

Abstract
Halohydrin dehalogenase from Agrobacterium radiobacter AD1 (HheC) shows great potential in producing valuable chiral epoxides and β-substituted alcohols. The wild-type (WT) enzyme displays a high R-enantiopreference toward most aromatic substrates, whereas no S-selective HheC has been reported to date. To obtain more enantioselective enzymes, seven noncatalytic active-site residues were subjected to iterative saturation mutagenesis (ISM). After two rounds of screening aspects of both activity and enantioselectivity (E), three outstanding mutants (Thr134Val/Leu142Met, Leu142Phe/Asn176His, and Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala mutants) with divergent enantioselectivity were obtained. The two double mutants displayed approximately 2-fold improvement in R-enantioselectivity toward 2-chloro-1-phenylethanol (2-CPE) without a significant loss of enzyme activity compared with the WT enzyme. Strikingly, the Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala mutant showed an inverted enantioselectivity (from an ER of 65 [WT] to an ES of 101) and approximately 100-fold-enhanced catalytic efficiency toward (S)-2-CPE. Molecular dynamic simulation and docking analysis revealed that the phenyl side chain of (S)-2-CPE bound at a different location than that of its R-counterpart; those mutations generated extra connections for the binding of the favored enantiomer, while the eliminated connections reduced binding of the nonfavored enantiomer, all of which could contribute to the observed inverted enantiopreference.
INTRODUCTION
The application of biocatalysts in the production of optically pure compounds has attracted much attention during the past few decades. Enantioselectivity (E) is one of the key parameters that define the usefulness of enzymes in the corresponding industrial synthesis of fine chemicals. Thus, identification of enzymes with high enantioselectivity for a desired transformation is important. In nature, biocatalysts with high enantioselectivity for specific industrial applications are not readily available. Directed evolution has become a routine approach for developing such novel biocatalysts, and a plethora of successful examples have been reported (1–6). A recent survey regarding the location of mutations that improve enzyme properties shows that to manipulate the enantioselectivity of enzymes, closer mutations are more effective than distant mutations (7). With the availability of structural information, the methodology of CASTing (combinatorial active-site saturation test) used in an iterative manner has been particularly successful in tailoring enzyme enantioselectivity (8–11).
Microbial halohydrin dehalogenases attract a great deal of attention for their ability to produce optically pure epoxides and halohydrins (12, 13). The enzymes which are involved in the biodegradation pathway of halopropanols catalyze the intramolecular nucleophilic displacement of a halogen by a vicinal hydroxyl group in halohydrins, producing epoxide and HCl. Halohydrin dehalogenase HheC has been studied extensively because of its high enzymatic activity and high R-enantioselectivity toward some short-chain aliphatic and aromatic substrates (14–17). The enzyme has been applied in the production of optically pure epoxides and most of the R-enantiomers of β-substituted alcohols through kinetic resolution, with a maximum yield of 50% (18–20). Although kinetic resolution has a maximum yield of 50%, both enantiomers could be obtained by the resolution of a racemic mixture using enantiocomplementary enzymes (21, 22). Directed evolution of enantiocomplementary enzymes has been investigated recently for the production of several optically active compounds (8, 9, 23). Wu et al. reported the evolution of enantiocomplementary Candida antarctica lipase B mutants for the hydrolytic kinetic resolution of p-nitro-phenyl-2-phenylpropanoate by applying iterative saturation mutagenesis (ISM) (9). In conjunction with the strategies for constructing high-quality libraries, van Leeuwen et al. also adopted the ISM strategy to obtain enantiocomplementary haloalkane dehalogenase variants that catalyze the conversion of 1,2,3-trichloropropane to enantiopure building blocks (23). These results indicated that the active-site residues are indeed hot spots for manipulating enzyme enantioselectivity, although the high risk of inactivation could not be excluded. However, such a limitation could be easily released by applying some on-line software products, such as HotSpots Wizard and ConSurf-HSSP (24, 25).
So far, only one S-selective mutant of halohydrin dehalogenase from Arthrobacter AD2 (HheA) has been evolved by using a semirational design approach, which can be used together with the R-selective HheC to produce optically pure halohydrins and epoxides in an enantioconvergent manner (26). However, the S-selective HheA mutant showed a very low catalytic efficiency in the dehalogenation of halohydrins. From this point of view, HheC could be a good starting point for evolution of efficient HheC variants because the wild-type (WT) enzyme displays much higher activity than HheA toward most halohydrins, especially for aromatic substrates and those with short-chain aliphatic substrates.
With the availability of the X-ray crystal structures of HheC (15, 27), it is possible to manipulate the enantioselectivity by exploring the active-site sequence space of the enzyme. The active site of HheC is located in a loop-rich region that forms the substrate-binding pocket and the halide-binding site. Inspection of the structures of HheC with both enantiomers of para-nitro-styrene oxide (PDB codes 1ZMT and 1ZO8) revealed that the epoxide ring of the unfavored S-enantiomer adopts a nonproductive binding mode with its Cβ atom rather than the epoxide oxygen connecting to the hydroxyl moieties of Ser132 and Tyr145. Thus, the high R-enantioselectivity is postulated to be mainly caused by the nonproductive binding of the unfavored S-enantiomer in the spatially limited substrate-binding pocket of HheC (27). Among all residues lining the substrate-binding pocket, the small bulge formed among the three active-site loop residues Thr134, Trp139, and Asn176 was proposed to be accountable for the high R-enantioselectivity toward disubstituted epoxides in the wild-type HheC (28). Moreover, the loop residue W139 was reported to be a critical obstacle for the binding of the S-enantiomer of bulky substrates, and the binding of the S-enantiomer was largely enhanced when Trp139 was replaced by a phenylalanine (29). These findings suggested that the enantioselectivity of HheC is amenable to being inverted by focused directed evolution.
In this study, seven active-site residues, including the above-mentioned three residues Thr134, Trp139, and Asn176, were subjected to iterative CASTing analysis aimed at evolving enantiocomplementary HheC for the conversion of 2-chloro-1-phenylethanol (2-CPE) into an optically active synthetic intermediate. After screening of approximately 7,000 colonies through two rounds of mutagenesis, three outstanding mutants (Thr134Val/Leu142Met, Leu142Phe/Asn176His, and Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala mutants) with divergent enantioselectivity were generated, by which both enantiomers of styrene oxide with a high enantiomeric excess (ee) were obtained. Most importantly, the catalytic efficiency of the S-selective mutant toward (S)-2-CPE is 100-fold higher than that of the wild-type HheC.
MATERIALS AND METHODS
Materials.
rac-2-Chloro-1-phenylethanol (rac-2-CPE), (R)-2-CPE, (S)-2-CPE, 1,3-dichloro-2-propanol (1,3-DCP), and (R)- and (S)-styrene oxides were purchased from Alfa Aesar (Ward Hill, MA). l-Arabinose was from Lancaster (Morecambe, United Kingdom). Phusion high-fidelity PCR kit, restriction enzyme DpnI, and DNA molecular weight standards were purchased from New England BioLabs (Ipswich, MA). Escherichia coli strain MC1061 which was kindly provided by Dick B. Janssen (Groningen University, Groningen, the Netherlands). All the other materials were purchased from local businesses.
Library construction.
All libraries were constructed by using a Phusion high-fidelity PCR kit (New England BioLabs). The primers used in this study were synthesized from Invitrogen (Shanghai, China) and are shown in Table S1 in the supplemental material. Libraries A to D were constructed by using the recombinant expression vector pBADHheC carrying as a template the wild-type hheC gene (GenBank accession number AF397296). The templates used for the construction of libraries E to G are shown in Table S2. All “small intelligent” degenerate codon sets rather than NNS (i.e., where N indicates A, T, C, or G and S indicates C or G) were designed by using the DC_Analyzer software tool (the software is available from http://cobi.uestc.edu.cn/resource/dc_analyzer/view) (30). Primers with the corresponding degenerated codons were synthesized independently and mixed with a ratio of their encoded amino acid numbers. The obtained primer mixtures were used for the following PCR processes. The 20-μl reaction mixture contained 1× high-fidelity buffer, 200 μM (each) deoxynucleoside triphosphate (dNTP), 1 mM Mg2+, 40 ng of template, 2 μM (each) mixed primers, and 0.01 U/μl of Phusion DNA polymerase. The modified PCR program used was 3 min at 98°C, followed by 10 cycles of 10 s at 98°C, 45 s at 58°C, and 2 min at 72°C and then 20 cycles of 10 s at 98°C, 45 s at 68°C, and 2 min at 72°C, ending with 10 min at 72°C. The modified PCR program was used for the construction of all paired-residue libraries, except for the library construction of P84/F86, which was performed according to the manual supplied with the Phusion high-fidelity PCR kit. The digestion of parental DNA by DpnI was carried out at 37°C for 2 h, after which the reaction mixtures were transformed into chemically competent E. coli MC1061.
Colorimetric assay for library screening.
All well-separated clones were picked from agar plates and transferred to 96-well plates containing 180 μl of LB in each well with 100 μg/ml of ampicillin and 0.05% (wt/vol) l-arabinose for protein expression induction. All libraries were screened for halohydrin dehalogenase activity using the previously developed colorimetric assay (31). The pH indicator-based assay is based on the absorbance change of phenol red in a weakly buffered system, due to the release of protons from the enzyme-catalyzed dehalogenation reactions. In the first round of mutagenesis, libraries were screened by using 1,3-DCP (5 mM) as a model substrate, while 5 mM (R)-2-CPE and (S)-2-CPE were used in the second round of mutagenesis for enantioselectivity screening.
Protein expression and purification.
The recombinant strain E. coli MC1061, carrying either the wild-type HheC or the mutant genes, was incubated overnight at 30°C in 200 ml of LB medium containing 100 μg/ml of ampicillin and 0.05% (wt/vol) l-arabinose to reach an optical density at 600 nm (OD600) of 1.7 to 2.0. Cells were harvested by centrifugation (5,000 × g for 50 min) at 4°C and washed once with 10 mM Tris-SO4 buffer (pH 8.0). Both wild-type HheC and its mutants were purified by using a Q-Sepharose (20 ml; GE) column as described before (32). The purity of the purified proteins was judged by SDS-PAGE (10% acrylamide gel). The concentrations of purified proteins were measured using Bradford's method.
Enzymatic kinetic resolutions.
The kinetic resolution experiments were performed using the purified enzyme in a sealed container at 30°C. The dehalogenation reactions (20-ml system) of 5 mM rac-2-CPE were performed in 200 mM Tris-SO4 (pH 8.0) and trigged by adding 120 μg of purified enzyme. All enzymatic reactions were validated by carrying out reactions in the absence of enzyme under the same conditions. The enzymatic conversion was monitored by periodically taking samples (0.25 ml) from the reaction mixture. The samples were extracted with 0.5 ml of methyl tert-butyl ether (MTBE) containing mesitylene as an internal standard. Gas chromatography (GC) analyses were preformed after the samples were dried with MgSO4. The enantiomeric excess (ee) of products was determined by chiral GC under the following conditions: 100°C for 6 min, 10°C/min to 170°C, and 15 min at 170°C. All separations were carried out using a chiral column β-dex 225 (30 m by 0.25 mm by 0.25 μm; Supelco). The ee values were calculated according to the method of Chen et al. (33). E values were calculated by using the program Selectivity (K. Faber and H. Hoenig [ftp://borgc185.kfunigraz.ac.at/pub/enantio/]).
Steady-state kinetic measurements.
Kinetic parameters of the wild-type HheC and its variants toward 1,3-DCP and both enantiomers of 2-CPE were taken in 50 mM Tris-SO4 buffer (pH 8.0) at 37°C by monitoring halide liberation using purified enzymes as described before (26, 34).
Molecular docking analysis.
Ligand Protein Data Bank (PDB) files were produced using the Prodrg Server. All structural models of HheC and its corresponding mutants were built by SWISS-MODEL. The crystal structure of HheC (PDB code 1ZMT) was used as a model template after removal of all crystallographic bound waters (27). Hydrogen atoms were added to the protein and residues were kept fixed in their crystallographic positions in all experiments. Gasteiger charges were added to the ligand. Proteins were prepared by the addition of the polar hydrogen atoms, followed by the addition of Kolloman charges. The rigid dockings between proteins and ligands were performed using the AutoDock Vina. The docking center was located at the catalytic residue Tyr145, and the docking box was settled to contain all desired active-site residues. Visualization and graphics were made using the PyMol software program.
MD simulation.
Molecular dynamic (MD) simulation analysis was performed to gain more information concerning the binding of (S)-2-CPE in the evolved S-selective Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala mutant. The model structure of the Pro84Vla/Phe86Pro/Thr134Ala/Asn176Ala mutant with substrate (S)-2-CPE docked in the active sites was used as the initial structure for simulation. The model structure of the mutant was built by using SWISS-MODEL, and the X-ray structure of HheC (PDB code 1ZMT) was used as a template (27). The whole simulation was implemented in NAMD 2.9. The AMBER 12 force field was chosen to parameterize the whole system, as it is an excellent force field for simulating biological macromolecules. The whole simulation process was conducted as follows: the system energy that contains a box of water molecules with restrained protein was minimized with a force constant of 5 kcal/mol for 2,000 steps; a full-scale minimization of the system for 8,000 steps was performed. To assess thermodynamic equilibrium, the widely used two-section protocol was adopted. A 9-ns MD simulation was performed (the system was heated to 20°C in 20 ps and a constant pressure equilibrium under 1 standard atmosphere pressure was conducted for 400 ps).
RESULTS AND DISCUSSION
Library design and preliminary screening.
A recent survey tracking the effect of the mutations on enzyme enantioselectivity revealed that in many cases closer mutations are more effective than distant ones (7). Based on the structural information and previous kinetic studies of HheC (14, 15, 27–29), seven noncatalytic active-site residues of HheC (Pro84, Phe86, Thr134, Leu142, Trp139, Asn176, and Tyr187) were targeted for the identification of key residues in controlling enzyme enantioselectivity. These residues, except for residue Leu142, are located at the three connected active-site loops (residues 74 to 99, loop 1; residues 132 to 140, loop 2; and residues 175 to 188, loop 3). According to the spatial location of these residues, 4 CASTing libraries named library A (Phe86/Leu142), library B (Thr134/Asn176), library C (Pro84/Phe86), and library D (Trp139/Tyr187) were constructed. To reduce amino acid bias, four “small intelligent” codon sets were designed by using the computer software DC_Analyzer (32). Theoretically, the resulting libraries contain 400 variants of each. For each library, 1,200 colonies should be screened to reach 95% coverage (35). To lower the experimental cost, 1,3-DCP was selected as a model substrate in the preliminary screening, which aimed to eliminate those variants that are inactive or less active than the wild-type enzyme. After screening approximately 4,800 colonies, 35 positive colonies with approximately 1.0- to 4.4-fold improvement in activity toward 1,3-DCP were selected (see Table S3 in the supplemental material). Sequencing results showed that in some randomized positions, the substituted amino acids are generally clustered into a specific group. Strikingly, in library C, nonpolar small residues showed localization preference at positions 84 and 86 for improved activity; most active variants contain a proline residue at either position 84 or 86. The existence of the distinctive cyclic structure of the side chain of proline has been proven to affect protein conformational rigidity in many cases (36). It is worth noting that a conformational change around these active-site loop regions in HheC has proven to be an essential aspect of enzymatic activity. Thus, the results might indicate that a certain conformational configuration of loop 1 is crucial for the enzyme activity of HheC, probably facilitating the release of a halide ion, because such an event was identified as a rate-limiting step during the catalytic cycle of HheC (14).
Evolving enantiocomplementary enzymes.
To fully analyze the enantioselective properties of the active variants, 12 out of 35 positive mutants that cover all classes of mutations in each library were subjected to GC analysis. For this, 2-CPE was selected as a model substrate mainly because the two enantiomers of this substrate are commercially available and the three-dimensional (3D) structures of HheC in complex with both enantiomers of the aromatic substrate have been solved (15, 27). The GC results showed that no mutant with an excellent enantioselectivity was obtained. This is not surprising, because those mutants were selected based on their catalytic ability toward 1,3-DCP. As shown in Table 1, all tested mutants in library A displayed enantioproperties similar to those of the wild-type enzyme; libraries B to D contained numerous HheC variants with decreased enantioselectivity toward 2-CPE. In particular, three mutants (Asn176Ala, Pro84Ala, and Thr134Ala mutants) displayed a significant decrease in enantioselectivity, retaining only 1.5 to 12.3% of the value of the wild-type enzyme (ER = 65 for the wild type). This result indicated that the side chains of these three residues in the wild-type HheC might be important for maintaining the R-enantiopreference of the enzyme. Moreover, the Thr134Vla and Asn176His replacements in library B did not show any negative effect on the enantioselectivity of HheC. Taking these results together, we speculated that the high R-enantiopreference of HheC toward these aromatic substrates is more likely due to steric constraints on the binding of S-enantiomers imposed by the side chains of these amino acids. Such a hypothesis was further supported by subsequent molecular docking analysis. With the Pro84Ala, Thr134Ala, and Asn176Ala mutants, (S)-2-CPE could be well fitted into the substrate-binding pocket of these mutant enzymes in a productive way (see Fig. S1 in the supplemental material), while a nonproductive binding mode was obtained when the (S)-2-CPE was docked with the wild-type HheC (see Fig. S2 in the supplemental material). Thus, residues in libraries B and C were mainly focused on evolution of S-selective HheC, while residues in libraries A and B were targeted for evolution of its counterpart R-selective HheC.
TABLE 1.
GC analysis of selected positive mutants from CASTing librariesa
| Library (residue) | Enzyme | % conversion (min) | eeP (%) | E valueb |
|---|---|---|---|---|
| HheC WT | 51 (20) | 91 | 65 | |
| A (F86/L142) | L142M mutant | 48 (30) | 95 | 66 |
| F86L/L142K mutant | 53 (35) | 96 | 78 | |
| L142F mutant | 46 (15) | 93 | 65 | |
| L142S mutant | 50 (25) | 90 | 58 | |
| B (T134/N176) | N176A mutant | 48 (20) | 30 | 1 |
| T134A mutant | 51 (25) | 73 | 8 | |
| T134E/N176F mutant | 48 (25) | 74 | 8 | |
| N176H mutant | 52 (20) | 95 | 72 | |
| T134V mutant | 47 (20) | 92 | 69 | |
| C (P84/F86) | P84V/F86P mutant | 48 (20) | 85 | 23 |
| P84A mutant | 55 (15) | 69 | 6 | |
| F86Y mutant | 45 (20) | 85 | 50 | |
| D (W139/Y187) | W139V/Y187F mutant | 64 (20) | 53 | 12 |
| W139V mutant | 54 (15) | 71 | 16 | |
| Y187F mutant | 47 (20) | 90 | 33 | |
| W139E mutant | 50 (25) | 85 | 26 |
The absolute configuration in all cases was R. eep, ee of the product.
E values were calculated using the program Selectivity (K. Faber and H. Hoenig [ftp://borgc185.kfunigraz.ac.at/pub/enantio/]).
Consequently, two combinatorial libraries, E (S-selective library) and F (R-selective library), were constructed (see Table S2 in the supplemental material). Because the variant diversities in the two libraries are only 45 and 60, respectively, the two libraries were directly subjected for enantioselectivity screening using both enantiomers of 2-CPE. After screening 180 colonies of each library, three positive colonies (Pro84Vla/Phe86Pro/Asn176Ala, Pro84Vla/Phe86Pro/Thr134Cys/Asn176Ala, and Pro84Vla/Phe86Pro/Thr134Ala/Asn176Ala colonies) from library E and two positive colonies (Thr134Vla/Leu142Met and Leu142Phe/Asn176His colonies) from library F were selected based on their activity difference toward the two enantiomers of 2-CPE. These three variants in library E displayed much higher activity toward (S)-2-CPE than toward its R-counterpart, while the other two variants in library F showed substantially diminished activity toward the two enantiomers of 2-CPE. It is probable that these variants are enantiocomplementary enzymes in the dehalogenation of rac-2-CPE.
Kinetic resolution studies.
To further investigate the enantioselectivity patterns of those variants, studies on the kinetic resolution of rac-2-CPE were performed using purified enzymes (Table 2). As expected, the Thr134Val/Leu142Met and Leu142Phe/Asn176His variants exhibited an improved R-enantioselectivity, with ER values of 107 and 132, respectively, compared with the ER value of the wild-type enzyme (ER = 65). The Pro84Val/Phe86Pro/Asn176Ala, Pro84Val/Phe86Pro/Thr134Cys/Asn176Ala, and Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala variants showed an inverted enantioselectivity, with ES values of 28, 54, and 101, respectively. Figure 1 showed the progress curves for the dehalogenation of rac-2-CPE catalyzed by both the wild-type HheC and the Leu142Phe/Asn176His and Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala mutants. No clear chemical reaction was observed during the reaction period, indicating that rac-2-CPE was chemically stable under our experimental conditions (Fig. 1B). In the dehalogenation reaction of rac-2-CPE catalyzed by the wild-type HheC and the S-selective Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala mutant (Fig. 1A), the reaction reached approximately 50% of the total conversion after 25 min, yielding ee values of 91% and 97% for the products of (R)- and (S)-styrene oxides, respectively (Table 2). With the same amount of enzyme, the Leu142Phe/Asn176His variant catalyzed the conversion 2-fold slower than the wild-type HheC, with a total conversion of 50% after 50 min, yielding R-product with an ee value of 98% (Table 2).
TABLE 2.
Characterization of the wild-type HheC and the enantioselective mutants obtained from combinatorial libraries
| Enzyme | Absolute configuration |
rac-2-Chloro-1-phenylethanol |
(R)-2-Chloro-1-phenylethanol |
(S)-2-Chloro-1-phenylethanol |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| eep (%) | % conversion (min) | E valuea | Km (mM) | kcat (s−1) | kcat/Km (s−1 M−1) | Km (mM) | kcat (s−1) | kcat/Km (s−1 M−1) | Ecalb | ||
| HheC WT | R | 91 | 51 (20) | 65 | 0.45 ± 0.04 | 14.83 ± 1.40 | 32,956 | 4.53 ± 0.46 | 2.37 ± 0.23 | 523 | 63 |
| P84V/F86P mutant | R | 85 | 48 (20) | 23 | 0.72 ± 0.13 | 25.1 ± 1.89 | 34,861 | 2.19 ± 0.61 | 4.24 ± 1.11 | 1,936 | 18 |
| P84V/F86P/N176A mutant | S | 84 | 50 (7.5) | 28 | 2.54 ± 0.43 | 6.57 ± 0.85 | 2,587 | 0.60 ± 0.09 | 29.98 ± 2.1 | 49,967 | 19 |
| P84V/F86P/T134C/N176A mutant | S | 90 | 51 (25) | 54 | 6.73 ± 0.58 | 3.54 ± 0.32 | 526 | 0.52 ± 0.11 | 13.56 ± 1.21 | 26,077 | 50 |
| P84V/F86P/T134A/N176A mutant | S | 97 | 49 (20) | 101 | 8.02 ± 1.03 | 2.77 ± 0.20 | 345 | 0.42 ± 0.06 | 15.06 ± 1.03 | 35,857 | 104 |
| T134V/L142M mutant | R | 97 | 47 (40) | 107 | 0.23 ± 0.07 | 5.61 ± 0.63 | 24,391 | 9.70 ± 1.18 | 2.19 ± 0.17 | 226 | 108 |
| L142F/N176H mutant | R | 98 | 57 (50) | 132 | 0.19 ± 0.05 | 4.45 ± 0.37 | 23,421 | 10.31 ± 1.01 | 1.57 ± 0.09 | 162 | 152 |
| P84V/F86P/T134A/W139Q/N176A/Y187F mutant | S | 94 | 36 (40) | 54 | —c | — | — | — | — | — | — |
E values were calculated by using the program Selectivity (K. Faber and H. Hoenig [ftp://borgc185.kfunigraz.ac.at/pub/enantio/]).
The Ecal value was calculated using the equation Ecal = (kcat/Km)R/(kcat/Km)S (R preference mutants) or Ecal = (kcat/Km)S/(kcat/Km)R (S preference mutants).
—, not determined.
FIG 1.
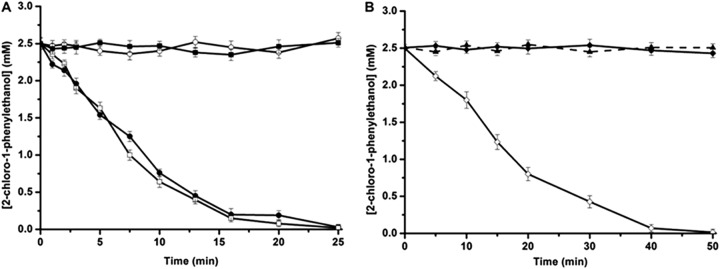
Progress curves of the dehalogenation of 5 mM rac-2-CPE in 20 ml of Tris-SO4 (200 mM; pH 8.0) at 37°C. Enantiomers of 2-CPE are shown as open symbols for S and solid symbols for R. (A) Reactions were carried out with wild-type HheC (circles) and P84V/F86P/T134A/N176A (squares). (B) The reaction was catalyzed by the L142F/N176H mutant (diamonds). We used 120 μg of purified enzymes in all cases. The chemical background was established in the absence of enzyme and is indicated by the solid triangle. The error bars indicate standard deviations from triplicate results.
In our previous studies, the active-site residue Trp139 also represented a critical obstacle for the binding of the S-enantiomer of bulky substrates. To further improve the S-enantioselectivity of HheC, mutations on position 139 were introduced in library G that were constructed by using the mixture of the two S-selective Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala and Pro84Val/Phe86Pro/Thr134Cys/Asn176Ala mutants as a template. However, only one mutant, the Pro84Val/Phe86Pro/Thr134Ala/Trp139Gln/Asn176Ala/Tyr187Phe mutant, obtained an enantioselectivity similar to that of the parental enzyme, indicating that the bulky side chain of Trp139 is no longer an obstacle for the binding of (S)-2-CPE in the case that such a binding obstacle was eliminated in these two S-selective mutants.
Steady-state kinetic analysis.
To gain insight into the mechanism of the improved and inverted enzyme enantioselectivity by those mutations in HheC, steady-state kinetic parameters for both the wild-type enzyme and the HheC variants toward each enantiomer of 2-CPE were determined using purified enzymes (Table 2). The purity of the purified variants is about 80% as judged by SDS-PAGE (see Fig. S3 in the supplemental material). Steady-state kinetic results showed a decreased effect on the kcat values toward both enantiomers of 2-CPE, while an opposite effect on the Km values by these mutations was observed in the two R-preference Thr134Val/Val142Met and Leu142Phe/Asn176His double mutants. Thus, the effect on Km values by these mutations could be the main determinant of the improved R-enantioselectivity with these two R-selective mutants.
All three S-selective mutants showed an approximately 10-fold-lower Km value and 6- to 12-fold-higher kcat values for (S)-2-CPE than those of the wild-type HheC, resulting in a 50- to 100-fold improvement in catalytic efficiency. In contrast, these mutations showed a negative effect on both Km and kcat values for (R)-2-CPE. The catalytic efficiency of these S-selective mutants toward (R)-2-CPE was greatly decreased (approximately 15- to 100-fold) by these mutations compared with that of the wild-type enzyme. The results indicated that the effects of these mutations on both substrate binding and catalytic activity all contribute to the inverted enantioselectivity with these S-selective mutants. It is worth noting that the catalytic efficiency of the highly S-selective mutant (the P84V/F86P/T134A/N176A mutant) is 32-fold higher than that of our previously evolved S-selective Asn178Ala HheA mutant (28), and also higher than that of the wild-type HheC toward its preferred substrate, (R)-2-CPE. Thus, the P84V/F86P/T134A/N176A mutant could be an efficient catalyst for producing (S)-styrene oxide.
The observed opposite effect of these mutations on Km values with these S-selective mutants might be due to the elimination of steric constraints on the binding of the S-enantiomer of 2-CPE imposed by the side chains of those active-site residues. The results correlate well with structural information suggesting that the high R-enantioselectivity is mainly caused by the nonproductive binding of the unfavored S-enantiomer in the spatially limited substrate-binding pocket of HheC (29). Moreover, it was found that the R-selective variants tend to adopt amino acids with a relatively large side chain at positions 134 and 176, while the two most S-selective mutants tend to adopt amino acids with small side chains, such as alanine, at these two positions. Therefore, these results imply that these two residues might play key roles in enantiomer recognition toward rac-2-CPE in HheC.
In addition to the opposite effect on Km values, these mutations also displayed a positive effect on the kcat value toward the S-enantiomer of 2-CPE and a negative effect toward its R-counterpart. The results imply that a different kinetic mechanism may exist for the dehalogenation of the two enantiomers by HheC. Such a conclusion was supported by our previous pre-steady-state kinetic studies for HheC (14), in which a pre-steady-state burst phase during conversion of (R)-para-nitro-2-bromo-1-phenyl ethanol [(R)-PNSHH] was observed; no such burst was observed with the S-enantiomer. This finding indicated that a product release step is rate limiting for the R-enantiomer but not for the S-enantiomer. In comparison with the kcat value of the Pro84Val/Phe86Pro mutant, the introduction of the mutation Asn176Ala showed a large increase in kcat value toward (S)-2-CPE. However, previous studies showed that the Asn176Ala mutation alone did not show any significant increased effect on the enzyme activity (17). Inspection of the X-ray structure of HheC (PDB code 1ZMT) revealed that these three residues are located at two connected active-site loop regions (15), implying that a cooperative interaction between the two loops might play a crucial role in modulating the catalytic properties of HheC toward (S)-2-CPE. For further exploration of the kinetic mechanism of HheC in the dehalogenation of (S)-2-CPE, more pre-steady-state kinetic studies are needed. Moreover, the Ecal values calculated on the basis of steady-state kinetic parameters are comparable to those obtained from kinetic resolution studies, indicating that these two experiments were conducted under similar conditions.
Structural characterization of mutants by computer modeling.
To further unveil the enantioselective nature of HheC toward 2-CPE, molecular docking of both enantiomers of 2-CPE with the two best R-selective and the two best S-selective HheC mutants was carried out using the program AutoDock Vina (Fig. 2; see also Fig. S4 in the supplemental material). In HheC, the enzyme uses a conserved catalytic triad (Ser132/Tyr145/Arg149) to catalyze the dehalogenation of halohydrins via a general base catalytic mechanism. Thus, the selection criteria for hits are as follows: the secondary hydroxyl moiety of Cα in the substrate was positioned within hydrogen bonding distance of the OH groups of the catalytic residues Tyr145 and Ser132; the halide group of Cβ was placed in the direction of the halide-binding site of HheC. In the selected binding modes for (R)-2-CPE with the two R-selective mutants (Fig. 2), the substrate docks in the same binding pocket as for the wild-type HheC, with a slight difference in the binding orientation of both the phenyl group and chloride atom (see Fig. S2B in the supplemental material). Moreover, these binding modes of (R)-2-CPE are similar to that of (R)-1-para-nitro-phenyl-2-azidoethanol in the active site of the wild-type HheC, as revealed from the 3D structure of the HheC·azidoalcohol complex (15), suggesting that these docking results are reliable and can be used for the following analysis. In these two cases, the chloride atom interacts mainly with the phenyl groups of Phe12 and Phe186 and the peptide oxygen of Pro175. In addition to the catalytic residues Ser132 and Tyr145, the hydroxyl group of the substrate of (R)-2-CPE also connects with the side chain of Vla134 and Thr134, while such connections were eliminated in the S-selective mutants in which residue 134 was replaced by Ala and Cys. Inspection of the binding pattern of the phenyl group in the hydrophobic pocket revealed a new van der Waals connection between the side chain of residue 142 and the phenyl group of (R)-2-CPE in the two R-selective mutants. In addition, the Leu142Phe/Asn176His mutant also forms one hydrogen bond between the hydroxyl group of the substrate and the Nδ1 atom of His176, which could well explain the facts that the two R-selective mutants showed lower Km values toward (R)-2-CPE than the wild-type HheC and that the Leu142Phe/Asn176His mutant had the lowest Km value among the three enzymes. These results emphasized that the three randomized residues 134, 142, and 176 all contribute to the binding of (R)-2-CPE.
FIG 2.

Representation of the structural models of two R-selective HheC mutants, the T134V/L142M (A) and L142F/N176H (B) mutants, with (R)-2-CPE docked in the active site. The catalytic triad Ser132/Tyr145/Arg149 and the mutated residues are shown in stick representation. Hydrogen bonds are indicated as dashed lines.
With the two S-selective Pro84Val/Phe86Pro/Thr134Cys/Asn176Ala and Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala mutants, (S)-2-CPE could be fitted well into the substrate-binding pocket in a productive way, as expected. Compared with the binding mode of (R)-2-CPE, (S)-2-CPE was accommodated into the same binding pocket, with a slightly different binding orientation for both the chloride atom and the phenyl group. However, no clear pattern could be found for how the enzyme controls its enantioselective preference from such a similar binding mode for the two enantiomers of 2-CPE.
To further unveil the source of inverted enantioselectivity induced by these mutations in the dehalogenation of 2-CPE, the binding mode of (S)-2-CPE in the best S-selective mutant (the Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala mutant) was optimized by performing MD simulation (Fig. 3). For this, a 10-ns MD simulation was conducted. The representative binding mode of (S)-2-CPE used for further analysis was obtained from the last 2 ns of MD simulation trajectory where the whole system is fairly stable. In this binding mode, the chloride atom of the substrate points to the halide-binding site of the enzyme and forms contacts with the side chain of Pro175 and Leu178 and the Cα atom of Ala176. Strikingly, the phenyl group of (S)-2-CPE points into the hydrophobic pocket bordered by residues Ala134, Trp139, Ala176, and Tyr187, forming connections with the side chain of Ala134 and Ala176, the hydroxyl group of Tyr187, and the indole group of Trp139. The results indicated that these mutations in the S-selective Pro84Val/Phe86Pro/Thr134Ala/Asn176Ala mutant greatly facilitated the binding of (S)-2-CPE. Such a binding mode is completely different from that for the phenyl group of (R)-2-CPE (binding pocket bordered by residues Pro84, Phe86, Trp139, Leu142, Phe186, and Tyr187). A similar binding mode for the two enantiomers of para-nitro-2-chloro-1-phenyl ethanol as the one we obtained in HheC was also reported with HheA, another microbial halohydrin dehalogenase originating from Arthrobacter AD2 (37). Strikingly, in HheA the aromatic side chain of the S-enantiomer of the substrate was docked in the pocket formed by residues Leu141, Asn144, and Asn178, among which Leu141 and Asn178 are at the equivalent positions of Trp139 and Asn176 in HheC. By manipulating these active-site residues, a variant with an inverted enantioselectivity (from ES of 21.7 to ER of 13) and an S-selective HheA variant toward 2-CPE have been developed (Asn178Ala) (26). The results indicated that the two enzymes might share similar mechanisms for controlling enantioselectivity. In the case of HheA, the catalytic efficiency of the highly S-selective mutant toward (S)-2-CPE is 32-fold lower than that of the S-selective P84V/F86P/T134A/N176A mutant.
FIG 3.

Representation of the structural model of the P84V/F86P/T134A/N176A mutant with (S)-2-CPE in the active site obtained by molecule dynamic (MD) simulation. The catalytic triad Ser132/Tyr145/Arg149 and the mutated residues are shown in stick representation. The hydrogen bonds are indicated as dashed lines.
These MD simulation and docking results emphasized that these amino acid residues that differed in the two substrate-binding pockets are more likely to cause the opposite substrate orientations of the two enantiomers of 2-CPE and that residues at positions 134 and 176 play dual roles in the binding of two enantiomers of 2-CPE. Amino acids with small side chains at these two positions are suitable for the binding of (S)-2-CPE, while those with large side chains were preferred to be adopted for those R-selective mutants. This notion is also in agreement with the observed Km values toward both enantiomers of 2-CPE and could explain the reversed enantioselectivity preference of those mutants. Taken together, the molecular dynamic simulation analysis and substrate docking study provided structural details for understanding the observed enantiopreference of HheC. In conjunction with in vitro kinetic analysis, the results enable us to propose that amino acids lining the two substrate-binding pockets are hot spots for controlling the enantioselectivity of HheC and that the randomization on these targeting sites ultimately led to success.
Conclusions.
Halohydrin dehalogenase HheC of bacterial origin represents a valuable multifunctional biocatalyst in producing optically pure epoxides and a series of β-substituted alcohols, which are valuable building blocks in synthetic organic chemistry and biotechnology. Although HheC has a remarkably high R-enantioselectivity toward aromatic halohydrins, its structural basis and the mechanism on controlling enantioselectivity is limited. In this study, iterative saturation mutagenesis of seven active-site residues of HheC yielded evolution of five enantioselective mutants in the dehalogenation of 2-CPE, two of which displayed an improved R-enantioselectivity while the other three enzymes showed inverted enantioselectivity, with ES values of 50 to 102. Most importantly, the best S-selective mutant exhibited an approximately 100-fold-higher catalytic efficiency toward-(S)-2-CPE than the wild-type HheC; such a catalytic efficiency is at the same level as that of the wild-type HheC toward (R)-2-CPE. The evolved enantiocomplementary HheC enzymes proved to be valuable tools in producing both enantiomers of optically pure styrene oxide. Our results demonstrated that this active-site focused mutagenesis strategy, ISM, could serve as a valuable method to develop such enantiocomplementary enzymes. Molecular modeling in combination with in vitro kinetic assays provided useful mechanistic details governing the enzymatic enantioselectivity of HheC, which will greatly facilitate the ability to rationally predict mutations that improve the enantioselectivity of an enzyme.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (no. 21342005) and a subproject under the National Science and Technology Major Project on Water Pollution Prevention and Control (no. 2012ZX07203-003).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.04153-14.
REFERENCES
- 1.Arnold FH, Volkov AA. 1999. Directed evolution of biocatalysts. Curr Opin Chem Biol 3:54–59. doi: 10.1016/S1367-5931(99)80010-6. [DOI] [PubMed] [Google Scholar]
- 2.Turner NJ. 2009. Directed evolution drives the next generation of biocatalysts. Nat Chem Biol 5:567–573. doi: 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]
- 3.Reetz MT, Wang LW, Bocola M. 2006. Directed evolution of enantioselective enzymes: iterative cycles of casting for probing protein-sequence space. Angew Chem Int Ed Engl 45:1236–1241. doi: 10.1002/anie.200502746. [DOI] [PubMed] [Google Scholar]
- 4.Alvizo O, Nguyen LJ, Savile CK, Bresson JA, Lakhapatri SL, Solis EO, Fox RJ, Broering JM, Benoit MR, Zimmerman SA, Novick SJ, Liang J, Lalonde JJ. 2014. Directed evolution of an ultrastable carbonic anhydrase for highly efficient carbon capture from flue gas. Proc Natl Acad Sci U S A 111:16436–16441. doi: 10.1073/pnas.1411461111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox RJ, Huisman GW. 2008. Enzyme optimization: moving from blind evolution to statistical exploration of sequence-function space. Trends Biotechnol 26:132–138. doi: 10.1016/j.tibtech.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A, Singh S. 2013. Directed evolution: tailoring biocatalysts for industrial applications. Crit Rev Biotechnol 33:365–378. doi: 10.3109/07388551.2012.716810. [DOI] [PubMed] [Google Scholar]
- 7.Morley KL, Kazlauskas RJ. 2005. Improving enzyme properties: when are closer mutations better? Trends Biotechnol 23:231–237. doi: 10.1016/j.tibtech.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Zhang ZG, Lonsdale R, Sanchis J, Reetz MT. 14 November 2014. Extreme synergistic mutational effects in the directed evolution of a Baeyer-Villiger monooxygenase as catalyst for asymmetric sulfoxidation. J Am Chem Soc doi: 10.1021/ja5098034. [DOI] [PubMed] [Google Scholar]
- 9.Wu Q, Soni P, Reetz MT. 2013. Laboratory evolution of enantiocomplementary Candida antarctica lipase B mutants with broad substrate scope. J Am Chem Soc 135:1872–1881. doi: 10.1021/ja310455t. [DOI] [PubMed] [Google Scholar]
- 10.Zheng H, Reetz MT. 2010. Manipulating the stereoselectivity of limonene epoxide hydrolase by directed evolution based on iterative saturation mutagenesis. J Am Chem Soc 132:15744–15751. doi: 10.1021/ja1067542. [DOI] [PubMed] [Google Scholar]
- 11.Prasad S, Bocola M, Reetz MT. 2011. Revisiting the lipase from Pseudomonas aeruginosa: directed evolution of substrate acceptance and enantioselectivity using iterative saturation mutagenesis. Chemphyschem 12:1550–1557. doi: 10.1002/cphc.201100031. [DOI] [PubMed] [Google Scholar]
- 12.Haak RM, Tarabiono C, Janssen DB, Minnaard AJ, de Vries JG, Feringa BL. 2007. Synthesis of enantiopure chloroalcohols by enzymatic kinetic resolution. Org Biomol Chem 5:318–323. doi: 10.1039/b613937j. [DOI] [PubMed] [Google Scholar]
- 13.Lutje Spelberg JH, Tang L, van Gelder M, Kellogg RM, Janssen DB. 2002. Exploration of the biocatalytic potential of a halohydrin dehalogenase using chromogenic substrates. Tetrahedron Asymmetry 13:1083–1089. doi: 10.1016/S0957-4166(02)00222-7. [DOI] [Google Scholar]
- 14.Tang L, Lutje Spelberg JH, Fraaije MW, Janssen DB. 2003. Kinetic mechanism and enantioselectivity of halohydrin dehalogenase from Agrobacterium radiobacter. Biochemistry 42:5378–5386. doi: 10.1021/bi0273361. [DOI] [PubMed] [Google Scholar]
- 15.de Jong RM, Tiesinga JJ, Rozeboom HJ, Kalk KH, Tang L, Janssen DB, Dijkstra BW. 2003. Structure and mechanism of a bacterial haloalcohol dehalogenase: a new variation of the short-chain dehydrogenase/reductase fold without an NAD(P)H binding site. EMBO J 22:4933–4944. doi: 10.1093/emboj/cdg479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schallmey M, Floor RJ, Hauer B, Breuer M, Jekel PA, Wijma HJ, Dijkstra BW, Janssen DB. 2013. Biocatalytic and structural properties of a highly engineered halohydrin dehalogenase. Chembiochem 14:870–881. doi: 10.1002/cbic.201300005. [DOI] [PubMed] [Google Scholar]
- 17.Tang L, Torres Pazmiño DE, Fraaije MW, Janssen DB. 2005. Improved catalytic properties of halohydrin dehalogenase by modification of the halide-binding site. Biochemistry 44:6609–6618. doi: 10.1021/bi047613z. [DOI] [PubMed] [Google Scholar]
- 18.Fox RJ, Davis SC, Mundorff EC, Newman LM, Gavrilovic V, Ma SK, Chung LM, Ching C, Tam S, Muley S, Grate J, Gruber J, Whitman JC, Sheldon RA, Huisman GW. 2007. Improving catalytic function by ProSAR-driven enzyme evolution. Nat Biotechnol 25:338–344. doi: 10.1038/nbt1286. [DOI] [PubMed] [Google Scholar]
- 19.You Z-Y, Liu Z-Q, Zheng Y-G. 2013. Properties and biotechnological applications of halohydrin dehalogenases: current state and future perspectives. Appl Microbiol Biotechnol 97:9–21. doi: 10.1007/s00253-012-4523-0. [DOI] [PubMed] [Google Scholar]
- 20.Molinaro C, Guilbault AA, Kosjek B. 2010. Resolution of 2,2-disubstituted epoxides via biocatalytic azidolysis. Org Lett 12:3772–3775. doi: 10.1021/ol101406k. [DOI] [PubMed] [Google Scholar]
- 21.Mugford PF, Wagner UG, Jiang Y, Faber K, Kazlauskas RJ. 2008. Enantiocomplementary enzymes: classification, molecular basis for their enantiopreference, and prospects for mirror-image biotransformations. Angew Chem Int Edit Engl 47:8782–8793. doi: 10.1002/anie.200705159. [DOI] [PubMed] [Google Scholar]
- 22.Scheller PN, Fademrecht S, Hofelzer S, Pleiss J, Leipold F, Turner NJ, Nestl BM, Hauer B. 2014. Enzyme toolbox: novel enantiocomplementary imine reductases. Chembiochem 15:2201–2204. doi: 10.1002/cbic.201402213. [DOI] [PubMed] [Google Scholar]
- 23.van Leeuwen JG, Wijma HJ, Floor RJ, van der Laan JM, Janssen DB. 2012. Directed evolution strategies for enantiocomplementary haloalkane dehalogenases: from chemical waste to enantiopure building blocks. Chembiochem 3:137–148. [DOI] [PubMed] [Google Scholar]
- 24.Pavelka A, Chovancova E, Damborsky J. 2009. HotSpot Wizard: a web server for identification of hot spots in protein engineering. Nucleic Acids Res 37:W376–W383. doi: 10.1093/nar/gkp410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glaser F, Rosenberg Y, Kessel A, Pupko T, Ben-Tal N. 2005. The ConSurf-HSSP database: the mapping of evolutionary conservation among homologs onto PDB structures. Proteins 58:610–617. doi: 10.1002/prot.20305. [DOI] [PubMed] [Google Scholar]
- 26.Tang L, Zhu X, Zheng H, Jiang R, Elenkov MM. 2012. Key residues for controlling enantioselectivity of halohydrin dehalogenase from Arthrobacter sp. AD2 revealed by structure-guided directed evolution. Appl Environ Microbiol 78:2631–2637. doi: 10.1128/AEM.06586-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Jong RM, Tiesinga JJW, Villa A, Tang L, Janssen DB, Dijkstr BW. 2005. Structural basis for the enantioselectivity of epoxide ring opening reactions catalyzed by haloalcohol dehalogenase HheC. J Am Chem Soc 127:13338–13343. doi: 10.1021/ja0531733. [DOI] [PubMed] [Google Scholar]
- 28.Elenkov MM, Wolfgang Hoeffken H, Tang L, Hauer B, Janssen DB. 2007. Enzyme-catalyzed nucleophilic ring opening of epoxides for the preparation of enantiopure tertiary alcohols. Adv Synth Catal 349:2279–2285. doi: 10.1002/adsc.200700146. [DOI] [Google Scholar]
- 29.Tang L, van Merode AEJ, Lutje Spelberg JH, Fraaije MW, Janssen DB. 2003. Steady-state kinetics and tryptophan fluorescence properties of halohydrin dehalogenase from Agrobacterium radiobacter: roles of W139 and W249 in the active site and halide-induced conformational change. Biochemistry 42:14057–14065. doi: 10.1021/bi034941a. [DOI] [PubMed] [Google Scholar]
- 30.Tang L, Gao H, Zhu X, Wang X, Zhou M, Jiang R. 2012. Construction of “small-intelligent” focused mutagenesis libraries using well-designed combinatorial degenerate primers. Biotechniques 52:149–158. doi: 10.2144/000113820. [DOI] [PubMed] [Google Scholar]
- 31.Tang L, Li Y, Wang X. 2010. A high-throughput colorimetric assay for screening halohydrin dehalogenase saturation mutagenesis libraries. J Biotechnol 147:164–168. doi: 10.1016/j.jbiotec.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 32.van Hylckama Vlieg JET, Tang L, Lutje Spelberg JH, Smilda T, Poelarends GJ, Bosma T, van Merode AEJ, Fraaije MW, Janssen DB. 2001. Halohydrin dehalogenases are structurally and mechanistically related to short-chain dehydrogenases/reductases. J Bacteriol 183:5058–5066. doi: 10.1128/JB.183.17.5058-5066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen CS, Fujimoto Y, Girdaukas G, Sih CJ. 1982. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J Am Chem Soc 104:7294–7299. doi: 10.1021/ja00389a064. [DOI] [Google Scholar]
- 34.Bergmann JG, Sanik J. 1957. Determination of trace amounts of chlorine in naphtha. Anal Chem 29:241–243. doi: 10.1021/ac60122a018. [DOI] [Google Scholar]
- 35.Reetz MT, Wu S. 2008. Greatly reduced amino acid alphabets in directed evolution: making the right choice for saturation mutagenesis at homologous enzyme positions. Chem Commun (Camb) 2008(43):5499–5501. doi: 10.1039/b813388c. [DOI] [PubMed] [Google Scholar]
- 36.Kamal MZ, Ahmad S, Molugu TR, Vijayalakshmi A, Deshmukh MV, Sankaranarayanan R, Rao NM. 2011. In vitro evolved non-aggregating and thermostable lipase: structural and thermodynamic investigation. J Mol Biol 413:726–741. doi: 10.1016/j.jmb.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 37.de Jong RM, Kalk KH, Tang L, Janssen DB, Dijkstra BM. 2006. The X-ray structure of the haloalcohol dehalogenase HheA from Arthrobacter sp. strain AD2: insight into enantioselectivity and halide binding in the haloalcohol dehalogenase family. J Bacteriol 188:4051–4056. doi: 10.1128/JB.01866-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.