

Abstract
In the United States, Shiga toxin (Stx)-producing Escherichia coli (STEC) is the most frequent infectious cause of hemorrhagic colitis. Hemolytic uremic syndrome (HUS) is a serious sequela that may develop after STEC infection that can lead to renal failure and death in up to 10% of cases. STEC can produce one or more types of Stx, Stx1 and/or Stx2, and Stx1 and Stx2 are responsible for HUS-mediated kidney damage. We previously generated two monoclonal antibodies (MAbs) that neutralize the toxicity of Stx1 or Stx2. In this study, we evaluated the protective efficacy of human/mouse chimeric versions of those monoclonal antibodies, named cαStx1 and cαStx2. Mice given an otherwise lethal dose of Stx1 were protected from death when injected with cαStx1 either 1 h before or 1 h after toxin injection. Additionally, streptomycin-treated mice fed the mouse-lethal STEC strain B2F1 that produces the Stx2 variant Stx2d were protected when given a dose of 0.1 mg of cαStx2/kg of body weight administered up to 72 h post-oral bacterial challenge. Since many STEC strains produce both Stx1 and Stx2 and since either toxin may lead to the HUS, we also assessed the protective efficacy of the combined MAbs. We found that both antibodies were required to protect mice from the presence of both Stx1 and Stx2. Pharmacokinetic studies indicated that cαStx1 and cαStx2 had serum half-lives (t1/2) of about 50 and 145 h, respectively. We propose that cαStx1 and cαStx2, both of which have been tested for safety in humans, could be used therapeutically for prevention or treatment early in the development of HUS.
INTRODUCTION
Shiga toxin (Stx)-producing Escherichia coli (STEC) causes both outbreaks and sporadic cases of bloody diarrhea and hemolytic uremic syndrome (HUS) in the United States as well as in other developed countries. The most prevalent serotype of STEC in the United States is O157:H7 (1); however, non-O157 strains represent half or more of all STEC infections (1–4). The number of E. coli O157 infections rose in the United States in 2005 and 2006 to roughly the levels found in 1996 to 1998, with some fluctuations between those time periods, remained stable through 2008 (3), and dropped slightly in 2012 (5). Approximately 25% of those U.S. O157 infections are associated with outbreaks, while the rest are found in sporadic cases (3). A serious sequela of STEC infection, the HUS, occurs in 4% to 15% of STEC infections (1, 6) and is characterized by thrombocytopenia, microangiopathic hemolytic anemia, and renal failure. The incidence of HUS in the United States in 2007 in children less than 5 years of age was 1.75/100,000 (3); this value varies by country from relatively low in Austria (0.51/100,000 [7]), Italy (0.75/100,000 [8]), and Japan (0.88/100,000 [9]) to levels similar to those in the United States in Australia (1.35/100,000 [10]), Germany (1.71/100,000 [7]), the United Kingdom and Ireland (1.54/100,000 [11]), and France (1.87/100,000 [12]) to a high in Argentina (1 to 12/100,000 [13]). There is presently no treatment that specifically addresses an STEC infection or the HUS. In the United States, antibiotics are not a recommended treatment for O157 infection because they do not appear to benefit the patient and may increase the risk of HUS (reviewed in reference 14). Medical intervention for patients with HUS is, therefore, primarily supportive. While intravenous delivery of solutions to expand blood volume appears to help protect children from oligoanuric HUS (15), that treatment does not prevent the HUS from occurring (15). Recently, eculizumab, a monoclonal antibody against the C5 component of complement, was used in some patients during the outbreak in Germany of an Stx2a-positive (Stx2a+) enteroaggregative E. coli strain that resulted in more than 800 HUS cases (16, 17). Although eculizumab is successful at improving the outcome in atypical or familial HUS (18), the efficacy of eculizumab during the outbreak was not clear, as a randomized controlled trial was not done, and patients were given multiple and different interventions concurrently (19–21).
The Shiga toxins (Stxs) are the major virulence factors of STEC that contribute to the development of the HUS. Two types of Stx may be found in E. coli: Stx1 and Stx2 (see review [22]). The Stx/Stx1 group consists of the prototype Stx from Shigella dysenteriae type 1 and Stx1 of E. coli. The Stx2 group from E. coli contains several subtypes that are associated with human disease, the most important of which are Stx2c and Stx2d (23, 24). Because both Stx1 and Stx2 have subtypes, the prototype toxins from those groups are now called Stx1a and Stx2a, respectively (25), but we maintain the designations of Stx1 and Stx2 in this study when we refer to the groups as a whole and use the specific name when we mean the prototype in particular. The two toxin groups have the same structure and enzymatic activity; however, the two groups are antigenically distinct. Epidemiological evidence suggests that the STEC strains that make Stx2a alone are approximately 15 or 6 times more likely to lead to the HUS than strains that produce Stx1a alone or strains that produce both Stx1a and Stx2a (24, 26). However, clinical data demonstrate that STEC strains that make Stx1a or Stx2a alone or in combination have the capacity to lead to the HUS (10, 24, 27) and that Stx of Shigella dysenteriae type 1 is linked to the HUS as well (28, 29).
Murine monoclonal antibodies that neutralize the cytotoxicity and animal lethality of each of the toxins were generated in the 1980s in our laboratory (30, 31). Although murine monoclonal antibodies or polyclonal antisera generated in animals are used in humans, chimeric human/mouse or fully humanized antibodies are preferred for use in people due to the potential for an antibody response to the constant region of the antibody (32). The murine monoclonal antibodies specific for Stx1 and Stx2 were made into human/mouse chimeras through genetic techniques (33). Preliminary testing of the human/mouse chimeric anti-Stx1 and anti-Stx2 antibodies, designated cαStx1 and cαStx2, showed that they neutralized the cytotoxicity of Stx1a and of both Stx2a and Stx2d, respectively, for Vero cells and were protective in mice (33). Furthermore, the antibodies were shown to be safe in humans (34, 35). In this study, we refined the doses of each antibody individually required for protection in mice, examined the protective efficacy of the combination of the antibodies against both toxins, and determined the pharmacokinetics of the antibodies in mice.
MATERIALS AND METHODS
Antibodies, toxins, and STEC strain B2F1.
Chimeric human/murine anti-Stx1 and anti-Stx2 antibodies were generated by genetic methods from the DNA of the hybridoma cell lines that produce murine antibodies 13C4 (anti-Stx1, cαStx1) and 11E10 (anti-Stx2, cαStx2) as described previously (33) and expressed in Chinese hamster ovary (CHO) cells. The predicted amino acid sequences of the chimeric antibodies were confirmed by N-terminal amino acid sequencing. The chimeric antibodies were produced and purified by Goodwin Biotechnology Inc. (Plantation, FL). Purified Stx1a and Stx2a were produced from culture supernatants of DH5α transformed with pLPSH3 (Stx1a) or pJES120 (Stx2a), and the cytotoxicity of the toxin preparations was determined on Vero cells as described previously (36, 37). STEC strain B2F1 (O91:H21; produces Stx2d [ATCC 51435]) was originally provided to us by M. A. Karmali. Stx2d is differentiated from other subtypes not just in sequence but also by toxicity. The Stx2d subtype becomes significantly more toxic after treatment with intestinal mucus, and strains that produce Stx2d are associated with HUS in humans and are more virulent in streptomycin (Str)-treated mice than strains that produce other subtypes of Stx2 (38, 39).
Mouse models.
All animal studies were approved by the Institutional Animal Care and Use Committee of the Uniformed Services University of the Health Sciences and were conducted in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals (40).
(i) Mouse model to measure the efficacy of cαStx1.
Adult male or female CD-1 mice weighing approximately 20 g were observed for 5 to 7 days after arrival in the animal facility. At the end of the quarantine period, the mice were weighed and assigned by weight classification to groups of 5 mice each. The mice were injected intraperitoneally with 2 50% lethal doses (LD50s) of Stx1a (250 ng). At either 1 h before or 1 h after toxin injection, the mice were given various doses (indicated in Table 1) of cαStx1 in buffer via the tail vein. The mice were observed for 2 weeks for mortality.
TABLE 1.
Protective efficacy of cαStx1 in CD-1 mice injected with 2 LD50s of Stx1a
| Dose of cαStx1 (mg/kg)a | Timing (h) of cαStx1 injection relative to Stx1a injectionb | No. of surviving mice/total no. of mice |
|
|---|---|---|---|
| Males | Females | ||
| 0 | −1 | 0/5 | 0/5 |
| 0.005 | −1 | 0/5 | 0/5 |
| 0.02 | −1 | 0/5 | 0/5 |
| 0.05 | −1 | 0/5 | 5/5 |
| 0.5 | −1 | 5/5 | 5/5 |
| 0 | +1 | 0/5 | 0/5 |
| 0.05 | +1 | 1/5 | 1/5 |
| 0.2 | +1 | 2/5 | 5/5 |
| 0.5 | +1 | 3/5 | 5/5 |
| 0.75 | +1 | 5/5 | 4/5 |
Antibody was administered intravenously.
Stx1a was given intraperitoneally.
(ii) Mouse models to measure the efficacy of cαStx2.
We used two mouse models to evaluate the protective efficacy of cαStx2. In the first model, male or female CD-1 mice were injected with approximately 2 LD50s of Stx2 intraperitoneally and 1 h later given cαStx2 at 0, 0.5, 0.75, 1.0, 3.0, or 5.0 mg/kg of body weight via the tail vein. For the second assessment of cαStx2, we utilized the orally infected Str-treated mouse model of STEC infection (38, 41). Briefly, 20-g CD-1 male mice were given drinking water with 5 g/liter Str after a 5-to-7-day quarantine period and fasted overnight. The mice were then fed a 25-μl droplet with approximately 106 CFU B2F1 Strr in 20% sucrose with a pipette. (To prepare the B2F1 Strr inoculum, the bacteria were grown in Luria Bertani broth overnight with shaking at 37°C. After overnight growth, the bacterial culture was collected by centrifugation, and the pellet was resuspended in 20% sucrose. Appropriate dilutions in sucrose were made to achieve an inoculum of 106 CFU/25 μl.) The mice were injected with a single dose of cαStx2 via the tail vein or intramuscularly into the thigh at various times relative to infection with B2F1. Preliminary protection data were reported for intravenous injection of cαStx2 in this mouse model (33). However, the data described for this model involved several changes to the original protocol, including use of a single dose of antibody, a higher inoculation level of B2F1 Strr, a determination of mouse weight on the day of antibody administration, and infection by a droplet administered from a pipette.
(iii) Mouse models to test the neutralization capacity of cαStx1 and cαStx2 against Stx1a and Stx2a in mice.
Two mouse models were used to test the neutralization capacity of cαStx1 and cαStx2 against Stx1a and Stx2a. In one model, approximately 2 LD50s of either toxin or both toxins was injected intraperitoneally into male CD-1 mice. The antibodies (5 mg/kg) were given alone or in combination intravenously 1 h before toxin was injected. In the other model, the antibodies were combined with 2 LD50s of each of the toxins, incubated together for 1 h at 37°C, and then injected intraperitoneally into male CD-1 mice. Specifically, cαStx1 (200 μg) and/or cαStx2 (400 μg) was combined with Stx1a (250 ng) and/or Stx2a (2.5 ng) in the presence of bovine serum albumin (BSA) (200 μg). The BSA was added to prevent loss of cytotoxicity of the toxins under conditions of incubation at 37°C. As a control, each toxin alone or in combination or the antibodies in combination were treated the same way and then injected into the mice. Mice were observed for morbidity and mortality for 2 weeks and weighed on day 1 and at death or at the study endpoint, day 14.
Pharmacokinetic analysis of cαStx1 or cαStx2 in mice. (i) cαStx1 alone.
Adult male or female CD-1 mice weighing approximately 20 g were observed for 5 to 7 days prior to the start of the experiment. At the start of the study, each mouse was weighed and the mice were placed into groups of 5 mice. The mice were intravenously injected with 1 mg/kg cαStx1. Two blood samples were collected at different times from groups of 5 male or 5 female mice each. The timings of blood collection for the groups were as follows: for group 1, 5 min and 24 h; for group 2, 15 min and 48 h; for group 3, 30 min and 72 h; for group 4, 1 and 96 h; for group 5, 2 and 120 h; for group 6, 4 h and 1 week; and for group 8, 8 h and 2 weeks. The first sample for each pair of sampling times was taken from the tail vein, and the second sample was collected at terminal exsanguination. Serum was prepared from each blood sample and the cαStx1 concentration determined by enzyme-linked immunosorbent assay (ELISA) as described below.
Noncompartmental methods were used to calculate the pharmacokinetic parameters. The parameters were determined for the male and female mice separately using the mean concentrations at each sampling time. Serum concentrations reported to be below the limit of detection (605 ng/ml) were assumed to be zero for the calculations. For most of the sampling times, the interanimal variation in concentration was relatively modest; however, there were some sampling times with values that appeared to be outlier values. The suspect values were tested by calculating the difference between the suspect value and the mean of the other four values. If this difference was greater than 3 times the standard deviation of the other four values, the suspect value was considered to be an outlier and excluded from the calculations. For the male mice, one value at 2 h was determined to be an outlier value. At the sampling times of 2, 4, 72, 120, and 168 h, one value at each time was determined to be an outlier value for the female mice. For male mice, the maximum concentration of drug in serum (Cmax) was determined as the time to maximum concentration of drug in serum (Tmax) at the time of the injection, since all concentrations for samples taken after the 5-min intervals were less than the values at 5 min.
(ii) cαStx2 in infected and uninfected mice.
For the two-pronged pharmacokinetic study of cαStx2, two groups of 40 CD-1 mice were injected with 15 mg/kg cαStx2. The mice in one of the groups of 40 mice were infected with B2F1 as described above just prior to injection with cαStx2. Blood was collected from one set of 5 mice from each group (infected or uninfected) at 1, 24, 48, or 72 h or at 1, 2, 3, or 4 weeks postinjection with cαStx2. Blood was collected as a terminal bleed rather than from the same animals over time to avoid the possibility that multiple bleeds would enhance any disease processes in the infected mice. The blood was processed to serum, and the levels of cαStx2 were determined by ELISA. The serum concentrations of cαStx2 were averaged at each time point to create a pharmacokinetic curve for infected animals and a separate pharmacokinetic curve for noninfected animals. Noncompartmental pharmacokinetic analysis was performed on these curves using WinNonlin version 1.1. (The reason for the larger dose of cαStx2 in this pharmacokinetic study than in the pharmacokinetic study for cαStx1 is that, before the above-described changes in the protection protocol for cαStx2, a higher dose of cαStx2 was used in the protection studies and the pharmacokinetic study was performed with the same large dose of cαStx2.)
ELISA to measure the level of cαStx1 or cαStx2 in mouse serum. (i) ELISA to detect cαStx1 in mouse serum.
Microtiter plates were coated with 1 μg/ml purified Stx1a. The wells were blocked with 0.01 M Tris–phosphate-buffered saline (TPBS) with 1% gelatin, washed, and overlaid with the serum samples. Serum samples were tested in triplicate. A sample of cαStx1 was used as a positive control. After 1 h of incubation, the wells were washed and then peroxidase-conjugated goat anti-human IgG (Bio-Rad, Hercules, CA)–TPBS–1% gelatin was added at a 1:1,000 dilution. After 30 min, the wells were washed and 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) was added. After 25 min, stop solution (1% sodium dodecyl sulfate [SDS]) was added and the plates were read at an optical density of 405 nm (OD405).
(ii) ELISA to detect cαStx2 in mouse serum.
The ELISA to detect cαStx2 in mouse serum was done essentially as described for cαStx1 except that the microtiter wells were coated with 2 μg/ml purified Stx2a and cαStx2 was used as the positive control.
Statistical analyses.
Survival curves and proportions were compared using the log-rank (Mantel-Cox) test and Fisher's exact test, respectively, through the application of GraphPad Prism version 6.03 software.
RESULTS
Protective efficacy of cαStx1 in mice.
We do not have a murine model in which infection with an Stx1a-producing STEC strain causes morbidity or mortality. Therefore, we evaluated the protective efficacy of cαStx1 in female and male CD-1 mice injected with two LD50s of Stx1a (Table 1). We found that cαStx1 given either 1 h before or after toxin injection protected mice, although higher doses of antibody were required to protect when the doses were given subsequent to toxin administration. Specifically, as little as 0.05 mg cαStx1/kg of body weight given 1 h before Stx1a administration protected the female mice from death, whereas 0.5 mg of the antibody/kg was needed to protect male mice in the same time frame. When we administered the antibody 1 h after toxin injection, 0.2 mg or 0.75 mg of the antibody/kg was required to protect the female or male mice, respectively. Although there appeared to be a heightened protective efficacy of cαStx1 in the female mice, a much larger study would be required to determine if there was a statistically significant difference between the dose necessary to protect male mice and that required to protect female mice.
The pharmacokinetic parameters of cαStx1 in mice.
To determine the serum half-life (t1/2) of cαStx1 in mice, a pharmacokinetic study was done in male and female CD-1 mice. The elimination half-life values were 52 h and 50 h for male and female mice, respectively (Table 2). The values for clearance were 1.9 and 1.75 ml/(h · kg) for male and female mice, respectively, findings that indicate that most of the cαStx1 was retained within the blood volume but that there was some distribution to tissues.
TABLE 2.
Pharmacokinetic parameters for cαStx1 after intravenous administration to mice
| Parametera | Value |
|
|---|---|---|
| Males | Females | |
| Cmax (μg/ml) | 22.68 | 19.39 |
| Tmax (h) | 0b | 1 |
| AUC0–24 (μg · h/ml) | 206.4 | 224.4 |
| AUC0–336 (μg · h/ml) | 532.1 | 578.0 |
| AUC0–∞ (μg · h/ml) | 527.1 | 572.8 |
| ke (h−1) | 0.0133 | 0.014 |
| t1/2 (h) | 51.9 | 49.6 |
| Cl (ml/h · kg) | 1.91 | 1.75 |
| VZ (ml/kg) | 142.2 | 124.8 |
The definitions of the parameter terms are as follows: Cmax, maximum concentration of drug in serum; Tmax, time to maximum concentration of drug in serum; AUC0–24, area under the plasma concentration-time curve from 0 to 24 h; AUC0–336, area under the plasma concentration-time curve from 0 to 336 h; AUC0–∞, area under the plasma concentration-time curve from 0 to infinity; ke, elimination rate constant; t1/2, plasma half-life; Cl, drug clearance rate; VZ, volume of distribution based on terminal elimination phase.
The first sampling time was 5 min. Tmax was extrapolated to the zero time point because all values after 5 min were below that of the 5-min sample.
The protective efficacy of cαStx2 in Stx2a-injected or B2F1-infected mice. (i) Protection by cαStx2 in Stx2a-injected mice.
We evaluated the capacity of cαStx2 to protect male and female mice that were injected with Stx2a (Fig. 1A and B, respectively). We found that compared to the buffer-treated animals, cαStx2 was protective in the male mice at all doses tested. For the female mice, a dose of 3 mg/kg cαStx2 was necessary for efficacy above that of the control. However, as was the case with cαStx1, a much larger study would be required to determine if there is a difference between the dose efficacy for the antibodies in male mice and the dose efficacy in female mice.
FIG 1.
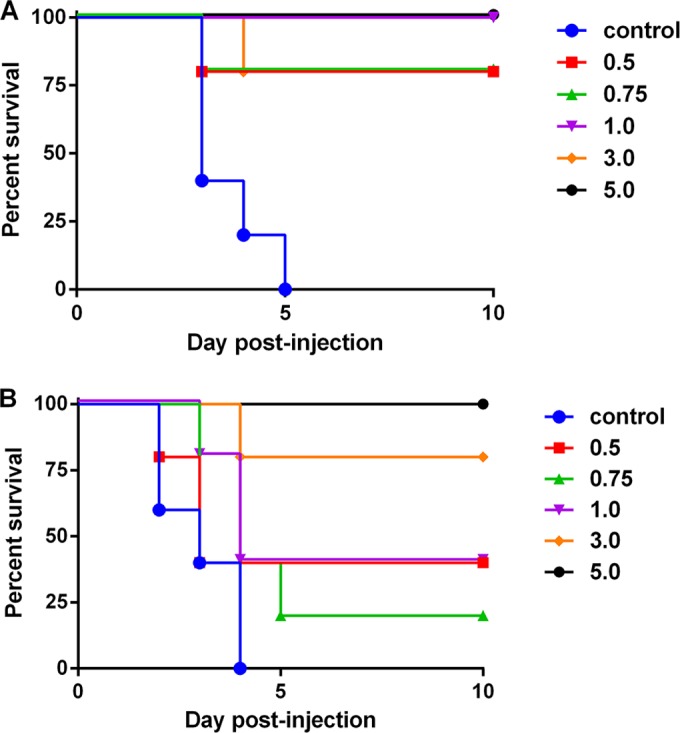
Protection by cαStx2 in mice injected with Stx2a. Male (A) or female (B) mice were given 3 ng Stx2a intraperitoneally, and then, 1 h later, buffer or cαStx2 was administered intravenously at the doses listed in mg/kg. n = 5 mice/group. For panel A, P ≤ 0.016 for the survival curve from all groups compared to the survival curve for the control. For panel B, P ≤ 0.008 for the 3 and 5 mg/kg survival curves compared to the survival curve for the control.
(ii) The protective efficacy of cαStx2 in B2F1-infected mice.
We tested the protective efficacy of cαStx2 in Str-treated mice infected with STEC strain B2F1. We gave mice infected with an otherwise lethal dose (106 CFU) of STEC strain B2F1 (Stx2d producer) various doses of cαStx2 at 24, 48, or 72 h postinfection (Fig. 2). We found that 0.1 mg/kg of cαStx2 protected the mice when the dose was administered 24 or 48 h postinfection, whereas doses of 0.01 mg/kg or below were not protective. At the 72-h time point, we found that some mice given saline solution alone survived. This finding that may indicate a positive effect of fluid at that time point in some animals. However, we did not observe this slight protective effect of saline solution at the 24-h or 48-h time point. At 72-h postinfection, protection was observed at 0.1 and 1 mg/kg cαStx2 compared to the 0.001 mg/kg antibody dose.
FIG 2.
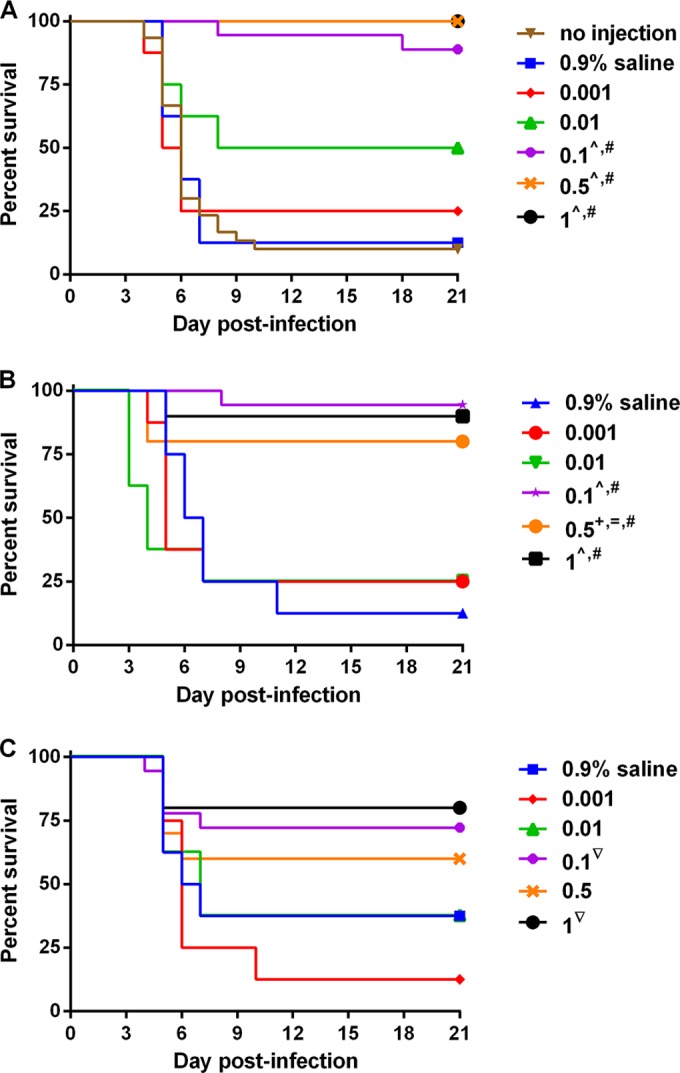
Protective efficacy of cαStx2 in mice infected with B2F1. Str-treated mice were infected with B2F1 and then given no treatment, 0.9% saline solution, or cαStx2 administered intravenously at the listed doses (mg/kg) 24 (A), 48 (B), or 72 (C) h later. The median time to death for untreated mice (n = 30) was 6 days. n = 8 for the mice in the 0.9% saline solution and 0.001 and 0.01 mg/kg cαStx2 groups, 18 for the mice in the 0.1 mg/kg cαStx2 groups, and 10 for the mice in the 0.5 and 1.0 mg/kg cαStx2 groups. The statistics that follow represent comparisons at the same time point. The ^ symbol indicates P ≤ 0.005 compared to the 0.9% saline solution or 0.001 mg/kg cαStx2. The # symbol indicates P ≤ 0.01 compared to 0.01 mg/kg cαStx2. The + symbol indicates P = 0.015 compared to 0.9% saline solution. The = symbol indicates P = 0.048 compared to 0.001 mg/kg. The ▽ symbol indicates P = 0.01 compared to 0.001 mg/kg cαStx2.
We next asked if cαStx2 administered intramuscularly could protect Str-treated, B2F1-infected mice. We found that intramuscularly delivered cαStx2 given either 24 h before or 24 h after infection protected infected mice (Fig. 3). There was no dose-specific difference in the protective responses except that, with cαStx2 given 24-h postinfection, the 0.01 mg/kg dose was not protective.
FIG 3.
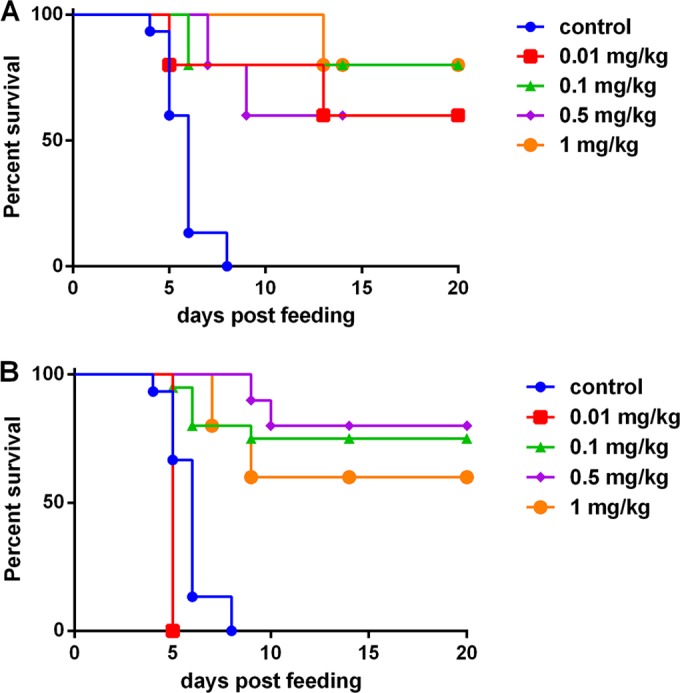
Protective efficacy of cαStx2 given intramuscularly to B2F1-infected mice. Str-treated, B2F1-infected mice were given cαStx2 intramuscularly either 24 h before (A) or 24 h after (B) infection. The same control animals were used for the experiments whose results are shown in panels A and B; n = 15 for the control group and the 0.1 mg/kg group administered cαStx2 before infection; n = 10 for the 0.5 and 1.0 mg/kg doses given before or after infection; n = 5 for the 0.01 mg/kg dose given before or after infection. The antibody provided protection better than that seen with the control (no injection) for all doses except 0.001 mg/kg cαStx2 given after infection. P < 0.001 for the 0.1, 0.5, and 1.0 mg/kg doses given either before or after infection; P = 0.022 for the 0.1 mg/kg dose given before infection.
The pharmacokinetic parameters of cαStx2 in mice.
We conducted a pharmacokinetic study to determine the serum half-life of cαStx2 in CD-1 mice infected or not infected with B2F1. After a 15-mg/kg intravenous dose of cαStx2 antibody, mean serum concentrations of the antibody and the resultant pharmacokinetic parameters were found to be similar in healthy and B2F1-infected mice (Table 3). Clearance was relatively slow and, consequently, the half-life was long, as would be expected for a monoclonal antibody. Although the calculated Cmax appeared to be slightly lower in noninfected mice than in B2F1-infected mice, the difference is most likely not significant because of variability between mice in the same dose group. Also, the serum half-life (t1/2) and the volume of distribution based on the terminal elimination phase (VZ) appeared to be greater in the noninfected mice than in the B2F1-infected mice, but these small differences are also likely to have been due to interanimal variability within each group. Because serum was collected from each animal at only one time point, the data do not allow a statistical assessment of these possible differences. The results suggest that the pharmacokinetic behavior of cαStx2 antibody is not altered appreciably by infection with STEC strain B2F1.
TABLE 3.
Pharmacokinetic parameters for cαStx2 antibody in uninfected and B2F1-infected, Str-treated mice
| Parametera | Value |
|
|---|---|---|
| Uninfected mice | B2F1-infected mice | |
| Cmax (μg/ml)b | 119.5 | 137.0 |
| AUC0–t (μg · h/ml) | 6,637.3 | 6,774.0 |
| AUC0–∞ (μg · h/ml) | 6,846.2 | 7,046.6 |
| t1/2 (h) | 145.7 | 109.1 |
| Cl (ml/h · kg) | 2.19 | 2.13 |
| Vz (ml/kg) | 460.7 | 335.0 |
Definitions are as in the footnotes of Table 2. AUC0–t, area under the plasma concentration-time curve from time zero to the last measurable concentration.
Cmax was calculated by extrapolation of the serum antibody-concentration time curve back to time zero, the time of antibody administration.
Protection of mice by cαStx1 and cαStx2 from challenge with Stx1a and Stx2a.
To determine whether mice can be protected from both Stx1a and Stx2a by the combination of cαStx1 and cαStx2, we injected CD-1 mice with approximately 2 LD50s of both toxins and gave them one or both antibodies 1 h before toxin injection. We found that both cαStx1 and cαStx2 were required to protect mice from injection with both Stx1a and Stx2a, with 70% survival of the mice when the antibodies were given 1 h before intoxication (Table 4). No heterologous protection was observed. This observation was additionally substantiated in a model in which the toxin(s) and antibody(ies) were premixed and incubated for 1 h at 37°C in vitro. The mixtures of toxin(s) and antibody(ies) were then injected into mice intraperitoneally. Mice were protected from the combination of Stx1a and Stx2a only when both cαStx1 and cαStx2 were present to neutralize the toxins prior to administration to the mice (Table 5). Mice were weighed at the start of the study and upon death or at the study endpoint. We found that the mice that died had lost an average of about 4 g whereas the surviving mice had gained about 6 g.
TABLE 4.
Protective efficacy of cαStx1 and cαStx2 in mice injected with Stx1a and Stx2a
| No. of Stx1 LD50s | No. of Stx2 LD50s | caStx1 dose (mg/kg) | caStx2 dose (mg/kg) | Timing (h) of buffer or antibody dose relative to toxin dose | No. of surviving mice/total no. of mice |
|---|---|---|---|---|---|
| 2 | 2 | 0 | 0 | −1 | 0/10 |
| 2 | 2 | 5 | 0 | −1 | 0/10 |
| 2 | 2 | 0 | 5 | −1 | 0/10 |
| 2 | 2 | 5 | 5 | −1 | 7/10 |
TABLE 5.
Neutralization capacity of cαStx1 and cαStx2 for Stx1a and Stx2a in vivo
| Material(s) injecteda | No. of surviving mice/total no. of mice |
|---|---|
| Stx1a | 1/10 |
| Stx2a | 0/10 |
| Stx1a/Stx2a/cαStx1 | 0/10 |
| Stx1a/Stx2a/cαStx2 | 1/10 |
| Stx1a/Stx2a/cαStx1/cαStx2 | 9/10 |
The toxin(s) or toxin/antibody combinations were mixed in vitro and injected as a mixture intraperitoneally. For Stx1a, 250 ng = 2 LD50s; for Stx2a, 2.5 ng = 2 LD50s; for cαStx1, 200 μg = 10 mg/kg; for cαStx2, 400 μg = 20 mg/kg.
DISCUSSION
The chimeric anti-Stx1 and anti-Stx2 antibodies showed protective efficacy in mice. Of particular note, both antibodies protected mice even after toxin exposure: cαStx1 efficacy was noted 1 h post-Stx1 toxin injection, and efficacy was seen with cαStx2 administered up to 72 h postinfection with B2F1. We found protective doses for cαStx2 given intramuscularly to be similar in efficacy to doses administered intravenously 24 h postinfection. When cαStx1 and cαStx2 were used in combination, the antibodies neutralized the effect of the two toxins coadministered in the mouse model.
Other groups have developed humanized anti-Stx antibodies. For example, Mukherjee et al. produced humanized anti-Stx1 B subunit monoclonal antibodies that protected mice at a dose of 2.5 mg/kg given 18 h prior to an otherwise lethal dose of Stx1 but did not report on lower doses of antibody or administration postintoxication (42). Our observation of protection by cαStx1 even at 1 h post-toxin injection is also in contrast to the lack of protection seen at 60 min post-toxin injection with polyclonal anti-Stx1 egg yolk antibody (43). Two other humanized Stx2 antibodies developed for use as therapeutics, TMA-15 and 5C12, are also protective in the B2F1 infection model (44–46). However, the dose of TMA-15 or 5C12 required to protect 80% or more of the animals 24 h post-B2F1 infection was 0.5 or 2.1 mg/kg, respectively (45, 46), whereas we observed similar levels of protection at 0.1 mg/kg, a finding that suggests that the potency of cαStx2 was ≥5-fold greater than that of TMA-15 and 5C12 in mice.
The chimeric antibodies described in this study showed good stability in vivo, with clearance rates that are typical for human/mouse hybrid molecules in mice. Furthermore, both cαStx1 and cαStx2, given either alone or in combination at doses up to 3 mg/kg of each antibody or 10 mg/kg for cαStx2 alone, were found to be safe in phase 1 clinical trials (34, 35). The potential doses of 3 and 10 mg/kg for cαStx1 and cαStx2, respectively, are well below those used for palivizumab (Synagis), a monoclonal antibody approved for use at multiple doses of 15 mg/kg in infants at risk for respiratory syncytial virus (47). TMA-15 (renamed urtoxazumab), which targets only Stx2, has also completed a phase 1 safety trial (48). The results of this study suggest that the two antibodies are able to neutralize their respective targets when both Stx1 and Stx2 are present.
We found relatively high VZ values for both cαStx1 and cαStx2 in mice, with the VZ higher for cαStx2 than for cαStx1. The high VZ numbers suggest that the antibodies distribute primarily to the tissues in mice. The (3-fold) higher VZ for cαStx2 than for cαStx1 may have been due, at least in part, to the higher dose of cαStx2 than cαStx1 used in the pK studies (15 mg/kg compared to 1 mg/kg).
Other approaches to the STEC problem include the attempt to eliminate STEC strains from the food supply or source animal. However, STEC organisms are hardy and estimates of the infectious dose suggest that fewer than 100 organisms are required for infection (49, 50). Two vaccines developed to remove E. coli O157 from the cattle reservoir show only partial reduction in colonization and shedding by E. coli O157 (see the reviews in references 51 and 52). In addition, other reservoirs for STEC exist, including deer, pigs, birds, rabbits, and possibly cats or dogs, so the problem of exposure to STEC in humans will not be eliminated even if E. coli O157 is eradicated in cattle. Finally, since non-O157 serogroups account for up to half of all STEC infections, elimination of E. coli O157 alone from the food supply would not be sufficient to prevent hemorrhagic colitis and the HUS in people.
ACKNOWLEDGMENTS
We thank Cara Olsen for facilitation of statistical analyses and Edda Twiddy for toxin purification. From Thallion Pharmaceuticals, we appreciate the review of the experimental design by Marc Rivière, Mariam Mehran, and Ruth Poole.
This work was supported by National Institutes of Health grant R37 AI020148 to A.D.O.
The opinions or assertions presented here are our private ones and are not to be construed as official or reflecting the views of the Department of Defense, the Uniformed Services University of the Health Sciences, or the National Institutes of Health.
REFERENCES
- 1.Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, Griffin PM, Tauxe RV. 1999. Food-related illness and death in the United States. Emerg Infect Dis 5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hedican EB, Medus C, Besser JM, Juni BA, Koziol B, Taylor C, Smith KE. 2009. Characteristics of O157 versus non-O157 Shiga toxin-producing Escherichia coli infections in Minnesota, 2000–2006. Clin Infect Dis 49:358–364. doi: 10.1086/600302. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. 2009. Preliminary FoodNet Data on the incidence of infection with pathogens transmitted commonly through food—10 states, 2008. MMWR Morb Mortal Wkly Rep 58:333–337. [PubMed] [Google Scholar]
- 4.Gould LH, Mody RK, Ong KL, Clogher P, Cronquist AB, Garman KN, Lathrop S, Medus C, Spina NL, Webb TH, White PL, Wymore K, Gierke RE, Mahon BE, Griffin PM; Emerging Infections Program Foodnet Working Group. 2013. Increased recognition of non-O157 Shiga toxin-producing Escherichia coli infections in the United States during 2000–2010: epidemiologic features and comparison with E. coli O157 infections. Foodborne Pathog Dis 10:453–460. doi: 10.1089/fpd.2012.1401. [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2013. Incidence and trends of infection with pathogens transmitted commonly through food—foodborne diseases active surveillance network, 10 U.S. sites, 1996–2012. MMWR Morb Mortal Wkly Rep 62:283–287. [PMC free article] [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention. 2006. Ongoing multistate outbreak of Escherichia coli serotype O157:H7 infections associated with consumption of fresh spinach—United States, September 2006. MMWR Morb Mortal Wkly Rep 55:1045–1046. [PubMed] [Google Scholar]
- 7.Gerber A, Karch H, Allerberger F, Verweyen HM, Zimmerhackl LB. 2002. Clinical course and the role of Shiga toxin-producing Escherichia coli infection in the hemolytic-uremic syndrome in pediatric patients, 1997–2000, in Germany and Austria: a prospective study. J Infect Dis 186:493–500. doi: 10.1086/341940. [DOI] [PubMed] [Google Scholar]
- 8.Tozzi AE, Caprioli A, Minelli F, Gianviti A, De Petris L, Edefonti A, Montini G, Ferretti A, De Palo T, Gaido M, Rizzoni G. 2003. Shiga toxin-producing Escherichia coli infections associated with hemolytic uremic syndrome, Italy, 1988–2000. Emerg Infect Dis 9:106–108. doi: 10.3201/eid0901.020266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawasaki Y, Suyama K, Maeda R, Yugeta E, Takano K, Suzuki S, Sakuma H, Nemoto K, Sato T, Nagasawa K, Hosoya M. 2014. Incidence and index of severity of hemolytic uremic syndrome in a 26 year period in Fukushima Prefecture, Japan. Pediatr Int 56:77–82. doi: 10.1111/ped.12193. [DOI] [PubMed] [Google Scholar]
- 10.Elliott EJ, Robins-Browne RM, O'Loughlin EV, Bennett-Wood V, Bourke J, Henning P, Hogg GG, Knight J, Powell H, Redmond D. 2001. Nationwide study of haemolytic uraemic syndrome: clinical, microbiological, and epidemiological features. Arch Dis Child 85:125–131. doi: 10.1136/adc.85.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynn RM, O'Brien SJ, Taylor CM, Adak GK, Chart H, Cheasty T, Coia JE, Gillespie IA, Locking ME, Reilly WJ, Smith HR, Waters A, Willshaw GA. 2005. Childhood hemolytic uremic syndrome, United Kingdom and Ireland. Emerg Infect Dis 11:590–596. doi: 10.3201/eid1104.040833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Espié E, Grimont F, Mariani-Kurkdjian P, Bouvet P, Haeghebaert S, Filliol I, Loirat C, Decludt B, Minh NN, Vaillant V, de Valk H. 2008. Surveillance of hemolytic uremic syndrome in children less than 15 years of age, a system to monitor O157 and non-O157 Shiga toxin-producing Escherichia coli infections in France, 1996–2006. Pediatr Infect Dis J 27:595–601. doi: 10.1097/INF.0b013e31816a062f. [DOI] [PubMed] [Google Scholar]
- 13.Rivas M, Sosa-Estani S, Rangel J, Caletti MG, Valles P, Roldan CD, Balbi L, Marsano de Mollar MC, Amoedo D, Miliwebsky E, Chinen I, Hoekstra RM, Mead P, Griffin PM. 2008. Risk factors for sporadic Shiga toxin-producing Escherichia coli infections in children, Argentina. Emerg Infect Dis 14:763–771. doi: 10.3201/eid1405.071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahn CK, Holt NJ, Tarr PI. 2009. Shiga-toxin producing Escherichia coli and the hemolytic uremic syndrome: what have we learned in the past 25 years? Adv Exp Med Biol 634:1–17. doi: 10.1007/978-0-387-79838-7_1. [DOI] [PubMed] [Google Scholar]
- 15.Ake JA, Jelacic S, Ciol MA, Watkins SL, Murray KF, Christie DL, Klein EJ, Tarr PI. 2005. Relative nephroprotection during Escherichia coli O157:H7 infections: association with intravenous volume expansion. Pediatrics 115:e673–e680. doi: 10.1542/peds.2004-2236. [DOI] [PubMed] [Google Scholar]
- 16.Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, Bernard H, Fruth A, Prager R, Spode A, Wadl M, Zoufaly A, Jordan S, Kemper MJ, Follin P, Müller L, King LA, Rosner B, Buchholz U, Stark K, Krause G, HUS Investigation Team. 2011. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med 365:1771–1780. doi: 10.1056/NEJMoa1106483. [DOI] [PubMed] [Google Scholar]
- 17.Karch H, Denamur E, Dobrindt U, Finlay BB, Hengge R, Johannes L, Ron EZ, Tonjum T, Sansonetti PJ, Vicente M. 2012. The enemy within us: lessons from the 2011 European Escherichia coli O104:H4 outbreak. EMBO Mol Med 4:841–848. doi: 10.1002/emmm.201201662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keating GM. 2013. Eculizumab: a review of its use in atypical haemolytic uraemic syndrome. Drugs 73:2053–2066. doi: 10.1007/s40265-013-0147-7. [DOI] [PubMed] [Google Scholar]
- 19.Porubsky S, Federico G, Muthing J, Jennemann R, Gretz N, Buttner S, Obermuller N, Jung O, Hauser IA, Grone E, Geiger H, Grone HJ, Betz C. 2014. Direct acute tubular damage contributes to Shigatoxin-mediated kidney failure. J Pathol 234:120–133. doi: 10.1002/path.4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menne J, Nitschke M, Stingele R, Abu-Tair M, Beneke J, Bramstedt J, Bremer JP, Brunkhorst R, Busch V, Dengler R, Deuschl G, Fellermann K, Fickenscher H, Gerigk C, Goettsche A, Greeve J, Hafer C, Hagenmuller F, Haller H, Herget-Rosenthal S, Hertenstein B, Hofmann C, Lang M, Kielstein JT, Klostermeier UC, Knobloch J, Kuehbacher M, Kunzendorf U, Lehnert H, Manns MP, Menne TF, Meyer TN, Michael C, Munte T, Neumann-Grutzeck C, Nuernberger J, Pavenstaedt H, Ramazan L, Renders L, Repenthin J, Ries W, Rohr A, Rump LC, Samuelsson O, Sayk F, Schmidt BM, Schnatter S, Schocklmann H, Schreiber S, von Seydewitz CU, et al. 2012. Validation of treatment strategies for enterohaemorrhagic Escherichia coli O104:H4 induced haemolytic uraemic syndrome: case-control study. BMJ 345:e4565. doi: 10.1136/bmj.e4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kielstein JT, Beutel G, Fleig S, Steinhoff J, Meyer TN, Hafer C, Kuhlmann U, Bramstedt J, Panzer U, Vischedyk M, Busch V, Ries W, Mitzner S, Mees S, Stracke S, Nurnberger J, Gerke P, Wiesner M, Sucke B, Abu-Tair M, Kribben A, Klause N, Schindler R, Merkel F, Schnatter S, Dorresteijn EM, Samuelsson O, Brunkhorst R. 2012. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC-HUS registry. Nephrol Dial Transplant 27:3807–3815. doi: 10.1093/ndt/gfs394. [DOI] [PubMed] [Google Scholar]
- 22.Melton-Celsa AR, Smith MJ, O'Brien DA. 2005. Shiga toxins: potent poisons, pathogenicity determinants, and pharmacological agents. EcoSal Plus doi: 10.1128/ecosalplus.8.7.8. [DOI] [PubMed] [Google Scholar]
- 23.Bielaszewska M, Friedrich AW, Aldick T, Schurk-Bulgrin R, Karch H. 2006. Shiga toxin activatable by intestinal mucus in Escherichia coli isolated from humans: predictor for a severe clinical outcome. Clin Infect Dis 43:1160–1167. doi: 10.1086/508195. [DOI] [PubMed] [Google Scholar]
- 24.Friedrich AW, Bielaszewska M, Zhang WL, Pulz M, Kuczius T, Ammon A, Karch H. 2002. Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J Infect Dis 185:74–84. doi: 10.1086/338115. [DOI] [PubMed] [Google Scholar]
- 25.Scheutz F, Teel LD, Beutin L, Pierard D, Buvens G, Karch H, Mellmann A, Caprioli A, Tozzoli R, Morabito S, Strockbine NA, Melton-Celsa AR, Sanchez M, Persson S, O'Brien AD. 2012. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J Clin Microbiol 50:2951–2963. doi: 10.1128/JCM.00860-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ostroff SM, Tarr PI, Neill MA, Lewis JH, Hargrett-Bean N, Kobayashi JM. 1989. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J Infect Dis 160:994–998. doi: 10.1093/infdis/160.6.994. [DOI] [PubMed] [Google Scholar]
- 27.Kim YB, Okuda J, Matsumoto C, Morigaki T, Asai N, Watanabe H, Nishibuchi M. 1998. Isolation of an Escherichia coli O157:H7 strain producing Shiga toxin 1 but not Shiga toxin 2 from a patient with hemolytic uremic syndrome in Korea. FEMS Microbiol Lett 166:43–48. doi: 10.1111/j.1574-6968.1998.tb13181.x. [DOI] [PubMed] [Google Scholar]
- 28.Koster F, Levin J, Walker L, Tung KS, Gilman RH, Rahaman MM, Majid MA, Islam S, Williams RC Jr. 1978. Hemolytic-uremic syndrome after shigellosis. Relation to endotoxemia and circulating immune complexes. N Engl J Med 298:927–933. [DOI] [PubMed] [Google Scholar]
- 29.Bhimma R, Rollins NC, Coovadia HM, Adhikari M. 1997. Post-dysenteric hemolytic uremic syndrome in children during an epidemic of Shigella dysentery in Kwazulu/Natal. Pediatr Nephrol 11:560–564. doi: 10.1007/s004670050338. [DOI] [PubMed] [Google Scholar]
- 30.Perera LP, Marques LR, O'Brien AD. 1988. Isolation and characterization of monoclonal antibodies to Shiga-like toxin II of enterohemorrhagic Escherichia coli and use of the monoclonal antibodies in a colony enzyme-linked immunosorbent assay. J Clin Microbiol 26:2127–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strockbine NA, Marques LR, Holmes RK, O'Brien AD. 1985. Characterization of monoclonal antibodies against Shiga-like toxin from Escherichia coli. Infect Immun 50:695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang WY, Foote J. 2005. Immunogenicity of engineered antibodies. Methods 36:3–10. doi: 10.1016/j.ymeth.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Edwards AC, Melton-Celsa AR, Arbuthnott K, Stinson JR, Schmitt CK, Wong HC, O'Brien AD. 1998. Vero cell neutralization and mouse protective efficacy of humanized monoclonal antibodies against Escherichia coli toxins Stx1 and Stx2, p 388–392. In Kaper JB, O'Brien AD (ed), Escherichia coli O157:H7 and other Shiga toxin-producing E. coli strains. ASM Press, Washington, DC. [Google Scholar]
- 34.Bitzan M, Poole R, Mehran M, Sicard E, Brockus C, Thuning-Roberson C, Riviere M. 2009. Safety and pharmacokinetics of chimeric anti-Shiga toxin 1 and anti-Shiga toxin 2 monoclonal antibodies in healthy volunteers. Antimicrob Agents Chemother 53:3081–3087. doi: 10.1128/AAC.01661-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dowling TC, Chavaillaz PA, Young DG, Melton-Celsa A, O'Brien A, Thuning-Roberson C, Edelman R, Tacket CO. 2005. Phase 1 safety and pharmacokinetic study of chimeric murine-human monoclonal antibody c alpha Stx2 administered intravenously to healthy adult volunteers. Antimicrob Agents Chemother 49:1808–1812. doi: 10.1128/AAC.49.5.1808-1812.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Melton-Celsa AR, O'Brien DA. 2000. Shiga toxins of Shigella dysenteriae and Escherichia coli, p 385–406. In Aktories K, Just I (ed), Handbook of experimental pharmacology, vol 145 Springer-Verlag, Berlin, Germany. [Google Scholar]
- 37.Schmitt CK, McKee ML, O'Brien AD. 1991. Two copies of Shiga-like toxin II-related genes common in enterohemorrhagic Escherichia coli strains are responsible for the antigenic heterogeneity of the O157:H- strain E32511. Infect Immun 59:1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindgren SW, Melton AR, O'Brien AD. 1993. Virulence of enterohemorrhagic Escherichia coli O91:H21 clinical isolates in an orally infected mouse model. Infect Immun 61:3832–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melton-Celsa AR, Rogers JE, Schmitt CK, Darnell SC, O'Brien AD. 1998. Virulence of Shiga toxin-producing Escherichia coli (STEC) in orally-infected mice correlates with the type of toxin produced by the infecting strain. Jpn J Med Sci Biol 51(Suppl):S108–S114. [DOI] [PubMed] [Google Scholar]
- 40.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed The National Academies Press, Washington, DC. [Google Scholar]
- 41.Wadolkowski EA, Burris JA, O'Brien AD. 1990. Mouse model for colonization and disease caused by enterohemorrhagic Escherichia coli O157:H7. Infect Immun 58:2438–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mukherjee J, Chios K, Fishwild D, Hudson D, O'Donnell S, Rich SM, Donohue-Rolfe A, Tzipori S. 2002. Production and characterization of protective human antibodies against Shiga toxin 1. Infect Immun 70:5896–5899. doi: 10.1128/IAI.70.10.5896-5899.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franchini M, Zaffanello M, Veneri D. 2006. Advances in the pathogenesis, diagnosis and treatment of thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Thromb Res 118:177–184. doi: 10.1016/j.thromres.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 44.Kimura T, Co MS, Vasquez M, Wei S, Xu H, Tani S, Sakai Y, Kawamura T, Matsumoto Y, Nakao H, Takeda T. 2002. Development of humanized monoclonal antibody TMA-15 which neutralizes Shiga toxin 2. Hybrid Hybridomics 21:161–168. doi: 10.1089/153685902760173872. [DOI] [PubMed] [Google Scholar]
- 45.Sheoran AS, Chapman S, Singh P, Donohue-Rolfe A, Tzipori S. 2003. Stx2-specific human monoclonal antibodies protect mice against lethal infection with Escherichia coli expressing Stx2 variants. Infect Immun 71:3125–3130. doi: 10.1128/IAI.71.6.3125-3130.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamagami S, Motoki M, Kimura T, Izumi H, Takeda T, Katsuura Y, Matsumoto Y. 2001. Efficacy of postinfection treatment with anti-Shiga toxin (Stx) 2 humanized monoclonal antibody TMA-15 in mice lethally challenged with Stx-producing Escherichia coli. J Infect Dis 184:738–742. doi: 10.1086/323082. [DOI] [PubMed] [Google Scholar]
- 47.Lambert M. 2014. AAP issues updated guidance on palivizumab prophylaxis for RSV infection. Am Fam Physician 90:867–868. [Google Scholar]
- 48.López EL, Contrini MM, Glatstein E, González Ayala S, Santoro R, Allende D, Ezcurra G, Teplitz E, Koyama T, Matsumoto Y, Sato H, Sakai K, Hoshide S, Komoriya K, Morita T, Harning R, Brookman S. 2010. Safety and pharmacokinetics of urtoxazumab, a humanized monoclonal antibody, against Shiga-like toxin 2 in healthy adults and in pediatric patients infected with Shiga-like toxin-producing Escherichia coli. Antimicrob Agents Chemother 54:239–243. doi: 10.1128/AAC.00343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hara-Kudo Y, Takatori K. 2011. Contamination level and ingestion dose of foodborne pathogens associated with infections. Epidemiol Infect 139:1505–1510. doi: 10.1017/S095026881000292X. [DOI] [PubMed] [Google Scholar]
- 50.Tilden J Jr, Young W, McNamara AM, Custer C, Boesel B, Lambert-Fair MA, Majkowski J, Vugia D, Werner SB, Hollingsworth J, Morris JG Jr. 1996. A new route of transmission for Escherichia coli: infection from dry fermented salami. Am J Public Health 86:1142–1145. doi: 10.2105/AJPH.86.8_Pt_1.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Snedeker KG, Campbell M, Sargeant JM. 2012. A systematic review of vaccinations to reduce the shedding of Escherichia coli O157 in the faeces of domestic ruminants. Zoonoses Public Health 59:126–138. doi: 10.1111/j.1863-2378.2011.01426.x. [DOI] [PubMed] [Google Scholar]
- 52.Varela NP, Dick P, Wilson J. 2013. Assessing the existing information on the efficacy of bovine vaccination against Escherichia coli O157:H7—a systematic review and meta-analysis. Zoonoses Public Health 60:253–268. doi: 10.1111/j.1863-2378.2012.01523.x. [DOI] [PubMed] [Google Scholar]