

Abstract
AdVAV is a replication-deficient adenovirus type 5-vectored vaccine expressing the 83-kDa protective antigen (PA83) from Bacillus anthracis that is being developed for the prevention of disease caused by inhalation of aerosolized B. anthracis spores. A noninferiority study comparing the efficacy of AdVAV to the currently licensed Anthrax Vaccine Absorbed (AVA; BioThrax) was performed in New Zealand White rabbits using postchallenge survival as the study endpoint (20% noninferiority margin for survival). Three groups of 32 rabbits were vaccinated with a single intranasal dose of AdVAV (7.5 × 107, 1.5 × 109, or 3.5 × 1010 viral particles). Three additional groups of 32 animals received two doses of either intranasal AdVAV (3.5 × 1010 viral particles) or intramuscular AVA (diluted 1:16 or 1:64) 28 days apart. The placebo group of 16 rabbits received a single intranasal dose of AdVAV formulation buffer. All animals were challenged via the inhalation route with a targeted dose of 200 times the 50% lethal dose (LD50) of aerosolized B. anthracis Ames spores 70 days after the initial vaccination and were followed for 3 weeks. PA83 immunogenicity was evaluated by validated toxin neutralizing antibody and serum anti-PA83 IgG enzyme-linked immunosorbent assays (ELISAs). All animals in the placebo cohort died from the challenge. Three of the four AdVAV dose cohorts tested, including two single-dose cohorts, achieved statistical noninferiority relative to the AVA comparator group, with survival rates between 97% and 100%. Vaccination with AdVAV also produced antibody titers with earlier onset and greater persistence than vaccination with AVA.
INTRODUCTION
Bacillus anthracis is a Gram-positive, rod-shaped, spore-forming bacterial pathogen and is the etiological agent of anthrax disease. Inhalation of the B. anthracis spores is associated with high levels of mortality in exposed individuals and has led to the use of aerosolized B. anthracis spores as a bioterror weapon (1–3). Following spore inhalation, B. anthracis undergoes a transformation from quiescent spores into a vegetative state associated with active growth and release of two binary toxins that mediate the majority of the pathological effects of anthrax disease. These toxins, lethal toxin and edema toxin, are formed through the interaction of lethal factor (LF) and edema factor (EF) with protective antigen (PA) (4). Vaccines against anthrax are not intended to inhibit B. anthracis vegetative growth but rather are directed against PA and the central role this protein plays in the pathogenesis of anthrax disease. Previous studies have shown that neutralizing antibody to PA is correlated with protection from anthrax disease (5–8).
Vaccines derived from replication-deficient adenoviral vectors are deleted in E1 gene function, ensuring that replication can only be supported in specialized cell lines that provide the essential E1 function in trans. Frequently, one or more of the E3 gene products are also deleted in order to abolish the immunosuppressive activities of some E3 gene products (9) and to allow for insertion of larger or multiple transgenes. As vaccines, adenoviral expression vectors possess distinct advantages compared to protein-based vaccines, including intracellular expression of the immunogen leading to induction of robust cellular and humoral immunity, the ability to administer the vaccine through a variety of routes, including intranasal (10, 11), topical (12), oral (13), and traditional intramuscular routes (14, 15), and improved vaccine stability profiles. Intranasal administration of adenoviral-vectored vaccines is a simple, noninvasive method of vaccination that also has the potential to induce mucosal immunity at the site of vaccination to provide first-line defense against pathogens that enter the body through the respiratory tract (11, 16, 17). AdVAV, an adenoviral anthrax vaccine, is an E1- and E3-deleted adenoviral expression vector that expresses the B. anthracis PA gene. The intranasal vaccine vector is being developed for preexposure prophylaxis of anthrax-associated disease following exposure to aerosolized spores.
Anthrax Vaccine Adsorbed (AVA; BioThrax, Emergent BioSolutions, Gaithersburg, MD) is a protein-based vaccine currently licensed in the United States for general use prophylaxis for disease caused by B. anthracis. While effective in blocking anthrax-related mortality and morbidity, the current AVA vaccination regimen requires a three-dose primary immunization wherein intramuscular doses are administered at 0, 1, and 6 months followed by booster immunizations at 12 and 18 months, with yearly boosters thereafter. Subjects are not considered protected until they have completed the 6-month primary immunization schedule (18). In addition to the prolonged immunization schedule, vaccination with AVA is associated with reactogenicity at the site of administration, typically manifested as pain and tenderness, which may be exacerbated in persons with a history of anthrax disease (18). A number of new vaccine strategies for the prevention of anthrax associated with the inhalation of B. anthracis spores are being developed to reduce the lengthy immunization schedule and reactogenicity associated with AVA vaccination (19). The purposes of this study were to determine if a single intranasal vaccination with AdVAV anthrax vaccine confers statistically noninferior protection from lethal challenge in a rabbit model of inhalation anthrax using AVA as a comparator and to quantify the level of immunogenicity associated with both vaccines.
MATERIALS AND METHODS
Study design.
We sought to evaluate the efficacy of selected doses of AdVAV relative to the approved AVA vaccine in a rabbit challenge model of inhalation anthrax using noninferiority of survival rates as the metric. In this design, the AVA comparator arm received two intramuscular doses of AVA diluted 1:16. The AVA dose and schedule were based on the rabbit preexposure prophylaxis schedule developed by the National Institute of Allergy and Infectious Diseases in which vaccinations occur on days 0 and 28, with spore challenge on day 70 (20), and reports showing that a 1:16 dilution of AVA given on that schedule resulted in near complete survival following challenge (5, 21) and good correlation between the immunogenicity observed in the rabbit model and that of vaccinated humans (21). An additional AVA group that received the intramuscular vaccine at a 1:64 dilution on study days 0 and 28 was not part of the noninferiority analysis but was included to gain additional information on AVA vaccine response. For AdVAV vaccination, rabbits received a single intranasal vaccination of 7.5 × 107, 1.5 × 109, or 3.5 × 1010 viral particles (vp) on study day 0. A fourth AdVAV group received a second AdVAV dose of 3.5 × 1010 vp on study day 28. The placebo group received an intranasal dose of AdVAV formulation buffer on study day 0.
Animals.
Animal studies were conducted at the Battelle Biomedical Research Center (Battelle, West Jefferson, OH) in compliance with the Animal Welfare Act and followed the principles outlined in the National Research Council Guide for the Care and Use of Laboratory Animals. All animal protocols were approved by the Institutional Animal Care and Use Committee. Male and female New Zealand White rabbits weighing 2 to 3 kg (∼20 to 21 weeks of age) were procured from Covance Research Products (Denver, PA). Rabbits were quarantined for 7 days prior to study initiation with observations performed a minimum of twice daily. Two hundred eight rabbits were randomized by animal identification number into six groups of 32 rabbits each and one control group of 16 rabbits (see Table 1). Group 1 contained 8 males and 8 females. Groups 2 through 7 each contained 16 males and 16 females. Rabbits were also randomized to one of eight initial vaccination days such that 26 animals were vaccinated each day, with equal numbers of rabbits from each group per day (four/group/day for the vaccine groups; two/group/day for the control group). Study day 0 for all groups was the day of the first vaccination. Rabbits were randomized to a challenge order within each day. Rabbits were observed twice daily postchallenge until the morning of study day 91 for clinical signs of illness due to anthrax infection (moribund, respiratory distress, decreased appetite, decreased activity, and seizures) and survival. Observed signs of illness and survival status (live/found dead) were recorded at the time of observation.
TABLE 1.
Group mortality and survival data and time to death
| Group | Product | Dose or dilution (day[s] administered)a | No. of survivors/total no. | % survival | Time to death (days) |
||
|---|---|---|---|---|---|---|---|
| Avg | SD | Range | |||||
| 1 | Buffer | NAb | 0/16 | 0 | 4.6 | 1.4 | 3.1–8.7 |
| 2 | AdVAV | 7.5 × 107 vp (0) | 29/32 | 91 | 5.2 | 0.6 | 4.8–5.8 |
| 3 | AdVAV | 1.5 × 109 vp (0) | 31/32 | 97 | 4.1 | NA | NA |
| 4 | AdVAV | 3.5 × 1010 vp (0) | 32/32 | 100 | NA | NA | NA |
| 5 | AdVAV | 3.5 × 1010 vp (0, 28) | 31/32 | 97 | 6.1 | NA | NA |
| 6 | AVA | 1:16 (0, 28) | 31/32 | 97 | 3.0 | NA | NA |
| 7 | AVA | 1:64 (0, 28) | 25/32 | 78 | 4.5 | 0.8 | 3.8–6.1 |
The vaccine dose is shown as viral particles (vp) for AdVAV or as a dilution for the AVA vaccine.
NA, not applicable.
Temperature monitoring.
Body temperatures were monitored daily for all animals (groups 1 to 7) between study days −7 and 7 and for animals in groups 5 to 7 between study days 21 and 35 via an implantable programmable temperature transponder (IPTT-300; Bio Medic Data Systems, Seaford, DE). Each rabbit had two transponders injected subcutaneously (one between the shoulder blades and one in the rump) on study day −7. Temperatures were recorded at the time of implantation to verify the function of both transponders. Baseline temperatures were recorded for all animals from study days −6 through 0 (prior to first vaccination) and for animals in groups 5 to 7 from study days 21 through 28 (prior to second vaccination). Postvaccination temperatures were recorded from study days 1 through 7 and for animals in groups 5 to 7 from study days 29 through 35. On days when animals were sedated with acepromazine, temperatures were recorded prior to sedation.
Vaccines.
AdVAV is a replication-deficient, E1- and E3-deleted adenovirus type 5 vector that contains a human codon-optimized gene encoding the 83-kDa PA protein of B. anthracis in which the endogenous leader sequence has been replaced with the human tissue plasminogen activator leader sequence under the control of the cytomegalovirus immediate early promoter/enhancer. The vector was plaque purified and grown in the E1-complementing PER.C6 cell line. Growth of E1-deleted adenoviruses in PER.C6 cells instead of in other E1-complementing cells lines, like HEK293, eliminates the formation of replication-competent adenovirus arising from homologous recombination between the E1 region contained in the cell line and the adenoviral vector backbone (22). AdVAV was purified over a CsCl gradient and extensively dialyzed against a formulation buffer containing 10 mM Tris at a pH of 7.4, 75 mM NaCl, 1 mM MgCl2, 10 mM histidine, 5% (wt/vol) sucrose, 0.02% polysorbate-80 (wt/vol), 0.1 mM EDTA, and 0.5% (vol/vol) ethanol and was stored frozen at −65°C. AdVAV was plaque purified and manufactured at Aeras (Rockville, MD). AVA, a licensed vaccine for preexposure prophylaxis of anthrax, is made from cell-free filtrates of microaerophilic cultures of an avirulent nonencapsulated strain of Bacillus anthracis. The sterile filtrate culture fluid contains proteins, including the 83-kDa PA protein, released during the growth period and contains no dead or live bacteria. The final product is formulated to contain 1.2 mg/ml aluminum, added as aluminum hydroxide in 0.85% sodium chloride. AVA was obtained from the Centers for Disease Control and Prevention (Atlanta, GA) and stored at 2°C to 8°C.
Vaccine dose preparation and administration.
AdVAV, supplied at a concentration of 1.2 × 1011 viral particles (vp)/ml, was used at this strength for the high-dose group (3.5 × 1010 vp/dose, single and double doses) and was diluted in the sterile formulation buffer to produce the vaccine for the intermediate-dose (1.5 × 109 vp) and low-dose (7.5 × 107 vp) groups. For vaccination, rabbits were anesthetized with ketamine/xylazine, placed in dorsal recumbency, and inoculated intranasally with 0.304 ml of AdVAV vaccine (4 × 76 μl installations, alternating nares). The dosed animals remained in dorsal recumbency for 3 min prior to being placed back in their cages. AVA was diluted 1:16 and 1:64 in sterile 0.9% saline. For AVA administration, rabbits were sedated with acepromazine before 0.5 ml of diluted vaccine was injected intramuscularly into the hip/thigh region. The second vaccine dose of AVA was administered in the contralateral hip/thigh. All AdVAV dosing solutions were verified postdosing using the infectious unit (fluorescence focus unit [FFU]) assay, as the viral particle concentrations of the dosing solutions were below the limit of quantification (LOQ) of the high-performance liquid chromatography-based viral particle assay. The viral particle concentration was then calculated using the established vp-to-FFU ratio of 6 for this lot of AdVAV.
FFU assay.
Briefly, 293 HEK cells were plated in a 6-well plate format followed by inoculation of the appropriate dilutions of adenovirus control and test sample(s) onto triplicate wells. Following a 3-h adsorption at 37°C, the inoculum was removed and replaced with growth medium. The cells were incubated for 48 h to allow for virus replication. At the end of the infection period, media were removed and cells were fixed with cold methanol. Following drying and rinsing with phosphate-buffered saline, fluorescein isothiocyanate (FITC)-conjugated anti-adenovirus hexon was added to each well of cells and incubated at 37°C for 1 h. After removal of the antibody and additional phosphate-buffered saline washes, the plate wells were observed using a fluorescence microscope. The number of fluorescing cells in each well was counted, and the values from three replicate wells containing ≥10 to ≤300 positive cells were averaged to calculate the adenovirus titer.
Aerosol challenge and necropsy.
On study day 70, rabbits were placed individually into a plethysmography chamber in a class III biosafety cabinet system and challenged with aerosolized B. anthracis Ames spores at a targeted dose of 200 times the 50% lethal dose (LD50) (∼2.2 × 107 CFU; 1 LD50 = 1.05 × 105 CFU, as described in reference 23). B. anthracis Ames spores were aerosolized by a Collison nebulizer and delivered via a nose-only inhalation exposure chamber. Gross necropsy was performed on all rabbits that died. Sections of target tissues, including but not limited to brain including meninges, spinal cord, lungs, spleen, liver, and mediastinal lymph nodes, as well as all gross lesions were preserved in 10% neutral buffered formalin. Microscopic examination of hematoxylin and eosin (H&E)-stained tissue sections was performed by a board-certified pathologist on all animals that died postchallenge to confirm death or illness due to anthrax.
Toxin neutralization assay.
Toxin neutralizing antibody (TNA) assays were conducted according to a validated assay method. The assay colorimetrically determines cell viability using a tetrazolium salt, 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide (MTT), as the reporter. The antibody-mediated neutralization of anthrax lethal toxin (LT) manifests as a suppression of cytotoxicity, hence, preservation of cell viability. A reference standard was used in this study, and analysis was conducted using an SAS platform and Taylor code (24). Briefly, microtiter cell culture plates were seeded with J774A.1 cells (2.0 × 104 to 5.0 × 104 cells/well) and allowed to adhere. In separate microplates, six 2-fold serial dilutions (7-point dilution series) of the test sample and control sera were prepared. LT was added to the prep plate and incubated at 37°C for 30 min to allow for LT neutralization by neutralizing antibodies. The contents of the prep plate were then transferred to the cell plate and incubated to allow intoxication to proceed at 37°C for 4 h. MTT was then added to the cell plates to allow viable cells to reduce the MTT dye for 2 h at 37°C. Finally, a solubilization buffer was added to all of the wells, and the plate was incubated at 37°C for 16 to 20 h to lyse the remaining viable cells and to solubilize the metabolized MTT. The optical density (OD) values for each plate were read on a BioTek microplate reader at a wavelength of 570 nm using a 690-nm reference wavelength. A TNA assay SAS program was used to fit the 7-point serial dilutions of the reference serum standard and test sample serum OD values to a four-parameter logistic-log (4PL) function and to calculate the reportable values. The primary assay endpoints are the 50% effective dilution (ED50) and the 50% neutralization factor (NF50). The ED50 is the reciprocal of the dilution of a serum sample that results in 50% neutralization of anthrax lethal toxin. The NF50 is the ratio of the ED50 of the test sample and the ED50 of the reference serum. The LOQ for the ED50 is 39 and for the NF50 is 0.086, and values less than the LOQ were replaced with half of the LOQ (i.e., 19.5 for the ED50 and 0.043 for the NF50) for graphing and statistical analysis. The reference serum (BMI526) used for this assay is a pool of immune human sera with a mean ED50 of 673.09 and an assay acceptance range of 375 to 971.
Anti-PA IgG ELISA.
The rabbit anti-PA IgG enzyme-linked immunosorbent assay (ELISA) is a validated assay designed to quantify immunoglobulin subtype G (IgG) antibodies against anthrax PA using an ELISA in which purified recombinant PA (rPA) is used as the solid-phase immobilized antigen and an enzyme-conjugated anti-gamma chain secondary antibody is used as the reporter or signal system. The assay endpoint is reported as the serum mean concentration of anti-PA-specific IgG (μg/ml). The limit of detection (LOD) for the assay was 1 μg/ml, and the LOQ was 5 μg/ml. Samples with anti-PA IgG concentrations less than the LOQ were assigned a value of half of the LOQ (i.e, 2.5 μg/ml).
Circulating PA-ELISA.
Quantitative determination of circulating PA in the serum of animals exposed to B. anthracis was determined using a validated PA-ELISA. Sample values are calculated against a PA standard curve and reported as the serum concentration of PA in nanograms per milliliter (ng/ml). The LOD for the assay was 1.3 ng/ml, and the LOQ was 10.5 ng/ml. Samples with PA concentrations less than the LOQ were assigned a value of half of the LOQ (i.e., 5.25 ng/ml).
Clinical score.
The clinical score is a combined evaluation of the results of five assays/assessments which are typically normal prechallenge but abnormal postchallenge that can be used to provide a relative measure of infection/illness postchallenge. The five assays/assessments included in this analysis were clinical observations, bacteremia, C-reactive protein (CRP), white blood cell count, and the neutrophil-to-lymphocyte ratio. Normal results were assigned a value of 0 and abnormal results a value of 1 for each parameter on a given study day, resulting in a score for each animal that ranged between 0 and 5 for each day. Averaging these scores for a group provided an average clinical score for each group per postchallenge study day.
Anti-adenovirus ELISA.
A quantitative assay for antibody against the adenovirus type 5 vector was performed by qualified ELISA using an E1/E3-deleted adenovirus as the capture antigen (Ad5-CMV-Null; Vector Biolabs, Philadelphia, PA). After blocking with 5% nonfat dry milk in the assay buffer (Tris-buffered saline with 0.05% Tween 20; Thermo Scientific) and incubation with sample sera that had been serially diluted 2-fold in the assay buffer, bound anti-adenovirus antibody was detected with goat anti-rabbit IgG conjugated to horseradish peroxidase (Abcam) using 3,3′,5,5′-tetramethylbenzidine (KPL) at 450 nm. Commercial rabbit anti-adenovirus serum (Abcam) was used a reference control for system suitability. The assay cutoff was defined as the optical density three standard deviations above the average assay signal in the absence of serum. Sample titers were expressed as the reciprocal of the highest dilution with an optical density greater than the assay cutoff. The LOQ for the assay was 200. Samples with values below the LOQ were assigned a titer of half of the LOQ (i.e., 100) for graphical analysis.
Statistical analysis.
Within the context of the rabbit challenge model, we sought to evaluate the comparability between selected doses of AdVAV and AVA as assessed by noninferiority in survival rates following a challenge with lethal inhalation of B. anthracis spores. Assuming that the probability of survival was 99% in the AVA group vaccinated with the 1:16 dilution and 97% in the AdVAV vaccinated groups, 32 animals per group provided greater than 90% power to conclude that the survival probability in a single AdVAV-vaccinated group was not inferior to that in the AVA group vaccinated with the 1:16 dilution using the score test and a noninferiority margin of 20% with an α value of 0.05. A 99% survival proportion for the AVA comparator arm was assumed based on the published literature (5, 21) and on considerations relating to the power of the study. Unless stated otherwise, all results are reported at the 0.05 level of significance. Noninferiority tests were performed to determine if the survival proportion in each AdVAV-vaccinated group was noninferior to that in the AVA group treated with the 1:16 dilution. Specifically, the noninferiority criterion was as follows: The upper limit of the one-sided 95% Newcombe's hybrid score confidence interval with a continuity correction for the difference between the survival proportions (survivalAVA − survivalAdVAV) should not exceed 20 percentage points. Kaplan-Meier estimates were plotted for the time-to-death data observed in each group, and pairwise log-rank tests were performed to determine if time to death was significantly different among the groups. Geometric means with 95% confidence intervals for the TNA ED50, the TNA NF50, and the anti-PA IgG ELISA were calculated for each group and study time. Analysis of variance (ANOVA) models were fitted separately to the base-10 log-transformed TNA ED50, TNA NF50, and anti-PA IgG ELISA data at each study time point to determine if there were significant differences among the groups. A two-parameter logistic regression model was fitted to the survival data from the AdVAV- and AVA-vaccinated groups to determine the relationship between animal survival and antibody response levels at each prechallenge study time, as measured by the TNA NF50.
RESULTS
Survival.
Animals were challenged with an average dose of 223 ± 59 rabbit LD50 equivalents of B. anthracis Ames spores on study day 70 and were monitored for 3 weeks. The survival and time-to-death data for each group are shown in Table 1. All animals that died postchallenge had a combination of a positive bacteremia, gross lesions, and/or histopathologic lesions consistent with anthrax. The average time to death postchallenge for animals that died was similar across groups and ranged from 3.0 to 6.1 days. Despite a nearly 500-fold dose range of AdVAV, greater than 90% of all animals vaccinated with a single intranasal dose of AdVAV survived spore challenge, with 97% to 100% survival in the intermediate- and high-dose groups of AdVAV. As expected, two intramuscular vaccinations with a 1:16 dilution of AVA also resulted in nearly complete protection, with only a single animal succumbing to disease. Vaccination with a 4-fold lower dose of AVA resulted in decreased survival (78%). Challenge of the placebo group was uniformly lethal.
The upper limit of the one-sided 95% confidence interval for the difference in survival proportions (survivalAVA − survivalAdVAV) did not exceed 20 percentage points for groups 3 to 5 and only slightly exceeded 20 percentage points for group 2 (AdVAV, 7.5 × 107 vp). This indicates that a single intranasal vaccination of AdVAV was noninferior to two intramuscular vaccinations with AVA (1:16) when using a survival noninferiority margin of 20% (Table 2).
TABLE 2.
Descriptive statistics comparing survival between groups 2 through 5 (AdVAV) and group 6 (1:16 AVA)
| AdVAV group | Dose, vp (day[s] administered) | Estimated difference of survival proportions (95% CI)a | Upper limit of one-sided 95% CI for the estimated difference of survival proportionsa |
|---|---|---|---|
| 2 | 7.5 × 107 (0) | 0.06 (−0.10 to 0.23) | 0.20b |
| 3 | 1.5 × 109 (0) | 0.00 (−0.15 to 0.15) | 0.12c |
| 4 | 3.5 × 1010 (0) | −0.03 (−0.18 to 0.11) | 0.08c |
| 5 | 3.5 × 1010 (0, 28) | 0.00 (−0.15 to 0.15) | 0.12c |
Estimates are for the survival proportion induced by the 1:16 dilution of AVA (group 6) minus the survival proportion induced by the AdVAV group. CI, confidence interval. The 95% CIs were calculated using Newcombe's hybrid score interval with a continuity correction.
Value was rounded to the hundredth; actual value was greater than 0.20 (0.2039).
The upper limit of the one-sided 95% CI was less than 0.20; therefore, survival in the AdVAV group was noninferior to that in the 1:16 dilution AVA group (at the 0.05 level of significance).
Bacteremia and circulating PA protein.
Whole blood was collected for analysis of bacteremia on days 1, 2, 4, 7, 14, and 21 postchallenge. All 16 of the animals in the placebo vaccine group were bacteremic for B. anthracis at least 1 day postchallenge, with most animals showing positive cultures on multiple days. Similarly, each of the animals that died in the vaccine groups were bacteremic on at least 1 day postchallenge, with the exception of group 7 (AVA, 1:64), in which 6 of the 7 animals that died were positive for bacteremia on at least 1 day postchallenge. Of the animals surviving the challenge, none of the animals within the AdVAV vaccine groups were bacteremic at any time, whereas 1 of 31 surviving animals in the AVA 1:16 and 2 of 25 surviving animals in the AVA 1:64 group were bacteremic at some point postchallenge. Serum for circulating PA protein analysis was collected on days 2, 4, 7, and 21 postchallenge. The majority (12/16, 75%) of the individual animals in the placebo group had quantifiable levels of serum PA protein at some point postchallenge. Quantifiable levels of circulating PA levels occurred much less frequently in the vaccinated animals, and the geometric mean PA concentrations for each of the AdVAV and AVA vaccine groups were below the LOD for the assay (Table 3).
TABLE 3.
Circulating serum levels of PA following challenge
| Group | Product | Dose or dilution (day[s] administered) | Concn of circulating PA on SD74a (95% CI) |
|---|---|---|---|
| 1 | Buffer | NA | 33.3 (1.25–890) |
| 2 | AdVAV | 7.5 × 107 vp (0) | 0.0782 (0.0536–0.114) |
| 3 | AdVAV | 1.5 × 109 vp (0) | 0.0814 (0.0515–0.129) |
| 4 | AdVAV | 3.5 × 1010 vp (0) | 0.0650 (0.0650–0.0650) |
| 5 | AdVAV | 3.5 × 1010 vp (0, 28) | 0.0746 (0.0564–0.0986) |
| 6 | AVA | 1:16 (0, 28) | 0.0650 (0.0650–0.0650) |
| 7 | AVA | 1:64 (0, 28) | 0.113 (0.0600–0.212) |
Geometric mean concentrations are given. Sample concentrations below the assay LOD of 1.3 ng/ml were assigned a value of half LOD (0.065 ng/ml). SD74, study day 74.
Immunogenicity.
Two independent measures of serum anti-PA immunogenicity were evaluated: total serum antibody against PA, as determined by an anti-PA IgG ELISA, and PA neutralizing antibody, as determined by a toxin neutralization assay, a cell-based neutralization assay. The geometric mean anti-PA IgG titers and the geometric mean TNA titers, expressed as the neutralizing factor (NF50), for each of the groups over the study period are shown in Fig. 1 and 2, respectively. Overall, immunogenicity as measured by these two assays appeared to be dose dependent in both the AdVAV and AVA vaccine groups. Within group 5, the addition of a second AdVAV vaccination at study day 28 resulted in a boost in immunogenicity, with the geometric mean anti-PA IgG and TNA titers increasing approximately 2-fold before approximating the levels of the high-dose single vaccination group (group 4) at the time of the challenge (study day 69). No sex-related differences in AdVAV immunogenicity were observed. (P values of the likelihood ratio test were 0.8633, 0.9035, and 0.9100 for the TNA (ED50), the TNA (NF50) and the anti-PA IgG ELISA, respectively.)
FIG 1.

Time course of geometric mean anti-PA IgG levels (μg/ml) by group as determined by ELISA. All animals received the initial vaccination on study day 0. Groups receiving a second vaccination (groups 5, 6, and 7) had vaccine administered on study day 28. Values less than the limit of quantification (5 μg/ml) were assigned a value of 2.5 μg/ml. Error bars, 95% confidence intervals of the geometric means; arrows, times of vaccination (v) and challenge (c); GMT, geometric mean titer.
FIG 2.

Time course of geometric mean TNA levels (NF50) by group as determined by cell-based neutralization assay. All animals received the initial vaccination on study day 0. Groups receiving a second vaccination (groups 5, 6, and 7) had vaccine administered on study day 28. Values less than the limit of quantification (0.086 NF50) were assigned a value of 0.043. Error bars, 95% confidence interval of the geometric means; arrows, times of vaccination (v) and challenge (c). The reference serum (BMI526) used for this assay is a pool of immune human sera with a mean ED50 of 673.09 and an assay acceptance range of 375 to 971.
Important differences were noted in the kinetics of the immune response. Anti-PA IgG and TNA (ED50 and NF50) antibody levels rose earlier in all of the AdVAV groups compared to the AVA groups. In each of the AdVAV groups, the anti-PA IgG and the TNA antibody levels increased significantly between 7 and 14 days postvaccination, whereas none of the antibody levels in the AVA groups rose above the assay LOQ until after the second AVA vaccination on study day 28. Further, while peak levels of PA antibody overlapped between the AdVAV and AVA groups, the anti-PA IgG and TNA titers in each of the AdVAV groups remained essentially stable between study days 35 and 69, while they consistently declined after study day 35 in both of the AVA vaccine groups. As a result, at study day 69, just prior to challenge, all of the AdVAV dose groups had statistically higher anti-PA IgG levels than the AVA groups, and AdVAV groups 3, 4, and 5 had statistically higher TNA (ED50 and NF50) antibody levels than either of the AVA dose groups (analysis of variance, P < 0.05).
Following challenge with B. anthracis, a rise in the anti-PA IgG and TNA titers over the study day 69 prechallenge levels was observed in all of the vaccinated groups. Interestingly, 1 week following challenge, there appeared to be an inverse correlation between the level of the prechallenge immunogenicity and the strength of the anamnestic response. For example, both of the AVA dose groups had statistically higher anamnestic responses for TNA (ED50 and NF50) on study day 77 than the AdVAV dose groups, with the exception of the AdVAV group that received the lowest dose (group 2). This trend was maintained at the end of the study, on study day 91, when both of the AVA groups had higher postchallenge TNA (ED50 and NF50) levels than any of the AdVAV groups.
Logistic regression modeling of survival and immunogenicity.
There was a strong positive correlation between the log10-transformed measures of immunogenicity and the probability of survival following challenge with aerosolized B. anthracis spores. In this study, a 95% probability of survival following challenge was associated with anti-PA IgG levels of 45.2 μg/ml (95% CI, 25.2 to 137 μg/ml) and TNA (NF50) levels of 0.427 (95% CI, 0.273 to 1.02) on study day 69, 1 day prior to challenge. The corresponding TNA (ED50) level associated with 95% probability of survival on study day 69 was 340 (95% CI, 217 to 824). The degree of protection associated with this level of TNA is consistent with previous work in rabbits (5, 8). A graphical depiction of the TNA (NF50) and survival regression analysis is shown in Fig. 3.
FIG 3.
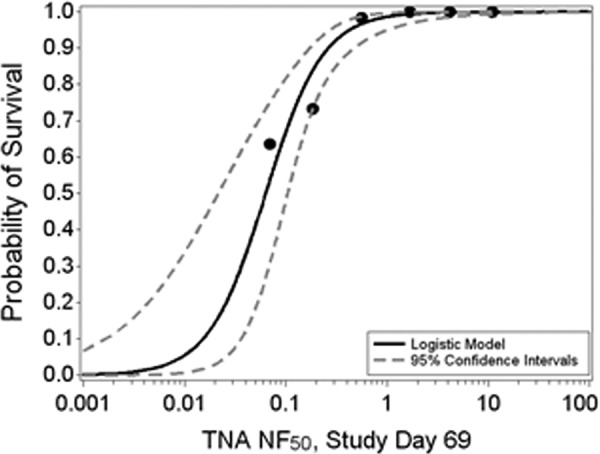
Two-parameter logistic regression analysis fitted to the probability of survival following challenge with Bacillus anthracis spores and the log10-transformed TNA (NF50) levels. Shown is the estimated regression line along with the 95% confidence intervals. Data from all AdVAV- and AVA-vaccinated animals were included in the analysis. The logistic regression curve is symmetric, and the extension to zero is based on the upper asymptote.
Clinical observations.
Vaccination with either vaccine was generally well tolerated, with infrequent clinical observations that included intermittent reduced food consumption, salivation, no stool, soft stool, diarrhea, mucoid stool, or reduced stool. These events appeared not to be related to the type of vaccine or the vaccine dose. Postchallenge, reduced food consumption and lethargy were the primary clinical observations, and these effects began to manifest 2 days postchallenge. These clinical observations occurred in 69% to 100% of the placebo group and at lower frequencies (28% to 78%) in the low-dose vaccine groups for both types of vaccine. Facial edema or swelling, respiratory abnormalities, and distress also occurred but at lower frequencies. Overall, the incidence of postchallenge clinical observations appeared to be inversely related to vaccine dose, with lower frequencies in the higher vaccine dose groups. No differences related to the type of vaccine were noted (data not shown).
Hematology, CRP, body temperature, and body weight. (i) Postvaccination effects.
Hematology was normal in the placebo group and in all of the vaccine groups; only occasionally did individual animals exhibit transient hematological parameters outside the normal range. C-reactive protein (CRP), a nonspecific marker for inflammation, was also normal in all vaccine groups, with no dose-related effects and only infrequent samples above the assay limit of detection. Body temperature in the vaccine groups appeared to have been elevated in the vaccinated animals between study days 1 to 7; 25% to 50% of each vaccine group, versus 19% of the placebo group, experienced an elevation in body temperature more than two standard deviations from baseline. These effects were largely transient and did not appear to be related to dose or type of vaccine. No vaccine-related effects were observed for body weight, which increased steadily throughout the prechallenge period.
(ii) Postchallenge effects.
Following challenge with B. anthracis spores, a pronounced inflammatory reaction consistent with bacteremia was observed in the placebo group and, to a lesser extent, in the vaccinated groups in an inverse dose-related manner. Beginning shortly after challenge, leukocytosis with neutrophilia was observed, especially in the placebo and low-dose vaccine groups (i.e., AdVAV 7.5 × 107 vp and AVA 1:64), followed by a general elevation in both lymphocyte and monocyte counts consistent with bacteremia. Two days postchallenge, significant elevations in the white blood cell counts, driven primarily by increases in neutrophils and monocytes, were noted in all groups, but the elevations were most striking in the placebo and low-dose vaccine groups. Platelets were significantly decreased in all groups 2 days postchallenge, with concomitant rebound 5 days later; the magnitude of these changes was inversely proportional to the vaccine dose. No consistent differences in the above parameters were noted between the two vaccines. A statistical decrease in hemoglobin was observed in all groups postchallenge, possibly as a result of the increased sampling frequency combined with iron sequestration by B. anthracis, but remained low only in the AVA groups (1:16 and 1:64 doses) at 2 weeks postchallenge and only in the AVA 1:64 dose group at 3 weeks postchallenge. Body weight was measured 2 weeks and 3 weeks postchallenge; since all of the placebo animals died within 2 weeks of the challenge, no postchallenge body weights were available for that group. Changes in postchallenge body weights in the remaining groups were generally less than 10% and included both weight gain and weight loss within each group. Body temperature was not measured postchallenge.
Clinical score.
The placebo group had the highest clinical scores, which escalated with time postchallenge until all animals had succumbed by study day 77 (Fig. 4). Clinical scores for the vaccinated groups peaked 4 days postchallenge (study day 74) and were generally similar, with the exception of the AVA 1:64 group, which had consistently higher scores over the postchallenge observation period than the other vaccine groups.
FIG 4.

Clinical score analysis. The clinical score is an aggregate assessment of postchallenge health indices related to anthrax disease. The health indices and their normal state or range were clinical observation (no observations), bacteremia (negative), CRP (<0.5 mg/dl), white blood cell count (2.90 × 103 to 8.10 × 103 cells/μl) and neutrophil/lymphocyte ratio (<0.5). Each animal was assigned a value of 0 for a normal result and a value of 1 for an abnormal result for each assay on each of the indicated study days. The individual scores within a group were averaged, and the standard error for the group was calculated. Prechallenge baseline indices for each of the five parameters were on study day 69 or study day 70. For this study, bacteremia analysis was not performed prior to study day 71, so the maximum clinical score at baseline was 4 instead of 5. Vertical dotted line, time of challenge; error bars, standard error of the mean.
Antivector immune response.
One of the advantages of adenoviral-vectored vaccines is that they can stimulate a local inflammatory response against the viral proteins and DNA, creating an adjuvant-like effect and boosting the immunogenicity of the expressed antigen (25–27). Conversely, antibodies directed against the major vector structural proteins could have an impact on the ability to administer more than a single dose or on the ability to reimmunize using the same vector. Sera were collected from each of the AdVAV-treated animals on study days 0 (preimmune), 28, and 56 for quantification of anti-adenovirus antibodies by an ELISA endpoint titration assay using E1/E3-deleted adenovirus as the capture antigen. Of the preimmune samples, 93% (28/30) were below the LOQ of the assay. The geometric mean titers (GMTs) for each AdVAV vaccinated group are shown in Fig. 5. Groups 2 and 3 (single low-dose and intermediate-dose AdVAV group, respectively) showed no consistent increase over the anti-adenovirus type 5 (anti-Ad5) preimmune titer following vaccination. The single and double high-dose AdVAV groups (groups 4 and 5, respectively) had increased anti-Ad5 titers over preimmune levels on study days 28 and 56. On study day 56, animals that received two high-dose vaccinations (group 5), had approximately three-fold higher anti-adenovirus titers than animals that received a single high-dose vaccination (group 4), though the titers even in those animals remained low (a GMT of 630 for group 5 compared to a GMT of 238 for group 4).
FIG 5.
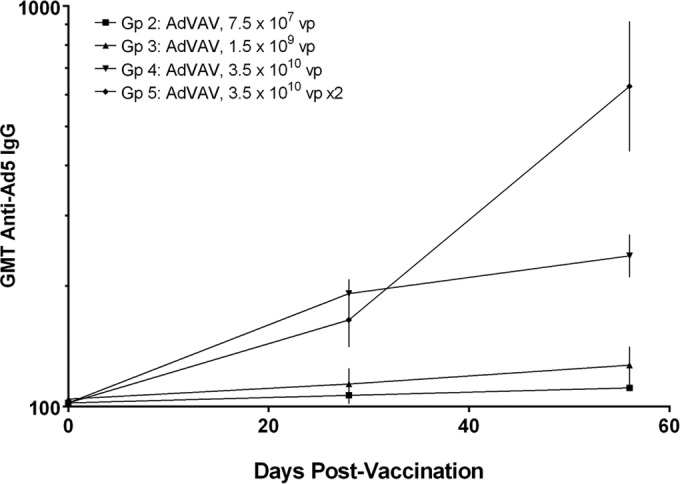
Time course of geometric mean anti-adenovirus type 5 antibody levels for the AdVAV vaccine groups as determined by endpoint dilution ELISA. Study day 0 samples were obtained prior to vaccination. Sample titers were calculated as the reciprocal of the highest dilution resulting in an optical density greater than the assay cutoff. Sample concentrations below the assay LOQ were assigned a value of half of the LOQ for calculating the GMT. Vertical bars represent the 95% confidence intervals of the geometric mean.
DISCUSSION
The purpose of this study was to determine if a single intranasal dose of AdVAV adenoviral-vectored anthrax vaccine was noninferior to two humanized doses of the licensed anthrax vaccine AVA. The primary endpoint for analysis was survival following lethal challenge with aerosolized B. anthracis spores. Immune response, defined as serum anti-PA IgG and TNA levels, was a secondary endpoint of the study. To demonstrate noninferiority, the upper limit of the one-sided 95% confidence interval for the difference between the survival proportions (survivalAVA − survivalAdVAV) should not exceed the noninferiority margin of 20 percentage points with a type 1 error (α) controlled at the 0.05 level. Noninferiority was established for single AdVAV doses of 3.5 × 1010 vp and 1.5 × 109 vp; the 7.5 × 107 vp dose nearly attained noninferiority, with an upper limit of the one-sided confidence interval for the difference in survival of 20.4%. Two vaccinations with AdVAV doses at 3.5 × 1010 vp given 1 month apart were also noninferior to two doses of AVA at 1:16. Both vaccines demonstrated a dose-proportional immune response, but the relative shallowness of the AdVAV dose response was surprising. For example, there was a 467-fold difference in the vaccine dose between the highest and lowest AdVAV dose groups but only 3.9-fold difference in the anti-PA IgG on study day 69. A qualitatively similar effect was observed with the TNA levels (ED50 and NF50). By comparison, a similar 4- to 5-fold drop in PA immunogenicity was noted between the AVA 1:16 and 1:64 groups at study day 69. It is reasonable to postulate that the AdVAV doses used here are near the upper asymptote of the dose-response relationship for AdVAV and that lower doses of the vaccine may be similarly efficacious.
Vaccination with AdVAV was associated with a rapid induction of immune response, with approximately 50% of the prechallenge peak anti-PA IgG level observed 14 days postvaccination. Induction of TNA occurred slightly later, with 50% prechallenge peak levels attained by 28 days postvaccination. In contrast, following vaccination with AVA, anti-PA IgG and TNA levels consistently above the assay LOQ were observed only after 5 weeks and administration of a second vaccine dose. Moreover, in the AdVAV-vaccinated animals, the peak levels of anti-PA IgG and TNA (both ED50 and NF50) were maintained at their plateau levels throughout the prechallenge period. This was in contrast to the consistent decline in protective antibody levels observed following vaccination with AVA. For example, 1 week after the second AVA vaccination at 1:16, the anti-PA IgG serum levels reached their peak geometric mean titer of 242.9 μg/ml and thereafter steadily declined to 40.75 μg/ml by study day 69, just prior to challenge. Vaccination with a single AdVAV dose of 3.5 × 1010 vp resulted in anti-PA IgG levels of 271.9 μg/ml on study day 28 that were maintained throughout the prechallenge period, with a titer of 299.2 μg/ml on study day 69. The vaccine-related difference in persistence of the immune response was also reflected in the TNA levels (ED50 and NF50). The duration of the immune response is a hallmark of AdVAV vaccine activity. When a single intranasal dose of AdVAV was used to vaccinate A/J mice, anti-PA IgG ELISA titers in the vaccinated mice were maintained at the 1-month postvaccination level for at least 1 year (10).
A greater anamnestic response of anti-PA IgG following challenge was observed in both AVA dose groups compared to all of the AdVAV dose groups, and it is possible that the preservation of peak antibody levels in the AdVAV groups sequestered the secreted PA released during germination of the spores and thereby blunted the ability of the immune system to mount a robust anamnestic response. This relationship was preserved for the TNA titers as well, with a significantly greater anamnestic response observed in both AVA groups compared to the AdVAV groups. While greater anamnestic responses were observed in the AVA dose groups, these responses did not appear to be correlated with survival, as the AVA 1:64 group had poorer survival than the other treatment groups despite having the greatest memory response.
The clinical and pathological sequelae following challenge with B. anthracis spores in this study were consistent with anthrax disease (23). Challenge of the placebo group was characterized by inflammation and elevation of CRP, bacteremia, and clinical observations consistent with anthrax disease, including lethargy and respiratory signs. The severity of the disease was less in the vaccinated groups than in the control group. Within the vaccinated groups, the severity of the illness was similar among the vaccinated groups with the exception of the low-dose AVA group (1:64), consistent with the lower level of protection provided in that group.
Intranasal vaccination with AdVAV was well tolerated in this study, consistent with previous data obtained in mice (10). Replication-deficient adenoviral vectors have been extensively studied as vehicles for gene therapy and vaccine delivery. In general, the vectors are safe, not widely disseminated following nonsystemic administration, and cleared quickly from the host tissues (28). In this context, it is also important to note that no behavioral issues have been reported following the intranasal administration of adenoviral vectors.
The immune response against the AdVAV vector was measured by ELISA using an E1/E3-deleted adenovirus as the capture reagent. Following intranasal administration, only low levels of antivector antibody were detected. AdVAV is deleted in the E3 gene, and given that several E3-encoded functions are known to be involved in suppression of the host immune response (9), the vector is designed to elicit a local inflammatory response. Of course, if the antibody response against the vector becomes excessive, the vector infectivity could be neutralized, negatively impacting the vaccine activity. In this study, the level of antivector immune response present at study day 28 did not block the activity of the second vaccination of group 5, as that group had a 2-fold boost in anti-PA IgG and TNA titers compared to the group that received a single vaccination at the same dose level (group 4). Other studies examining the role of preexisting antibodies on the activity of intranasally administered adenoviral vectors have been inconclusive, with at least one report showing that the intranasal route of administration, unlike the intramuscular and oral routes, is not affected by preexisting antibody (11, 29).
Currently, protection from anthrax disease following vaccination with AVA requires the completion of an immunization program of 3 vaccinations over 6 months, and the parenteral route of AVA administration requires the active participation of a health care worker for vaccination. Moreover, the complex and incompletely characterized nature of the AVA vaccine results in significant levels of reactogenicity in the majority of vaccinated individuals (19). If the rapid onset of immunity following a single, well-tolerated, and noninvasive vaccination with AdVAV is translated to clinical immunogenicity trials, AdVAV would represent an important advance toward the goal of an effective and easily administered prophylaxis against inhalation anthrax disease. In this context, it is important to note that a clinical dose of AdVAV associated with protective levels of immunogenicity has not yet been determined, and caution should be used in extrapolating the comparison between AdVAV and AVA beyond the specific model system used in this study. It is currently unknown whether the noninferiority of the single-dose AdVAV vaccine demonstrated in this study will translate to the clinical setting. Antibody levels at the time of challenge have recently been used to compare the immunogenicity of AVA vaccination in rabbits and nonhuman primates to that in humans (21), so it is interesting that, at the time of the challenge in this study, the immunogenicity induced by the lowest dose of AdVAV either approximated (TNA) or was greater than (anti-PA IgG) that of AVA diluted 1:16.
AdVAV is being developed for preexposure prophylaxis for individuals with an increased likelihood of being exposed to B. anthracis spores. The rapid immune response afforded by AdVAV could also be an important feature of an effective postexposure prophylaxis (PEP) indication. In this scenario, individuals who were recently exposed to B. anthracis spores would begin a course of antibiotic therapy (21, 30–32). Antibiotics, however, only affect the vegetative state of B. anthracis, so vaccination against PA would be prudent to protect against the germination of any cryptic spores after the cessation of antibiotic therapy (32). In this setting, a vaccine with a rapid onset of protective immunity could allow discontinuation of the antibiotic course sooner, with better patient adherence to the regimen and decreases in antibiotic-related adverse events, logistics, and cost.
In conclusion, we have shown that, on the basis of survival following lethal challenge with inhaled B. anthracis spores, a single intranasal dose of AdVAV was statistically noninferior to two intramuscular doses of AVA. Importantly, we have also shown that the vaccine-mediated protective immune response of AdVAV was both more rapid and more stable than that of AVA. These data warrant the continued development of AdVAV as a vaccine for preexposure general use prophylaxis of anthrax disease and suggest promising characteristics as a potential PEP vaccine.
ACKNOWLEDGMENTS
We thank AERAS CMO for the manufacture of AdVAV used in these studies, Battelle Biomedical Research Center (West Jefferson, OH) for technical support in the execution of the animal studies and supporting assays, Bill Enright, Richard Warren, and Tom Dreier for critical review of the manuscript, Victoria Haque for exceptional program management, and Tanya Krubit and Shretta Lawson for outstanding quality oversight.
V.K., B.H.A., C.S., J.Z., T.F., and M.S.R. are employees of Vaxin, Inc., and may have received company stock. M.D. is a paid consultant for Vaxin, Inc.
This project has been funded in part with federal funds from the Biomedical Advanced Research and Development Authority, Department of Health and Human Services, contract HHSO-100-2011-00032C.
REFERENCES
- 1.Dalton R. 2001. Genetic sleuths rush to identify anthrax strains in mail attacks. Nature 413:657–658. doi: 10.1038/35099687. [DOI] [PubMed] [Google Scholar]
- 2.Brachman PS. 1980. Inhalation anthrax. Ann N Y Acad Sci 353:83–93. doi: 10.1111/j.1749-6632.1980.tb18910.x. [DOI] [PubMed] [Google Scholar]
- 3.Pile JC, Malone JD, Eitzen EM, Friedlander AM. 1998. Anthrax as a potential biological warfare agent. Arch Intern Med 158:429–434. doi: 10.1001/archinte.158.5.429. [DOI] [PubMed] [Google Scholar]
- 4.Brossier F, Mock M. 2001. Toxins of Bacillus anthracis. Toxicon 39:1747–1755. doi: 10.1016/S0041-0101(01)00161-1. [DOI] [PubMed] [Google Scholar]
- 5.Pitt ML, Little SF, Ivins BE, Fellows P, Barth J, Hewetson J, Gibbs P, Dertzbaugh M, Friedlander AM. 2001. In vitro correlate of immunity in a rabbit model of inhalational anthrax. Vaccine 19:4768–4773. doi: 10.1016/S0264-410X(01)00234-1. [DOI] [PubMed] [Google Scholar]
- 6.Reuveny S, White MD, Adar YY, Kafri Y, Altboum Z, Gozes Y, Kobiler D, Shafferman A, Velan B. 2001. Search for correlates of protective immunity conferred by anthrax vaccine. Infect Immun 69:2888–2893. doi: 10.1128/IAI.69.5.2888-2893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Little SF, Ivins BE, Webster WM, Fellows PF, Pitt ML, Norris SL, Andrews GP. 2006. Duration of protection of rabbits after vaccination with Bacillus anthracis recombinant protective antigen vaccine. Vaccine 24:2530–2536. doi: 10.1016/j.vaccine.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 8.Little SF, Ivins BE, Fellows PF, Pitt ML, Norris SL, Andrews GP. 2004. Defining a serological correlate of protection in rabbits for a recombinant anthrax vaccine. Vaccine 22:422–430. doi: 10.1016/j.vaccine.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Efrat S, Fejer G, Brownlee M, Horwitz MS. 1995. Prolonged survival of pancreatic islet allografts mediated by adenovirus immunoregulatory transgenes. Proc Natl Acad Sci U S A 92:6947–6951. doi: 10.1073/pnas.92.15.6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Jex E, Feng T, Sivko GS, Baillie LW, Goldman S, Van Kampen KR, Tang DC. 2013. An adenovirus-vectored nasal vaccine confers rapid and sustained protection against anthrax in a single-dose regimen. Clin Vaccine Immunol 20:1–8. doi: 10.1128/CVI.00280-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemiale F, Kong WP, Akyurek LM, Ling X, Huang Y, Chakrabarti BK, Eckhaus M, Nabel GJ. 2003. Enhanced mucosal immunoglobulin A response of intranasal adenoviral vector human immunodeficiency virus vaccine and localization in the central nervous system. J Virol 77:10078–10087. doi: 10.1128/JVI.77.18.10078-10087.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi Z, Zeng M, Yang G, Siegel F, Cain LJ, van Kampen KR, Elmets CA, Tang DC. 2001. Protection against tetanus by needle-free inoculation of adenovirus-vectored nasal and epicutaneous vaccines. J Virol 75:11474–11482. doi: 10.1128/JVI.75.23.11474-11482.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexander J, Ward S, Mendy J, Manayani DJ, Farness P, Avanzini JB, Guenther B, Garduno F, Jow L, Snarsky V, Ishioka G, Dong X, Vang L, Newman MJ, Mayall T. 2012. Pre-clinical evaluation of a replication-competent recombinant adenovirus serotype 4 vaccine expressing influenza H5 hemagglutinin. PLoS One 7:e31177. doi: 10.1371/journal.pone.0031177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu S, Zhang Z, Yu R, Zhang J, Liu Y, Song X, Yi S, Liu J, Chen J, Yin Y, Xu J, Hou L, Chen W. 2014. Intramuscular delivery of adenovirus serotype 5 vector expressing humanized protective antigen induces rapid protection against anthrax that may bypass intranasally originated preexisting adenovirus immunity. Clin Vaccine Immunol 21:156–164. doi: 10.1128/CVI.00560-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan Y, Hackett NR, Boyer JL, Crystal RG. 2003. Protective immunity evoked against anthrax lethal toxin after a single intramuscular administration of an adenovirus-based vaccine encoding humanized protective antigen. Hum Gene Ther 14:1673–1682. doi: 10.1089/104303403322542310. [DOI] [PubMed] [Google Scholar]
- 16.Xu Q, Pichichero ME, Simpson LL, Elias M, Smith LA, Zeng M. 2009. An adenoviral vector-based mucosal vaccine is effective in protection against botulism. Gene Ther 16:367–375. doi: 10.1038/gt.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lobanova LM, Baig TT, Tikoo SK, Zakhartchouk AN. 2010. Mucosal adenovirus-vectored vaccine for measles. Vaccine 28:7613–7619. doi: 10.1016/j.vaccine.2010.09.055. [DOI] [PubMed] [Google Scholar]
- 18.Emergent BioSolutions. 2013. Biothrax package insert, revised May 2013 Emergent BioSolutions, Rockville, MD: http://www.biothrax.com/prescribinginformation_biothrax_us.pdf Accessed June 2014. [Google Scholar]
- 19.Wright JG, Plikaytis BD, Rose CE, Parker SD, Babcock J, Keitel W, El Sahly H, Poland GA, Jacobson RM, Keyserling HL, Semenova VA, Li H, Schiffer J, Dababneh H, Martin SK, Martin SW, Marano N, Messonnier NE, Quinn CP. 2014. Effect of reduced dose schedules and intramuscular injection of anthrax vaccine adsorbed on immunological response and safety profile: a randomized trial. Vaccine 32:1019–1028. doi: 10.1016/j.vaccine.2013.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.US Food and Drug Administration. Pathway to licensure for protective antigen-based anthrax vaccines for a postexposure prophylaxis indication using the animal rule. US Food and Drug Administration, Silver Spring, MD: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/VaccinesandRelatedBiologicalProductsAdvisoryCommittee/UCM232400.pdf Accessed January 2015. [Google Scholar]
- 21.Ionin B, Hopkins RJ, Pleune B, Sivko GS, Reid FM, Clement KH, Rudge TL Jr, Stark GV, Innes A, Sari S, Guina T, Howard C, Smith J, Swoboda ML, Vert-Wong E, Johnson V, Nabors GS, Skiadopoulos MH. 2013. Evaluation of immunogenicity and efficacy of anthrax vaccine adsorbed for postexposure prophylaxis. Clin Vaccine Immunol 20:1016–1026. doi: 10.1128/CVI.00099-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fallaux FJ, Bout A, van der Velde I, van den Wollenberg DJ, Hehir KM, Keegan J, Auger C, Cramer SJ, van Ormondt H, van der Eb AJ, Valerio D, Hoeben RC. 1998. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum Gene Ther 9:1909–1917. doi: 10.1089/hum.1998.9.13-1909. [DOI] [PubMed] [Google Scholar]
- 23.Zaucha GM, Pitt LM, Estep J, Ivins BE, Friedlander AM. 1998. The pathology of experimental anthrax in rabbits exposed by inhalation and subcutaneous inoculation. Arch Pathol Lab Med 122:982–992. [PubMed] [Google Scholar]
- 24.Li H, Soroka SD, Taylor TH Jr, Stamey KL, Stinson KW, Freeman AE, Abramson DR, Desai R, Cronin LX, Oxford JW, Caba J, Pleatman C, Pathak S, Schmidt DS, Semenova VA, Martin SK, Wilkins PP, Quinn CP. 2008. Standardized, mathematical model-based and validated in vitro analysis of anthrax lethal toxin neutralization. J Immunol Methods 333:89–106. doi: 10.1016/j.jim.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 25.Knowles MR, Hohneker KW, Zhou Z, Olsen JC, Noah TL, Hu PC, Leigh MW, Engelhardt JF, Edwards LJ, Jones KR, et al. . 1995. A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N Engl J Med 333:823–831. doi: 10.1056/NEJM199509283331302. [DOI] [PubMed] [Google Scholar]
- 26.Hermens WT, Verhaagen J. 1998. Suppression of inflammation by dexamethasone prolongs adenoviral vector-mediated transgene expression in the facial nucleus of the rat. Brain Res Bull 47:133–140. doi: 10.1016/S0361-9230(98)00042-2. [DOI] [PubMed] [Google Scholar]
- 27.Shifrin AL, Chirmule N, Zhang Y, Raper SE. 2005. Macrophage ablation attenuates adenoviral vector-induced pancreatitis. Surgery 137:545–551. doi: 10.1016/j.surg.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Sheets RL, Stein J, Bailer RT, Koup RA, Andrews C, Nason M, He B, Koo E, Trotter H, Duffy C, Manetz TS, Gomez P. 2008. Biodistribution and toxicological safety of adenovirus type 5 and type 35 vectored vaccines against human immunodeficiency virus-1 (HIV-1), Ebola, or Marburg are similar despite differing adenovirus serotype vector, manufacturer's construct, or gene inserts. J Immunotoxicol 5:315–335. doi: 10.1080/15376510802312464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Croyle MA, Patel A, Tran KN, Gray M, Zhang Y, Strong JE, Feldmann H, Kobinger GP. 2008. Nasal delivery of an adenovirus-based vaccine bypasses pre-existing immunity to the vaccine carrier and improves the immune response in mice. PLoS One 3:e3548. doi: 10.1371/journal.pone.0003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brookmeyer R, Johnson E, Bollinger R. 2003. Modeling the optimum duration of antibiotic prophylaxis in an anthrax outbreak. Proc Natl Acad Sci U S A 100:10129–10132. doi: 10.1073/pnas.1631983100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bossi P, Tegnell A, Baka A, Van Loock F, Hendriks J, Werner A, Maidhof H, Gouvras G. 2004. Bichat guidelines for the clinical management of anthrax and bioterrorism-related anthrax. Euro Surveill 9:E3-4. [DOI] [PubMed] [Google Scholar]
- 32.Friedlander AM, Welkos SL, Pitt ML, Ezzell JW, Worsham PL, Rose KJ, Ivins BE, Lowe JR, Howe GB, Mikesell P, et al. . 1993. Postexposure prophylaxis against experimental inhalation anthrax. J Infect Dis 167:1239–1243. doi: 10.1093/infdis/167.5.1239. [DOI] [PubMed] [Google Scholar]