evolution of the perA-ΔR* alleles appears particularly complex, considering the disparate phylogenies of the regions encoding A2 and T2-ΔR* (Fig. 6). The T2-ΔR*phylogeny placed perA and perA-ΔR* into separate clades, each independently reflecting relationships of the Epichloë species. The T2-ΔR* phylogeny suggested transspecies polymorphism (TSP), whereby the corresponding sequences in perA and perA-ΔR* diverged early during, or even before, evolution of the genus Epichloë. This pattern is similar to evidence of TSP in other systems, such as vertebrate major histocompatibility loci (30) and fungal vegetative incompatibility loci (31).
In contrast to the T2 phylogeny, the phylogeny of the A2-encoding sequences consistently grouped perA-ΔR* alleles with perA of the same or closely related species, although in most species, the separation of the perA-ΔR* and perA subclades seemed deeply rooted in the species. The disparity between the A2 and T2-ΔR* phylogenies suggests multiple recombination events. What appears to be the most recent example affected perA allele 2 in FaTG-2 G3 isolate NFe45115. The region encoding the A2 domain of this perA allele groups with the E. festucae and E. baconii perA-ΔR* alleles, whereas the T2-ΔR* sequence groups with E. festucae perA alleles (Fig. 6). The fact that multiple species clades exhibit the disparate A2 and T2-ΔR* phylogenies suggests that a recombination hot spot exists between these two portions of perA.
The only two hybrid isolates containing perA-ΔR* alleles were E. siegelii isolate e915 and Epichloë sp. e4768. This is perhaps surprising given the wide distribution seen in the sexual isolates (44% of isolates tested in this study) (Fig. 2) and the number of hybrid species we tested (21 isolates representing 10 species) that contain E. festucae (7 isolates) and E. typhina (14 isolates) ancestral progenitors.
Previous studies of Epichloë alkaloid biosynthesis loci, such as the ergot alkaloid (EAS), indole-diterpene (IDT/LTM), and loline (LOL) gene clusters, have shown the presence or absence of pathway-specific genes to be the primary factor determining chemotype diversity of these alkaloids (16, 32, 33). The IDT/LTM and EAS loci are localized to dynamic subterminal regions of chromosomes (16), and both these and the LOL gene cluster are closely associated with a variety of transposable elements (16, 17, 34). These factors provide mechanisms through which genes from these clusters, or even an entire gene cluster, may be lost via recombination when selective pressure for a cluster is reduced. In contrast, with the exception of perA-ΔR* alleles, full-length perA alleles have not been found in association with transposable elements (16). In the absence of selective pressure, perA is likely to be retained longer than genes from the other secondary metabolite gene clusters, and this could explain the observed increase of perA inactivation by nonsense, frameshift, or deletion mutations relative to gene loss events common to the EAS, IDT/LTM and LOL gene clusters.
Diagnostic PCR utilizing markers developed from sequences of housekeeping and secondary metabolite biosynthetic genes is an effective approach to identify and quantify potential contamination from mycotoxin-producing fungi within foodstuffs for human and animal consumption (35, 36). For example, multiplex PCRs have been successfully used to simultaneously detect multiple fungal genera found in cereals that are likely to produce ochratoxins and trichothecenes (37). A quantitative PCR (qPCR) assay that detects polymorphisms within TRI12 can identify different trichothecene genotypes within Fusarium species from field samples (38). Chemotype prediction using PCR to detect the presence of biosynthesis genes has also been very successful when evaluating Epichloë species for the ability to produce ergot alkaloids, indole-diterpenes, and lolines and provides insight into the bioactive potential of any given endophyte isolate (18, 19, 32, 39–41). In all of these approaches, the ability to directly analyze infected plant material by PCR provides rapid detection methods for a wide range of organisms and their biosynthetic potential. To determine whether an endophyte isolate is likely to produce peramine, we have refined the PCR approach described previously (18, 19, 39, 41) in order to identify the presence and integrity of all domains encoded by perA. In addition, sequence analysis of the regions encoding the A1 and A2 domains can be used to identify the most commonly found mutations. Using this pipeline, specific isolates with known and unknown peramine chemotypes were screened to identify perA-ΔR* alleles and other observable deletions, and sequence analysis was used to identify frameshift and nonsense mutations that would render perA nonfunctional. Although this method will not eliminate the need to evaluate peramine production, especially for determination of the levels of peramine produced by a given isolate, it does provide insight into the likelihood of peramine production. Evaluation of endophyte-infected plant germplasm for potential peramine producers as well as production of other bioactive alkaloids will help us understand the bioprotective potential of Epichloë species and facilitate investigation into the effects of different geographic and selective pressures on the evolution of this locus.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Ginger A. Swoboda (The Samuel Roberts Noble Foundation) for technical assistance, Pierre-Yves Dupont (Massey University) for assistance with phylogeny reconstruction methodology, and Wade Mace (AgResearch) for measurement of peramine concentrations. We thank Adrian Leuchtmann (ETH Zurich) for access to Epichloë bromicola (AL0434 and AL0426_2) genome sequences.
This research was supported by a Massey University Ph.D. scholarship and a grant from the Royal Society of New Zealand Marsden Fund (contract MAU1002). Genome sequencing was supported by United States Department of Agriculture grants 2012-67013-19384 and 2010-34457-21269, National Institutes of Health grants R01GM086888 and 2 P20 RR-16481, and The Samuel Roberts Noble Foundation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03721-14.
REFERENCES
- 1.Keller NP, Turner G, Bennett JW. 2005. Fungal secondary metabolism—from biochemistry to genomics. Nat Rev Microbiol 3:937–947. doi: 10.1038/nrmicro1286. [DOI] [PubMed] [Google Scholar]
- 2.Forseth RR, Amaike S, Schwenk D, Affeldt KJ, Hoffmeister D, Schroeder FC, Keller NP. 2013. Homologous NRPS-like gene clusters mediate redundant small-molecule biosynthesis in Aspergillus flavus. Angew Chem Int Ed Engl 52:1590–1594. doi: 10.1002/anie.201207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scharf DH, Heinekamp T, Brakhage AA. 2014. Human and plant fungal pathogens: the role of secondary metabolites. PLoS Pathog 10(1):e1003859. doi: 10.1371/journal.ppat.1003859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroken S, Glass NL, Taylor JW, Yoder OC, Turgeon BG. 2003. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc Natl Acad Sci U S A 100:15670–15675. doi: 10.1073/pnas.2532165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clay K, Schardl C. 2002. Evolutionary origins and ecological consequences of endophyte symbiosis with grasses. Am Nat 160:S99–S127. doi: 10.1086/342161. [DOI] [PubMed] [Google Scholar]
- 6.Siegel MR, Latch GCM, Bush LP, Fannin FF, Rowan DD, Tapper BA, Bacon CW, Johnson MC. 1990. Fungal endophyte-infected grasses—alkaloid accumulation and aphid response. J Chem Ecol 16:3301–3315. doi: 10.1007/BF00982100. [DOI] [PubMed] [Google Scholar]
- 7.Bush LP, Wilkinson HH, Schardl CL. 1997. Bioprotective alkaloids of grass-fungal endophyte symbioses. Plant Physiol 114:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bluett SJ, Thom ER, Clark DA, Macdonald KA, Minnee EMK. 2005. Effects of perennial ryegrass infected with either AR1 or wild endophyte on dairy production in the Waikato. N Z J Agric Res 48:197–212. doi: 10.1080/00288233.2005.9513650. [DOI] [Google Scholar]
- 9.Schardl CL, Grossman RB, Nagabhyru P, Faulkner JR, Mallik UP. 2007. Loline alkaloids: currencies of mutualism. Phytochemistry 68:980–996. doi: 10.1016/j.phytochem.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 10.Rowan DD, Hunt MB, Gaynor DL. 1986. Peramine, a novel insect feeding deterrent from ryegrass infected with the endophyte Acremonium loliae. J Chem Soc Chem Commun 1986:935–936. doi: 10.1039/C39860000935. [DOI] [PubMed] [Google Scholar]
- 11.Siegel MR, Bush LP. 1997. Toxin production in grass/endophyte associations, p 185–207. In Carroll G, Tudzynski P (ed), Plant relationships, vol 5 Springer, Berlin, Germany. [Google Scholar]
- 12.Tanaka A, Tapper BA, Popay A, Parker EJ, Scott B. 2005. A symbiosis expressed non-ribosomal peptide synthetase from a mutualistic fungal endophyte of perennial ryegrass confers protection to the symbiotum from insect herbivory. Mol Microbiol 57:1036–1050. doi: 10.1111/j.1365-2958.2005.04747.x. [DOI] [PubMed] [Google Scholar]
- 13.Liu XY, Walsh CT. 2009. Cyclopiazonic acid biosynthesis in Aspergillus sp.: characterization of a reductase-like R* domain in cyclopiazonate synthetase that forms and releases cyclo-acetoacetyl-l-tryptophan. Biochemistry (Wash) 48:8746–8757. doi: 10.1021/bi901123r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leuchtmann A, Bacon CW, Schardl CL, White JF Jr, Tadych M. 2014. Nomenclatural realignment of Neotyphodium species with genus Epichloë. Mycologia 106:202–215. doi: 10.3852/13-251. [DOI] [PubMed] [Google Scholar]
- 15.Moon CD, Craven KD, Leuchtmann A, Clement SL, Schardl CL. 2004. Prevalence of interspecific hybrids amongst asexual fungal endophytes of grasses. Mol Ecol 13:1455–1467. doi: 10.1111/j.1365-294X.2004.02138.x. [DOI] [PubMed] [Google Scholar]
- 16.Schardl CL, Young CA, Hesse U, Amyotte SG, Andreeva K, Calie PJ, Fleetwood DJ, Haws DC, Moore N, Oeser B, Panaccione DG, Schweri KK, Voisey CR, Farman ML, Jaromczyk JW, Roe BA, O'Sullivan DM, Scott B, Tudzynski P, An Z, Arnaoudova EG, Bullock CT, Charlton ND, Chen L, Cox M, Dinkins RD, Florea S, Glenn AE, Gordon A, Gueldener U, Harris DR, Hollin W, Jaromczyk J, Johnson RD, Khan AK, Leistner E, Leuchtmann A, Li C, Liu J, Liu J, Liu M, Mace W, Machado C, Nagabhyru P, Pan J, Schmid J, Sugawara K, Steiner U, Takach JE, Tanaka E, Webb JS, Wilson EV, Wiseman JL, Yoshida R, Zeng Z. 2013. Plant-symbiotic fungi as chemical engineers: multi-genome analysis of the Clavicipitaceae reveals dynamics of alkaloid loci. PLoS Genet 9(2):e1003323. doi: 10.1371/journal.pgen.1003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleetwood DJ, Khan AK, Johnson RD, Young CA, Mittal S, Wrenn RE, Hesse U, Foster SJ, Schardl CL, Scott B. 2011. Abundant degenerate miniature inverted-repeat transposable elements in genomes of epichloid fungal endophytes of grasses. Genome Biol Evol 3:1253–1264. doi: 10.1093/gbe/evr098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takach JE, Mittal S, Swoboda GA, Bright SK, Trammell MA, Hopkins AA, Young CA. 2012. Genotypic and chemotypic diversity of Neotyphodium endophytes in tall fescue from Greece. Appl Environ Microbiol 78:5501–5510. doi: 10.1128/AEM.01084-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charlton ND, Craven KD, Afkhami ME, Hall BA, Ghimire SR, Young CA. 2014. Interspecific hybridization and bioactive alkaloid variation increases diversity in endophytic Epichloë species of Bromus laevipes. FEMS Microbiol Ecol 90:276–289. doi: 10.1111/1574-6941.12393. [DOI] [PubMed] [Google Scholar]
- 20.Moon CD, Tapper BA, Scott B. 1999. Identification of Epichloë endophytes in planta by a microsatellite-based PCR fingerprinting assay with automated analysis. Appl Environ Microbiol 65:1268–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moon CD, Scott B, Schardl CL, Christensen MJ. 2000. The evolutionary origins of Epichloë endophytes from annual ryegrasses. Mycologia 92:1103–1118. doi: 10.2307/3761478. [DOI] [Google Scholar]
- 22.Rasmussen S, Lane GA, Mace W, Parsons AJ, Fraser K, Xue H. 2012. The use of genomics and metabolomics methods to quantify fungal endosymbionts and alkaloids in grasses. Methods Mol Biol 860:213–226. doi: 10.1007/978-1-61779-594-7_14. [DOI] [PubMed] [Google Scholar]
- 23.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 24.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamura K. 1992. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol Biol Evol 9:678–687. [DOI] [PubMed] [Google Scholar]
- 26.Panaccione DG, Johnson RD, Wang JH, Young CA, Damrongkool P, Scott B, Schardl CL. 2001. Elimination of ergovaline from a grass-Neotyphodium endophyte symbiosis by genetic modification of the endophyte. Proc Natl Acad Sci U S A 98:12820–12825. doi: 10.1073/pnas.221198698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young CA, Bryant MK, Christensen MJ, Tapper BA, Bryan GT, Scott B. 2005. Molecular cloning and genetic analysis of a symbiosis-expressed gene cluster for lolitrem biosynthesis from a mutualistic endophyte of perennial ryegrass. Mol Genet Genomics 274:13–29. doi: 10.1007/s00438-005-1130-0. [DOI] [PubMed] [Google Scholar]
- 28.Ekanayake PN, Rabinovich M, Guthridge KM, Spangenberg GC, Forster JW, Sawbridge TI. 2013. Phylogenomics of fescue grass-derived fungal endophytes based on selected nuclear genes and the mitochondrial gene complement. BMC Evol Biol 13:270. doi: 10.1186/1471-2148-13-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leuchtmann A, Schmidt D, Bush LP. 2000. Different levels of protective alkaloids in grasses with stroma-forming and seed-transmitted Epichloë/Neotyphodium endophytes. J Chem Ecol 26:1025–1036. doi: 10.1023/A:1005489032025. [DOI] [Google Scholar]
- 30.Cutrera AP, Lacey EA. 2007. Trans-species polymorphism and evidence of selection on class II MHC loci in tuco-tucos (Rodentia: Ctenomyidae). Immunogenetics 59:937–948. doi: 10.1007/s00251-007-0261-3. [DOI] [PubMed] [Google Scholar]
- 31.Muirhead CA, Glass NL, Slatkin M. 2002. Multilocus self-recognition systems in fungi as a cause of trans-species polymorphism. Genetics 161:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young CA, Tapper BA, May K, Moon CD, Schardl CL, Scott B. 2009. Indole-diterpene biosynthetic capability of Epichloë endophytes as predicted by ltm gene analysis. Appl Environ Microbiol 75:2200–2211. doi: 10.1128/AEM.00953-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schardl CL, Young CA, Pan J, Florea S, Takach JE, Panaccione DG, Farman ML, Webb JS, Jaromczyk J, Charlton ND, Nagabhyru P, Chen L, Shi C, Leuchtmann A. 2013. Currencies of mutualisms: sources of alkaloid genes in vertically transmitted epichloae. Toxins 5:1064–1088. doi: 10.3390/toxins5061064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young CA, Felitti S, Shields K, Spangenberg G, Johnson RD, Bryan GT, Saikia S, Scott B. 2006. A complex gene cluster for indole-diterpene biosynthesis in the grass endophyte Neotyphodium lolii. Fungal Genet Biol 43:679–693. doi: 10.1016/j.fgb.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Niessen L. 2007. PCR-based diagnosis and quantification of mycotoxin producing fungi. Int J Food Microbiol 119:38–46. doi: 10.1016/j.ijfoodmicro.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 36.Gong L, Jiang Y, Chen F. 25 September 2014. Molecular strategies for detection and quantification of mycotoxin-producing Fusarium species: a review. J Sci Food Agric doi: 10.1002/jsfa.6935. [DOI] [PubMed] [Google Scholar]
- 37.Vegi A, Wolf-Hall CE. 2013. Multiplex real-time PCR method for detection and quantification of mycotoxigenic fungi belonging to three different genera. J Food Sci 78:M70–M76. doi: 10.1111/j.1750-3841.2012.03008.x. [DOI] [PubMed] [Google Scholar]
- 38.Nielsen LK, Jensen JD, Rodriguez A, Jorgensen LN, Justesen AF. 2012. TRI12 based quantitative real-time PCR assays reveal the distribution of trichothecene genotypes of F. graminearum and F. culmorum isolates in Danish small grain cereals. Int J Food Microbiol 157:384–392. doi: 10.1016/j.ijfoodmicro.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Charlton ND, Craven KD, Mittal S, Hopkins AA, Young CA. 2012. Epichloë canadensis, a new interspecific epichloid hybrid symbiotic with Canada wildrye (Elymus canadensis). Mycologia 104:1187–1199. doi: 10.3852/11-403. [DOI] [PubMed] [Google Scholar]
- 40.Pan J, Bhardwaj M, Faulkner JR, Nagabhyru P, Charlton ND, Higashi RM, Miller A-F, Young CA, Grossman RB, Schardl CL. 2014. Ether bridge formation in loline alkaloid biosynthesis. Phytochemistry 98:60–68. doi: 10.1016/j.phytochem.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takach JE, Young CA. 2014. Alkaloid genotype diversity of tall fescue endophytes. Crop Sci 54:667–678. doi: 10.2135/cropsci2013.06.0423. [DOI] [Google Scholar]
- 42.Schardl CL, Young CA, Moore N, Krom N, Dupont P-Y, Pan J, Florea S, Webb JS, Jaromczyk J, Jaromczyk JW, Cox MP, Farman ML. 2014. Genomes of plant-associated Clavicipitaceae. Adv Bot Res 70:291–327. doi: 10.1016/B978-0-12-397940-7.00010-0. [DOI] [Google Scholar]
- 43.Christensen MJ, Leuchtmann A, Rowan DD, Tapper BA. 1993. Taxonomy of Acremonium endophytes of tall fescue (Festuca arundinacea), meadow fescue (F. pratensis) and perennial rye-grass (Lolium perenne). Mycol Res 97:1083–1092. doi: 10.1016/S0953-7562(09)80509-1. [DOI] [Google Scholar]
- 44.Schardl CL, Young CA, Faulkner JR, Florea S, Pan J. 2012. Chemotypic diversity of epichloae, fungal symbionts of grasses. Fungal Ecol 5:331–344. doi: 10.1016/j.funeco.2011.04.005. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.