

Abstract
Over the past decade, our understanding of cardiomyopathies has improved dramatically, due to improvements in screening and detection of gene defects in the human genome as well as a variety of novel animal models (mouse, zebrafish, and drosophila) and in silico computational models. These novel experimental tools have created a platform that is highly complementary to the naturally occurring cardiomyopathies in cats and dogs that had been available for some time. A fully integrative approach, which incorporates all these modalities, is likely required for significant steps forward in understanding the molecular underpinnings and pathogenesis of cardiomyopathies. Finally, novel technologies, including CRISPR/Cas9, which have already been proved to work in zebrafish, are currently being employed to engineer sarcomeric cardiomyopathy in larger animals, including pigs and non-human primates. In the mouse, the increased speed with which these techniques can be employed to engineer precise ‘knock-in’ models that previously took years to make via multiple rounds of homologous recombination-based gene targeting promises multiple and precise models of human cardiac disease for future study. Such novel genetically engineered animal models recapitulating human sarcomeric protein defects will help bridging the gap to translate therapeutic targets from small animal and in silico models to the human patient with sarcomeric cardiomyopathy.
Keywords: Cardiomyopathy, Animal models, In silico models, Genetics, Sarcomeres
1. Introduction
Cardiomyopathies are defined as myocardial disorders in which the heart muscle is structurally and functionally abnormal in the absence of coronary artery disease, hypertension, valvular disease, and congenital heart disease, sufficient to explain the observed myocardial abnormality.1 They are grouped into specific morphological and functional phenotypes, including hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy, dilated cardiomyopathy (DCM), and arrhythmogenic right ventricular cardiomyopathy (ARVC).
Animal models of cardiac hypertrophy and heart failure associated with ischaemic heart disease, chronic haemodynamic (volume and/or pressure) overload, and tachyarrhythmias have been available for >40 years, and have proved instrumental in advancing our understanding of pathophysiology and in developing novel therapies of hypertrophy and heart failure.2–6 In contrast, although some naturally occurring cardiomyopathies in cats and dogs had already been known for some time, animal models of cardiomyopathies have become available only recently with the advent of transgenesis and gene targeting (Table 1).
Table 1.
Animal models of sarcomeric cardiomyopathy
| Model | Breed | Phenotype | Affected gene | Mutations | Advantages | Disadvantages |
|---|---|---|---|---|---|---|
| Naturally occurring models | ||||||
| Cat7–12 | Maine Coon Ragdoll |
HCM | MYPBC3 | c.91G>C [p.Ala31Pro] c.2328C>T [p.Arg820Trp] |
|
|
| Dog13–25 | Port. Waterdogs Great Danes Doberman Boxers |
DCM DCM DCM DCM/ARVC |
Uncertain Uncertain Uncertain Uncertain |
Unknown Unknown Unknown Unknown |
|
- Genes and mutations presently unknown.- Gene targeting is difficult. |
| Genetically engineered models | ||||||
| Mouse26–40 | HCM HCM DCM |
Mybpc3, Myh6, Tnni3, Tnnt2, Tpm1 Mybpc3, Myh6, Tnnt2 Mlp/Csrp3 |
Transgenesis Knock-in/-out Knock-out |
|
|
|
| Rat41 | HCM | Tnnt2 | Transgenesis (truncated protein) |
|
|
|
| Rabbit42 | HCM | TNNI3 | Transgenesis |
|
||
| Zebrafish43 | DCM | Mybpc3 | [p.Val762Asp/p. Arg820Gln] |
|
|
|
| Drosophila44,45 | HCM RCM |
mybpc3 tnnt2 |
Wild-type, truncated up101 TnT1 |
|
|
|
HCM: hypertrophic cardiomyopathy; DCM: dilated cardiomyopathy; ARVC: arrhythmogenic right ventricular cardiomyopathy; RCM: restrictive cardiomyopathy; Gene abbreviations used: Mybpc3: cardiac myosin-binding protein C; Myh6: α-myosin heavy chain; Myh7: β-myosin heavy chain; Tnni3: cardiac troponin I; Tnnt2: cardiac troponin T; ilk: integrin-linked kinase; mlp/Csrp3: cysteine and glycine-rich protein; not3: CG8426 gene product from the transcript CG8426-RB.
The purpose of this review is to provide an overview of the various naturally occurring and genetically engineered animal models of cardiomyopathy that allow detailed and integrated physiological and molecular studies of cardiomyopathies, as well as the evaluation of therapy. We will end with a novel approach to integrating existing knowledge into an in silico model to understand the molecular basis of cardiomyopathies and to predict phenotypes and therapeutic targets.
2. Cat models of cardiomyopathy
HCM is the most common cardiac disease in domestic cats,7 and is characterized by left ventricular hypertrophy (LVH), particularly of the papillary muscles, systolic anterior motion, and myocardial disarray. It is a progressive disease that starts in the adolescence (generally after 6 months of age) and can result in heart failure, paralysis of the hind legs due to clot embolization originating in the heart, and sudden cardiac death.
HCM is transmitted in an autosomal-dominant trait in the Maine Coon and Ragdoll breeds.7,8 Two mutations in MYBPC3 have been identified so far. The first one, identified only in the Main Coon breed, is a c.91G>C missense mutation in exon 3, which gives rise to the p.Ala31Pro cardiac myosin-binding protein C (cMyBP-C) mutant in the linker region between the C0 and C1 domains of the protein.9,10 Some rare isolated cases of British Longhair, Ragdoll, or Siberian breeds also carry this mutation.10,11 The second one, identified only in the Ragdoll breed, is a c.2328C>T transition in exon 26, which results in the p.Arg820Trp cMyBP-C mutant in the C6 domain.8,10 Both heterozygous and homozygous cats for MYBPC3 mutations developed LVH (mainly concentric),12 but some heterozygotes do not exhibit clinical signs of HCM. On the other hand, whereas all homozygotes developed diastolic dysfunction, few heterozygotes developed minor regional myocardial diastolic dysfunction without LVH,12 suggesting that diastolic dysfunction could be the first feature of the disease, such as observed in heterozygous human patients and mouse model of HCM.26,47,48 Importantly, the c.91G>C mutation results in a lower amount of cMyBP-C protein in the heart in both heterozygous and homozygous Maine Coon cats,9 such as seen in human HCM.49–51 This suggests regulation of mutation expression by protein quality control mechanisms, such as the ubiquitin–protein system, which has been shown to be involved after MYBPC3 gene transfer in cardiac myocytes and in vivo in the Mybpc3-targeted knock-in mice.27,52–56
Cats with HCM represent therefore a good intermediary model between the many mouse models that have been made and humans to evaluate different causal therapeutic strategies to prevent the development of heart failure and/or sudden cardiac death or to rescue the phenotype in both heterozygotes and homozygotes for MYBPC3 mutations. Recent evidence that RNA-based therapies, such as exon skipping or trans-splicing, can repair Mybcp3 mRNA,57,58 and more recently, that Mybpc3 gene therapy long term prevents the development of the disease phenotype in Mybpc3-targeted knock-in mice59 paved the way to evaluate these strategies in cats.
3. Canine models of cardiomyopathy
Large animal models of inherited cardiomyopathies would be extremely useful for the evaluation of novel pharmacological, gene, cell, and device therapies. To our knowledge, there are no known porcine or canine genetically engineered models of cardiomyopathy. However, several naturally occurring forms of DCM have been described in dogs,13 and in fact constitute the most common form of heart disease in large- and giant-bred dogs.14 For example, DCM has been described in Portuguese waterdogs,15 Great Danes,16 Doberman Pinschers,17 and Boxers.18 The exact genetic basis in each of these breeds remains, however, incompletely understood, with reports showing an association with mutations in genes encoding for sarcomeric,19 desmosomal,18 or metabolic proteins.20–22 Similarly, an autosomal-dominant form of ARVC has been described in Boxers,23,24 and while no mutations in desmosomal genes (known to be associated with ARVC in humans) were found in these dogs, there was loss of gap junction plaques resembling the phenotype found in humans.25 These naturally occurring canine models of cardiomyopathy not only provide a model for testing novel therapies, but (as the gene mutations in these breeds appear different from known gene mutations in humans with DCM and ARVC) also provide an interesting target for genetic screening providing novel genes to be tested, in human forms of DCM and ARVC.
4. Genetic approaches for modelling disease in the mammalian heart
Approximately 30 years ago, techniques became available to perform directed genetic experiments on the mouse and rat using genetic engineering approaches in which direct modifications to the DNA coding sequences could be made. Using either gene targeting via homologous recombination or pronuclear injection of fertilized eggs, it became possible to engineer genetically defined changes in the mouse genome.
As first practiced, transgenesis remained a rather blunt instrument and it quickly became apparent that for meaningful questions to be asked and answered, more precise targeting of transgene's (TG's) expression would be needed. Promoter elements that were able to direct high levels of transcription only in cardiomyocytes were developed for the various stages of cardiac development. These promoters, which were derived from the alpha (Myh6) and beta (Myh7) myosin heavy chain (MHC) genes, were flanked with insulator sequences that shielded their transcriptional activity from the surrounding genetic contexts, making them position independent and copy number dependent in terms of their transcriptional activities. This allowed investigators to perform both developmental stage-specific and dose–response studies and provided the reagents needed to carry out precise TG's expression in the mouse heart28 and rat heart.41 Within months of the first reports of these reagents, other laboratories began to use them and, in the ensuing decade, literally hundreds of laboratories took advantage of these promoters to express proteins of their choice in either the fetal or adult cardiomyocyte populations (Figure 1). Using binary inducible systems, it became possible to reversibly activate a TG's expression in cardiomyocytes.60
Figure 1.
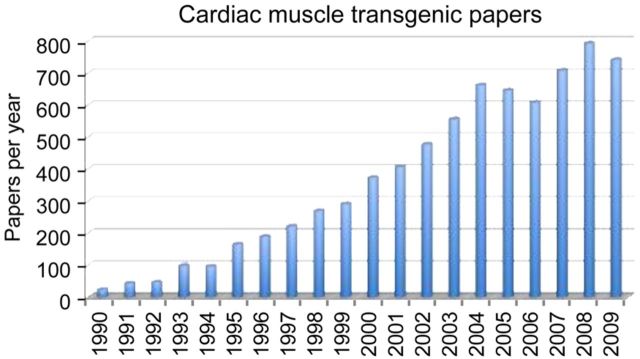
Graph showing the marked increase in the number of published articles using cardiac muscle genetic engineering technology.
While the ability to manipulate the cardiac protein complement with these techniques renders very significant advantages to the mouse as the species of choice for the overwhelming majority of TG's investigations,29–35 the mouse does have limitations in terms of its relevance to human disease. For example, the basic motor protein that underlies contractile force in the sarcomere, the MHC, differs between the mouse and human ventricle. The mouse heart, which beats at ∼600 bpm, expresses the fast cardiac MHC isoform, Myh6, whereas the human ventricle, which beats at approximately 1/10 the rate, expresses the ‘slow’ beta isoform of MHC, Myh7. While closely related, the two isoforms do differ in very fundamental ways and our investigations have shown important differences in the sequences that can limit the murine data's applicability to human cardiovascular function. Indeed, even the most ardent defenders of the mouse note that there are significant differences between the mouse models and human disease presentation. These have been best documented for HCM. While those models show some aspects of the human disease (e.g. premature death, cardiac myocyte disarray, interstitial fibrosis, and diastolic dysfunction), LVH, a defining aspect of human HCM, is rarely present.36 In contrast to regional ventricular systolic dysfunction with maintenance of normal ejection fraction characteristic of human disease, the mouse ventricle is globally impaired.37 In addition to the differences in the motor protein isoform content, the mouse heart is not representative of the human myocardium in other important ways. For example, relative to the mouse, human cardiomyocytes have an increased contribution from the sarcolemmal sodium–calcium exchanger for cytosolic Ca2+ removal, with a lesser contribution from the sarcoplasmic reticulum–Ca2+–ATPase (SERCA) pump.61,62 In contrast, smaller rodents rely more heavily on SERCA2a as the primary mode of Ca2+ removal.61,62
Because of these types of concerns, it became important to develop a larger animal model to validate some of the findings being made in the mouse models. The rabbit offers an experimental model with significant advantages for cardiovascular research. Compared with the mouse, the larger size and slower heart rate of the rabbit are advantageous for physiological analyses such as echocardiography and cardiac catheterization. Importantly, the rabbit ventricles, like the humans, express MYH7 and handle Ca2+ flux in much the same way as is observed for the human heart.
To establish the potential validity of TG methodology for remodelling a larger four-chambered heart, we explored cardiac-selective expression in TG rabbits. The rabbit MYH7 promoter was used to express a reporter gene, and TG's expression was quantified in cardiac, skeletal, and smooth muscles as well as in non-muscle tissues. The promoter was able to drive high levels of TG expression in the cardiac compartment and directed slow myocyte-specific expression, showing that TG manipulation of the cardiomyocyte was possible in the rabbit.42 We then explored the role of the two MHC isoforms in the rabbit ventricle, in order to understand the significance of the small (5–10%) amount of MYH6 that is present in the human ventricle but is down-regulated in heart failure.63 To test the effects of persistent MYH6 expression on the background of MYH7, we made TG rabbits that expressed rabbit MYH6 in the ventricle, so that the endogenous myosin was partially replaced by the transgenically encoded species. We hypothesized that Myh6's unique biochemical properties might offer functional advantages in a failing heart that is normally expressing Myh7. This hypothesis cannot be tested in mice or rats because both species express Myh6 as the predominant isoform.64 Molecular, histological, and functional analyses showed no significant baseline effects in the TG rabbits, compared with non-TG (NTG) littermates. Cohorts of TG and NTG rabbits were subsequently subjected to rapid ventricular pacing. Although both the TG and NTG rabbits developed DCM, the TG rabbits had a higher shortening fraction, less septal thinning, and more normal haemodynamics than paced NTG rabbits. Thus, in a ventricle whose protein complement closely reflects that of the human, MYH6 is cardioprotective in experimental tachycardia-induced cardiomyopathy. These results are now being translated into the clinic and, in a small compound screen, a candidate was selected that was able to specifically increase the ATPase activity of the cardiac myosin motor.65 A phase IIb clinical trial is now underway in which the therapeutic potential of the compound omecamtiv mecarbil® for directly affecting MHC's motor function is being tested.
While cardiomyocyte-specific transgenesis has been widely used, it is less precise than gene targeting. With over 1000 mutations characterized in 10 or more sarcomeric genes, the paucity of gene-targeted models is striking, with only a handful of the mutations being precisely inserted into the relevant gene being reported in the literature. Thus, gene-targeted models for mutations in the MHC,38 cardiac troponin T (cTNT),66 cardiac troponin I,39 and cMyBP-C26,27 have all been reported. For the sarcomeric proteins, transgenesis has proved to be an effective way of mimicking gene-targeted animals, as the cardiomyocyte has post-translational mechanisms for ensuring that sarcomeric protein stoichiometry is maintained. Thus, when a sarcomeric protein is transgenically overexpressed, the net effect is down-regulation and replacement of the normal protein with the transgenically encoded species.28 The relative (to transgenic animals) paucity of gene-targeted animals undoubtedly reflects the time (15–30 months or longer) and expense ($20 000–45 000). However, new technologies have recently become operational in a number of laboratories, and one of the authors' laboratory (J.R.) is now producing genetically modified animals using the CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 technology and is in the process of bringing TALEN methodology online.67,68 These have the significant advantage of being more cost effective and more rapid than the usual gene targeting via homologous recombination, in which the desired homologous recombination events occur extremely infrequently (1 in 106–109 cells).69 The new gene editing technologies use a series of RNA-guided endonucleases along with other components to greatly increase the efficiency of precisely targeted homologous recombination events,70 and mice carrying single-site modifications can be generated in ∼8 months and for a cost of about $4000. With these technologies now available, we think it likely that the number of cardiomyopathy mouse models carrying precisely targeted, single-site mutations will increase exponentially in the next 5 years.
5. The zebrafish as a model to study cardiomyopathies
In two studies published in 2002, zebrafish (Danio rerio) loss-of-function mutants were described in the sarcomeric genes coding for cTnT and titin, both well known for their role in HCM and DCM, respectively.71,72 Since the first description of these zebrafish models for cardiomyopathies a decade ago, the zebrafish has continued to demonstrate its value in studying the genetics of human cardiomyopathies.73–75 Zebrafish embryos have a number of advantages that make them particularly well suited to study the genetics of human cardiomyopathies such as large clutches of eggs that are fertilized externally and an extensive genetic toolbox available to the zebrafish researcher.
Sequencing of the zebrafish genome was completed in 2013, which revealed that 82% of the known human disease-related genes have an orthologous gene in the zebrafish genome.76 Forward genetic screens in zebrafish have been very useful in identifying novel genes required for cardiac development and function,77 among which was the integrin-linked kinase (ilk) gene required for maintaining cardiomyocyte integrity in the embryonic heart.73,78 ILK is located in the Z-disc of cardiomyocytes and when deleted in mouse cardiomyocytes results in DCM, while overexpressing ILK in rat cardiomyocytes can protect against DCM.79,80 Particularly advantageous is the use of zebrafish to decipher the consequence of genetic variants discovered in the genome of cardiomyopathy patients.43 In the case of ILK, for example, this was achieved by rescuing the zebrafish ilk mutant phenotype by injecting synthetic RNA encoding human ILK: while normal ILK RNA rescued efficiently, an ILK variant (c.785C>T [p.Ala262Val]) derived from a DCM patient failed to rescue the zebrafish ilk phenotype.78 Thus far, reverse genetic approaches to disrupt a particular gene-of-interest have employed transient techniques, such as antisense morpholino oligonucleotides.81 This technique relies on the specific blockage of protein translation or mRNA splicing, but can create artefacts due to off-target effects.82 Recently, high-throughput DNA sequencing with chemically induced mutagenesis techniques was initiated, resulting in the identification of potentially disruptive mutations in 38% of all known zebrafish protein-encoding genes.83
A major advantage of zebrafish, compared with other vertebrate models, is that the embryos are transparent allowing high-resolution in vivo observations of the heart.84,85 The transparency of the embryos, combined with genetic engineering approaches where fluorescent proteins are expressed in various cell types of the cardiovascular system, has resulted in improved knowledge of mechanisms that regulate how and when cardiomyocytes differentiate,86 acquire their typical shape,87 or how the myocardium is regenerated after damage.88 The advancement in new microscopy techniques such as light sheet planar illumination microscopy allows high-resolution imaging of the heart during normal or abnormal contraction cycles revealing details about the cellular and sub-cellular level.89 Automated image analysis techniques in combination with high-speed video imaging has been used to extract functional cardiac parameters such as heart rate, arrhythmia index, and ejection fraction from embryos.90
To study gene function in relation to cardiomyopathies, it would be very helpful to study the role of a particular gene only in the heart or even in only one cell type present in the heart, independently from its role in other tissues. Likely, the application of existing technologies such as Cre-recombinase in combination with the recently developed homologous recombination techniques to introduce elements such as loxP-sites at specific locations in the genome will fill in this need.91 The possibility to introduce specific DNA elements by homologous recombination using the TALEN or the CRISPR/Cas9 system opens unexplored territories: it will now be possible to introduce into the zebrafish genome genetic variants that were identified in patients with cardiomyopathies to create patient-specific disease models. These patient-specific models will allow researchers to study the mechanisms by which cardiomyopathies develop and, together with other animal models, will aid in bridging the current gap between patient genotyping and phenotyping. Furthermore, since zebrafish embryos are small (1–2 mm) and take up chemicals from the medium in which they are kept, they are well suited for in vivo chemical screens. In combination with newly developed zebrafish disease models, this opens new possibilities for identifying drugs that can restore cardiac function and may be of benefit to specific groups of patients with a well-defined genetic cardiomyopathy. These new developments ensure that zebrafish will remain a valuable model to study the genetics of cardiomyopathies in the future.
6. The Drosophila heart as a versatile model system to study cardiomyopathy
Since several years, the Drosophila heart has been used as a tool to study various aspects of the heart, such as identification of genes regulating heart development, but also unravelling mechanisms underlying pathophysiology of heart diseases (Figure 2). There are several reasons why the Drosophila heart is such an interesting tool. The Drosophila heart is a linear tube, reminiscent of the primitive vertebrate embryonic heart tube.92 Although the final heart structure in Drosophila is very different compared with that in vertebrates, the basic elements for heart development, function, and ageing are remarkably conserved.93 Heart development is regulated by an evolutionarily conserved gene regulatory network consisting of functional interconnections between myogenic transcription factors (NK-2, MEF2, GATA, Tbx, and Hand), their downstream target genes expressing contractile proteins, and upstream signalling pathways that direct cardiomyocyte differentiation and cardiac morphogenesis.92 In addition to conserved heart development between simple model organisms and vertebrates, an important advantage of Drosophila is the ability to manipulate gene expression in a highly precise spatial and temporal fashion, by the use of a UAS/GAL4 system.94,95 In Drosophila, the UAS/GAL4 system was successfully utilized to identify genes causing human cardiomyopathies.95 Moreover, new techniques such as optical coherence tomography allow accurate phenotyping of cardiac diseases (like stretch and arrhythmia) in flies.95,96 Because of the conserved heart development, the simplicity in structure and availability of powerful genetic tools, the Drosophila heart has emerged as a pioneering model system for unravelling the basic genetic and molecular mechanisms of cardiac development, function, and ageing.93 The Drosophila heart has proved to be a valuable asset to elucidate the pathophysiology of human cardiac diseases, including HCM and DCM, channelopathies, congenital heart disease, as well as cardiac tachycardia, such as atrial fibrillation (AF).44,95,97–99 In addition, the Drosophila heart has also been successfully used for drug and genome-wide screening assays, demonstrating the versatility of the Drosophila heart as a model system.
Figure 2.

Examples of studies utilizing the Drosophila heart model as a tool.
For example, the Drosophila heart has been used to identify druggable targets and to screen for novel small molecules to treat AF.100,101 AF is a serious and progressive tachycardia and is a major cause of stroke as well as a precursor for congestive heart failure and cardiomyopathy.102 AF progression is rooted in structural remodelling, especially sarcomeric protein damage.101,103 To uncover druggable targets against sarcomeric protein damage, the prepupae of Drosophila were subjected to tachypacing by placing them in an electric field.99,101 Tachypacing of the pupae induced structural damage and contractile dysfunction to the cardiomyocytes.99 In addition, the Drosophila heart was recently utilized to uncover the role of epigenetics in AF by screening various HDAC inhibitors.101 This screen revealed that HDAC6 is a druggable target, since the specific HDAC6 inhibitor tubacin protected against tachypacing-induced microtubuline network disruption and contractile dysfunction. Taken together, these studies demonstrate the potential of the Drosophila heart as a tool for druggable target identification and drug screen assays in heart diseases.
Next to the Drosophila as a tool in cardiomyopathies, and druggable target identification, the Drosophila heart has been exploited to verify the outcomes of a human genome-wide association study (GWAS) on genes related to heart rate.46 In this GWAS, 21 loci associated with the heart rate were identified. Experimental down-regulation of gene expression in Drosophila confirmed the relevance of 20 genes at 11 loci for heart rate regulation and highlighted a role for the involved signal transduction routes, embryonic cardiac development and the pathophysiology of DCM, congenital heart failure, and/or sudden cardiac death. The Drosophila findings provide additional mechanistic insights but also identified new therapeutic targets.
Finally, using cardiac-specific RNAi silencing in Drosophila, 7061 evolutionarily conserved genes were knocked down.45 In this way, a first global roadmap of pathways potentially playing conserved roles in the cardiovascular system was elucidated.45 One critical pathway identified was the CCR4-Not complex, which was found to play a key role in cardiac function. Silencing of CCR4-Not components in adult Drosophila resulted in myofibrillar disarray and DCM. Findings were expanded to mouse studies and humans. In Not3 knock-out mice, spontaneous impairment of cardiac contractility and increased susceptibility to heart failure was found. In humans, a common NOT3 SNP was found to correlate with altered cardiac QT intervals, a known cause of potentially lethal ventricular tachyarrhythmias.45 Thus, verification and application of genome-wide screens in Drosophila can identify candidate genes that translate into conserved mammalian genes involved in heart function.
7. Computational approaches in sarcomeric cardiomyopathies
Among the many model systems for studying complex genetic cardiomyopathies, computational approaches are relatively new. The use of computation has arisen in response to both a wide range of basic unanswered biological questions and rapidly growing genetic datasets. To understand the current use and promise of these varied methodologies, it is important to first define the precise role of each approach and where the resultant data fit in an integrative system to fully define the molecular pathogenesis of sarcomeric cardiomyopathies. As was noted in the recent ACC/AHA guidelines for the management of HCM (the most common genetic form of cardiomyopathy), the identity of individual sarcomeric mutations cannot be used in the clinical management of patients.104 For a disorder that was first identified as having a genetic cause in 1990, this is a sobering state of affairs and represents a crucial roadblock to the development of genotype-driven management.105 Although the issues that contribute to this limitation are multifactorial and beyond the scope of the current discussion (for review see ref.106), there is clearly a pressing need for novel approaches that directly address the primary mechanisms that define the molecular basis of this ‘disease of the cardiac sarcomere’. Given that >1400 mutations have been linked to sarcomeric cardiomyopathies to date, computational approaches are a compelling answer to this growing challenge.
The development of computational model systems has focused on several central unmet needs in the study of sarcomeric cardiomyopathies. In particular, the growing availability of genetic testing (leading to a rapidly growing number of putative disease mutations) and the fact that many sarcomeric mutations arise de novo combine to vastly complicate the most basic question of whether a single amino acid substitution in a protein component of the cardiac sarcomere is sufficient to cause disease in an affected individual. For the practicing cardiologist, this is the ‘raison d’être’ for submitting patient samples for testing. The sheer number of proteins affected, the many possible mutations and the limitations in identifying comprehensive, accessible and reproducible in vitro measures of protein function has led to the development of a host of algorithmic approaches for predicting allele ‘pathogenicity’.107 For sarcomeric cardiomyopathies, the two most commonly utilized are PolyPhen-2 and ‘Sorting Tolerant from Intolerant’ (SIFT).108,109 PolyPhen-2 is a robust algorithm that mines known protein databases for existing information regarding local structure, known, or predicted molecular function (e.g. binding domains, salt bridge formation, and hydrophobicity) and, combined with overall amino acid conservation maps, develops a template to determine the potential effects of amino acid substitutions. For proteins with known 3D structural information, it is possible to incorporate secondary and tertiary structures to assess allelic effects on intramolecular interactions, thus adding to the fidelity of the predictions. SIFT serves a similar role and utilizes a comprehensive approach that focuses on amino acid conservation compiled from known protein structure and a robust use of homology mapping to extend predictions beyond what is known for a given protein. Both algorithms have been optimized for use with large datasets and are also extensively used in industry and basic research to predict the structural and potentially functional effects of individual substitutions (and more recently indels) in sarcomeric proteins.
The application of the database-driven algorithms to predict primary pathogenicity is a reasonable approach; from the mechanistic standpoint, however, sarcomeric proteins present a unique challenge in that they are components of a complex multisubunit ‘machine’.106 Moreover, the basic function of this machine is highly dynamic and fully dependent on allosteric interactions, Ca2+ regulation, and load and post-translational modifications, conditions that cannot be easily incorporated into models based on static structures of single proteins. Thus, from the computational standpoint, it is necessary to develop more comprehensive, biologically complex models that can incorporate structural and functional information across multiple levels of experimental resolution with a goal of developing a coupled system that can be validated from the single molecule to the whole organ level.110 While multiscale modelling has yet to be applied to sarcomeric mutations, its promise was recently demonstrated by Sheikh et al.,111 where computational modelling of skinned fibre data suggested a novel two-tiered role for myosin light chain 2v phosphorylation in regulating cardiac muscle contraction and potentially modulating preclinical remodelling in a murine DCM model. As illustrated by this study, it is important to note that a major role for computational modelling going forward is to shed light on potential molecular mechanisms that would not be detected via standard, low-resolution experiments and provide unique, testable hypotheses for discovery and validation.
Given the complexity of the cardiac sarcomere, a crucial component of the multiscaled approach is the ability to define atomic-level changes in protein dynamics, both intra- and intermolecular. Alterations in protein dynamics likely represent an important proximal manifestation of sarcomeric mutations at the molecular level and can be addressed computationally via molecular dynamics (MD) simulations. MD is based on an atom-level determination of the time dependence of molecular interactions.112 It is well suited to the study of dynamic biological systems like the cardiac thin filament because of its scalability and the ability to map atomic motions over predicted trajectories for a given unit of time, resulting in a high-resolution ‘window’ into both the local and distant effects of contractile protein mutations. The first application of MD to sarcomeric cardiomyopathies was performed by Ertz-Berger et al.,113 and focused on the effects of known single amino acid substitutions at Residue 92 in cTnT. Results from this and a subsequent study revealed mutation-specific alterations in local protein flexibility and, importantly, for the first time established that thin filament mutations within alpha-helical regions can lead to a propagation of structural and presumably functional effects at a distance from the mutated residue.114 Similar MD results were observed by Li et al.115 for the tropomyosin Asp175Asn and Glu180Gly mutants, in that both were found to have effects on local and global flexibility. The findings regarding propagation effects are particularly important. First, the understanding that changes in structure and dynamics at the primary mutational site may not be the sole effect of a given mutation broadens our functional interpretation of mutational effects. Secondly, independent mutations at significant distances from each other may propagate structural effects to the same functional domain, thus providing a novel approach to define groups of mutations. The latter observation provides a framework for the design of therapeutic small molecules that can be used for an array of mutations.
There are many exciting new computational approaches on the horizon. In particular, as shown by Manning et al.,116 the development of a full all-atom model of the cardiac thin filament, coupled with advances in computing power, will allow for an ever more detailed understanding of the most proximal molecular causes of disease. As computational powers grow (for example, via the inclusion of graphic processing units or GPUs), it becomes conceivable that a fully atomistic model of the thin filament in explicit solvent will be available. That coupled with a more detailed understanding of chemistry in the thick filament will serve as an input to higher-level multiscale models that will provide an fully integrative, iterative, and eventually predictive approach to understanding these common genetic cardiomyopathies.
8. Concluding remarks and future perspectives
Over the past decade, our understanding of cardiomyopathies has improved dramatically, which is not only due to improvements in screening and detection of gene defects in the human genome, but also to the availability of a variety of novel animal and in silico computational models. These novel experimental tools have created a platform that is highly complementary to the naturally occurring cardiomyopathies in cats and dogs that had been available for some time. It is likely that a fully integrative approach, which incorporates all these modalities, is required for major steps forward into understanding the molecular underpinnings and pathogenesis of cardiomyopathies. Finally, the recent development of new technologies, such as CRISPR/Cas9, which has already been proved to work in zebrafish and in mice, is currently being employed to engineer sarcomeric cardiomyopathy in larger animals, including pigs and non-human primates. In the mouse, the increased speed with which these techniques can be employed to engineer precise ‘knock-in’ models that previously took years to make via multiple rounds of homologous recombination-based gene targeting promises multiple and precise models of human cardiac disease for future study. Such novel genetically engineered animal models recapitulating human sarcomeric protein defects will help bridging the gap to translate therapeutic targets from smaller animal and in silico models to the human patient with sarcomeric cardiomyopathy.
Funding
D.D. is supported by grants from the Netherlands CardioVascular Research Initiative (CVON-ARENA CVON2011-11) and from the European Commission (FP7-Health-2010; MEDIA-261409). J.B. is supported by the Netherlands CardioVascular Research Initiative grant (CVON-Hustcare CVON2011-12). B.B. is supported by the Dutch Heart Foundation (2013T096 and 2013T144) and LSH-Impulse grant (40-43100-98-008). J.R. is supported by the National Institutes of Health (grants P01HL69779, P01HL059408, R01HL05924, and R011062927) and by a Trans-Atlantic Network of Excellence grant from La Fondation Leducq (Research grant no. 11, CVD 04). J.T. is supported by the National Institutes of Health (grants HL075619 and HL107046). L.C. is supported by grants from the DZHK (German Centre for Cardiovascular Research) and the German Ministry of Research and Education (BMBF), La Fondation Leducq (Research grant no. 11, CVD 04), and Association Institut de Myologie (Paris).
Acknowledgements
J.B. thanks E. Noël and F. Tessadori for critical reading of the manuscript.
Conflict of interest: none declared.
References
- 1.Rapezzi C, Arbustini E, Caforio AL, Charron P, Gimeno-Blanes J, Helio T, Linhart A, Mogensen J, Pinto Y, Ristic A, Seggewiss H, Sinagra G, Tavazzi L, Elliott PM. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34:1448–1458. doi: 10.1093/eurheartj/ehs397. [DOI] [PubMed] [Google Scholar]
- 2.Dixon JA, Spinale FG. Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail. 2009;2:262–271. doi: 10.1161/CIRCHEARTFAILURE.108.814459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duncker DJ, Zhang J, Bache RJ. Coronary pressure-flow relation in left ventricular hypertrophy. Importance of changes in back pressure versus changes in minimum resistance. Circ Res. 1993;72:579–587. doi: 10.1161/01.res.72.3.579. [DOI] [PubMed] [Google Scholar]
- 4.Duncker DJ, Boontje NM, Merkus D, Versteilen A, Krysiak J, Mearini G, El-Armouche A, de Beer VJ, Lamers JM, Carrier L, Walker LA, Linke WA, Stienen GJ, van der Velden J. Prevention of myofilament dysfunction by beta-blocker therapy in postinfarct remodeling. Circ Heart Fail. 2009;2:233–242. doi: 10.1161/CIRCHEARTFAILURE.108.806125. [DOI] [PubMed] [Google Scholar]
- 5.Kuster DW, Merkus D, van der Velden J, Verhoeven AJ, Duncker DJ. ‘Integrative Physiology 2.0’: integration of systems biology into physiology and its application to cardiovascular homeostasis. J Physiol. 2011;589:1037–1045. doi: 10.1113/jphysiol.2010.201533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA, Koch WJ American Heart Association Council on Basic Cardiovascular Sciences CoCC, Council on Functional Genomics, Translational Biology. Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res. 2012;111:131–150. doi: 10.1161/RES.0b013e3182582523. [DOI] [PubMed] [Google Scholar]
- 7.Kittleson MD, Meurs KM, Munro KJ, Liu SK, Pion PD. Familial hypertrophic cardiomyopathy in Maine Coon cats: an animal model of human disease. Circulation. 1999;99:8. doi: 10.1161/01.cir.99.24.3172. [DOI] [PubMed] [Google Scholar]
- 8.Meurs KM, Norgard MM, Ederer MM, Hendrix KP, Kittleson MD. A substitution mutation in the myosin binding protein C gene in ragdoll hypertrophic cardiomyopathy. Genomics. 2007;90:5. doi: 10.1016/j.ygeno.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Meurs KM, Sanchez X, David RM, Bowles NE, Towbin JA, Reiser PJ, Kittleson JA, Munro MJ, Dryburgh K, Macdonald KA, Kittleson MD. A cardiac myosin binding protein C mutation in the Maine Coon cat with familial hypertrophic cardiomyopathy. Hum Mol Genet. 2005;14:3587–3593. doi: 10.1093/hmg/ddi386. [DOI] [PubMed] [Google Scholar]
- 10.Longeri M, Ferrari P, Knafelz P, Mezzelani A, Marabotti A, Milanesi L, Pertica G, Polli M, Brambilla PG, Kittleson M, Lyons LA, Porciello F. Myosin-binding protein C DNA variants in domestic cats (A31P, A74T, R820W) and their association with hypertrophic cardiomyopathy. J Vet Intern Med. 2013;27:275–285. doi: 10.1111/jvim.12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fries R, Heaney AM, Meurs KM. Prevalence of the myosin-binding protein C mutation in Maine Coon cats. J Vet Intern Med. 2008;22:893–896. doi: 10.1111/j.1939-1676.2008.0113.x. [DOI] [PubMed] [Google Scholar]
- 12.Carlos Sampedrano C, Chetboul V, Mary J, Tissier R, Abitbol M, Serres F, Gouni V, Thomas A, Pouchelon JL. Prospective echocardiographic and tissue Doppler imaging screening of a population of Maine Coon cats tested for the A31P mutation in the myosin-binding protein C gene: a specific analysis of the heterozygous status. J Vet Intern Med. 2009;23:91–99. doi: 10.1111/j.1939-1676.2008.0218.x. [DOI] [PubMed] [Google Scholar]
- 13.Sleeper MM, Bish LT, Sweeney HL. Gene therapy in large animal models of human cardiovascular genetic disease. ILAR J. 2009;50:199–205. doi: 10.1093/ilar.50.2.199. [DOI] [PubMed] [Google Scholar]
- 14.Tidholm A, Haggstrom J, Borgarelli M, Tarducci A. Canine idiopathic dilated cardiomyopathy. Part I: aetiology, clinical characteristics, epidemiology and pathology. Vet J. 2001;162:92–107. doi: 10.1053/tvjl.2001.0571. [DOI] [PubMed] [Google Scholar]
- 15.Dambach DM, Lannon A, Sleeper MM, Buchanan J. Familial dilated cardiomyopathy of young Portuguese water dogs. J Vet Intern Med. 1999;13:65–71. [PubMed] [Google Scholar]
- 16.Meurs KM, Miller MW, Wright NA. Clinical features of dilated cardiomyopathy in Great Danes and results of a pedigree analysis: 17 cases (1990–2000) J Am Vet Med Assoc. 2001;218:729–732. doi: 10.2460/javma.2001.218.729. [DOI] [PubMed] [Google Scholar]
- 17.Wess G, Schulze A, Butz V, Simak J, Killich M, Keller LJ, Maeurer J, Hartmann K. Prevalence of dilated cardiomyopathy in Doberman Pinschers in various age groups. J Vet Intern Med. 2010;24:533–538. doi: 10.1111/j.1939-1676.2010.0479.x. [DOI] [PubMed] [Google Scholar]
- 18.Meurs KM, Stern JA, Sisson DD, Kittleson MD, Cunningham SM, Ames MK, Atkins CE, DeFrancesco T, Hodge TE, Keene BW, Reina Doreste Y, Leuthy M, Motsinger-Reif AA, Tou SP. Association of dilated cardiomyopathy with the striatin mutation genotype in boxer dogs. J Vet Intern Med. 2013;27:1437–1440. doi: 10.1111/jvim.12163. [DOI] [PubMed] [Google Scholar]
- 19.O'Sullivan ML, O'Grady MR, Pyle WG, Dawson JF. Evaluation of 10 genes encoding cardiac proteins in Doberman Pinschers with dilated cardiomyopathy. Am J Vet Res. 2011;72:932–939. doi: 10.2460/ajvr.72.7.932. [DOI] [PubMed] [Google Scholar]
- 20.Meurs KM, Lahmers S, Keene BW, White SN, Oyama MA, Mauceli E, Lindblad-Toh K. A splice site mutation in a gene encoding for PDK4, a mitochondrial protein, is associated with the development of dilated cardiomyopathy in the Doberman pinscher. Hum Genet. 2012;131:1319–1325. doi: 10.1007/s00439-012-1158-2. [DOI] [PubMed] [Google Scholar]
- 21.Owczarek-Lipska M, Mausberg TB, Stephenson H, Dukes-McEwan J, Wess G, Leeb T. A 16-bp deletion in the canine PDK4 gene is not associated with dilated cardiomyopathy in a European cohort of Doberman Pinschers. Anim Genet. 2013;44:239. doi: 10.1111/j.1365-2052.2012.02396.x. [DOI] [PubMed] [Google Scholar]
- 22.Philipp U, Vollmar A, Haggstrom J, Thomas A, Distl O. Multiple loci are associated with dilated cardiomyopathy in Irish wolfhounds. PLoS ONE. 2012;7:e36691. doi: 10.1371/journal.pone.0036691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basso C, Fox PR, Meurs KM, Towbin JA, Spier AW, Calabrese F, Maron BJ, Thiene G. Arrhythmogenic right ventricular cardiomyopathy causing sudden cardiac death in boxer dogs: a new animal model of human disease. Circulation. 2004;109:1180–1185. doi: 10.1161/01.CIR.0000118494.07530.65. [DOI] [PubMed] [Google Scholar]
- 24.Meurs KM, Lacombe VA, Dryburgh K, Fox PR, Reiser PR, Kittleson MD. Differential expression of the cardiac ryanodine receptor in normal and arrhythmogenic right ventricular cardiomyopathy canine hearts. Hum Genet. 2006;120:111–118. doi: 10.1007/s00439-006-0193-2. [DOI] [PubMed] [Google Scholar]
- 25.Oxford EM, Everitt M, Coombs W, Fox PR, Kraus M, Gelzer AR, Saffitz J, Taffet SM, Moise NS, Delmar M. Molecular composition of the intercalated disc in a spontaneous canine animal model of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm. 2007;4:1196–1205. doi: 10.1016/j.hrthm.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fraysse B, Weinberger F, Bardswell SC, Cuello F, Vignier N, Geertz B, Starbatty J, Kramer E, Coirault C, Eschenhagen T, Kentish JC, Avkiran M, Carrier L. Increased myofilament Ca2+ sensitivity and diastolic dysfunction as early consequences of Mybpc3 mutation in heterozygous knock-in mice. J Mol Cell Cardiol. 2012;52:1299–1307. doi: 10.1016/j.yjmcc.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vignier N, Schlossarek S, Fraysse B, Mearini G, Kramer E, Pointu H, Mougenot N, Guiard J, Reimer R, Hohenberg H, Schwartz K, Vernet M, Eschenhagen T, Carrier L. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ Res. 2009;105:239–248. doi: 10.1161/CIRCRESAHA.109.201251. [DOI] [PubMed] [Google Scholar]
- 28.Robbins J. Remodeling the cardiac sarcomere using transgenesis. Annu Rev Physiol. 2000;62:261–287. doi: 10.1146/annurev.physiol.62.1.261. [DOI] [PubMed] [Google Scholar]
- 29.Carrier L, Knoll R, Vignier N, Keller DI, Bausero P, Prudhon B, Isnard R, Ambroisine ML, Fiszman M, Ross J, Jr, Schwartz K, Chien KR. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc Res. 2004;63:293–304. doi: 10.1016/j.cardiores.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 30.Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 2011;108:751–764. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.James J, Zhang Y, Osinska H, Sanbe A, Klevitsky R, Hewett TE, Robbins J. Transgenic modeling of a cardiac troponin I mutation linked to familial hypertrophic cardiomyopathy. Circ Res. 2000;87:805–811. doi: 10.1161/01.res.87.9.805. [DOI] [PubMed] [Google Scholar]
- 32.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1235–1244. doi: 10.1172/JCI7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muthuchamy M, Pieples K, Rethinasamy P, Hoit B, Grupp IL, Boivin GP, Wolska B, Evans C, Solaro RJ, Wieczorek DF. Mouse model of a familial hypertrophic cardiomyopathy mutation in alpha-tropomyosin manifests cardiac dysfunction. Circ Res. 1999;85:47–56. doi: 10.1161/01.res.85.1.47. [DOI] [PubMed] [Google Scholar]
- 34.Tardiff JC, Factor SM, Tompkins BD, Hewett TE, Palmer BM, Moore RL, Schwartz S, Robbins J, Leinwand LA. A truncated cardiac troponin T molecule in transgenic mice suggests multiple cellular mechanisms for familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:2800–2811. doi: 10.1172/JCI2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J Clin Invest. 1998;102:1292–1300. doi: 10.1172/JCI3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tardiff JC, Hewett TE, Palmer BM, Olsson C, Factor SM, Moore RL, Robbins J, Leinwand LA. Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J Clin Invest. 1999;104:469–481. doi: 10.1172/JCI6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. In vivo modeling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ Res. 1999;85:841–847. doi: 10.1161/01.res.85.9.841. [DOI] [PubMed] [Google Scholar]
- 38.Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–734. doi: 10.1126/science.272.5262.731. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Pinto JR, Solis RS, Dweck D, Liang J, Diaz-Perez Z, Ge Y, Walker JW, Potter JD. Generation and functional characterization of knock-in mice harboring the cardiac troponin I-R21C mutation associated with hypertrophic cardiomyopathy. J Biol Chem. 2012;287:2156–2167. doi: 10.1074/jbc.M111.294306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Javadpour MM, Tardiff JC, Pinz I, Ingwall JS. Decreased energetics in murine hearts bearing the R92Q mutation in cardiac troponin T. J Clin Invest. 2003;112:768–775. doi: 10.1172/JCI15967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frey N, Franz WM, Gloeckner K, Degenhardt M, Muller M, Muller O, Merz H, Katus HA. Transgenic rat hearts expressing a human cardiac troponin T deletion reveal diastolic dysfunction and ventricular arrhythmias. Cardiovasc Res. 2000;47:254–264. doi: 10.1016/s0008-6363(00)00114-0. [DOI] [PubMed] [Google Scholar]
- 42.Sanbe A, James J, Tuzcu V, Nas S, Martin L, Gulick J, Osinska H, Sakthivel S, Klevitsky R, Ginsburg KS, Bers DM, Zinman B, Lakatta EG, Robbins J. Transgenic rabbit model for human troponin I-based hypertrophic cardiomyopathy. Circulation. 2005;111:2330–2338. doi: 10.1161/01.CIR.0000164234.24957.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodatsu A, Konno T, Hayashi K, Funada A, Fujita T, Nagata Y, Fujino N, Kawashiri MA, Yamagishi M. Compound heterozygosity deteriorates phenotypes of hypertrophic cardiomyopathy with founder MYBPC3 mutation: evidence from patients and zebrafish models. Am J Physiol Heart Circ Physiol. 2014;307:H1594–H1604. doi: 10.1152/ajpheart.00637.2013. [DOI] [PubMed] [Google Scholar]
- 44.Viswanathan MC, Kaushik G, Engler AJ, Lehman W, Cammarato A. A Drosophila melanogaster model of diastolic dysfunction and cardiomyopathy based on impaired troponin-T function. Circ Res. 2014;114:e6–17. doi: 10.1161/CIRCRESAHA.114.302028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neely GG, Kuba K, Cammarato A, Isobe K, Amann S, Zhang L, Murata M, Elmen L, Gupta V, Arora S, Sarangi R, Dan D, Fujisawa S, Usami T, Xia CP, Keene AC, Alayari NN, Yamakawa H, Elling U, Berger C, Novatchkova M, Koglgruber R, Fukuda K, Nishina H, Isobe M, Pospisilik JA, Imai Y, Pfeufer A, Hicks AA, Pramstaller PP, Subramaniam S, Kimura A, Ocorr K, Bodmer R, Penninger JM. A global in vivo Drosophila RNAi screen identifies NOT3 as a conserved regulator of heart function. Cell. 2010;141:142–153. doi: 10.1016/j.cell.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.den Hoed M, Eijgelsheim M, Esko T, Brundel BJ, Peal DS, Evans DM, Nolte IM, Segre AV, Holm H, Handsaker RE, Westra HJ, Johnson T, Isaacs A, Yang J, Lundby A, Zhao JH, Kim YJ, Go MJ, Almgren P, Bochud M, Boucher G, Cornelis MC, Gudbjartsson D, Hadley D, van der Harst P, Hayward C, den Heijer M, Igl W, Jackson AU, Kutalik Z, Luan J, Kemp JP, Kristiansson K, Ladenvall C, Lorentzon M, Montasser ME, Njajou OT, O'Reilly PF, Padmanabhan S, St Pourcain B, Rankinen T, Salo P, Tanaka T, Timpson NJ, Vitart V, Waite L, Wheeler W, Zhang W, Draisma HH, Feitosa MF, Kerr KF, Lind PA, Mihailov E, Onland-Moret NC, Song C, Weedon MN, Xie W, Yengo L, Absher D, Albert CM, Alonso A, Arking DE, de Bakker PI, Balkau B, Barlassina C, Benaglio P, Bis JC, Bouatia-Naji N, Brage S, Chanock SJ, Chines PS, Chung M, Darbar D, Dina C, Dorr M, Elliott P, Felix SB, Fischer K, Fuchsberger C, de Geus EJ, Goyette P, Gudnason V, Harris TB, Hartikainen AL, Havulinna AS, Heckbert SR, Hicks AA, Hofman A, Holewijn S, Hoogstra-Berends F, Hottenga JJ, Jensen MK, Johansson A, Junttila J, Kaab S, Kanon B, Ketkar S, Khaw KT, Knowles JW, Kooner AS, Kors JA, Kumari M, Milani L, Laiho P, Lakatta EG, Langenberg C, Leusink M, Liu Y, Luben RN, Lunetta KL, Lynch SN, Markus MR, Marques-Vidal P, Mateo Leach I, McArdle WL, McCarroll SA, Medland SE, Miller KA, Montgomery GW, Morrison AC, Muller-Nurasyid M, Navarro P, Nelis M, O'Connell JR, O'Donnell CJ, Ong KK, Newman AB, Peters A, Polasek O, Pouta A, Pramstaller PP, Psaty BM, Rao DC, Ring SM, Rossin EJ, Rudan D, Sanna S, Scott RA, Sehmi JS, Sharp S, Shin JT, Singleton AB, Smith AV, Soranzo N, Spector TD, Stewart C, Stringham HM, Tarasov KV, Uitterlinden AG, Vandenput L, Hwang SJ, Whitfield JB, Wijmenga C, Wild SH, Willemsen G, Wilson JF, Witteman JC, Wong A, Wong Q, Jamshidi Y, Zitting P, Boer JM, Boomsma DI, Borecki IB, van Duijn CM, Ekelund U, Forouhi NG, Froguel P, Hingorani A, Ingelsson E, Kivimaki M, Kronmal RA, Kuh D, Lind L, Martin NG, Oostra BA, Pedersen NL, Quertermous T, Rotter JI, van der Schouw YT, Verschuren WM, Walker M, Albanes D, Arnar DO, Assimes TL, Bandinelli S, Boehnke M, de Boer RA, Bouchard C, Caulfield WL, Chambers JC, Curhan G, Cusi D, Eriksson J, Ferrucci L, van Gilst WH, Glorioso N, de Graaf J, Groop L, Gyllensten U, Hsueh WC, Hu FB, Huikuri HV, Hunter DJ, Iribarren C, Isomaa B, Jarvelin MR, Jula A, Kahonen M, Kiemeney LA, van der Klauw MM, Kooner JS, Kraft P, Iacoviello L, Lehtimaki T, Lokki ML, Mitchell BD, Navis G, Nieminen MS, Ohlsson C, Poulter NR, Qi L, Raitakari OT, Rimm EB, Rioux JD, Rizzi F, Rudan I, Salomaa V, Sever PS, Shields DC, Shuldiner AR, Sinisalo J, Stanton AV, Stolk RP, Strachan DP, Tardif JC, Thorsteinsdottir U, Tuomilehto J, van Veldhuisen DJ, Virtamo J, Viikari J, Vollenweider P, Waeber G, Widen E, Cho YS, Olsen JV, Visscher PM, Willer C, Franke L, Global BC, Consortium CA, Erdmann J, Thompson JR, Consortium PG, Pfeufer A, Consortium QG, Sotoodehnia N, Consortium Q-I, Newton-Cheh C, Consortium C-A, Ellinor PT, Stricker BH, Metspalu A, Perola M, Beckmann JS, Smith GD, Stefansson K, Wareham NJ, Munroe PB, Sibon OC, Milan DJ, Snieder H, Samani NJ, Loos RJ. Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat Genet. 2013;45:621–631. doi: 10.1038/ng.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michels M, Soliman OI, Kofflard MJ, Hoedemaekers YM, Dooijes D, Majoor-Krakauer D, ten Cate FJ. Diastolic abnormalities as the first feature of hypertrophic cardiomyopathy in Dutch myosin-binding protein C founder mutations. JACC Cardiovasc Imaging. 2009;2:58–64. doi: 10.1016/j.jcmg.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 48.Ho CY, Carlsen C, Thune JJ, Havndrup O, Bundgaard H, Farrohi F, Rivero J, Cirino AL, Andersen PS, Christiansen M, Maron BJ, Orav EJ, Kober L. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:314–321. doi: 10.1161/CIRCGENETICS.109.862128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 50.van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 51.van Dijk SJ, Paalberends ER, Najafi A, Michels M, Sadayappan S, Carrier L, Boontje NM, Kuster DW, van Slegtenhorst M, Dooijes D, dos Remedios C, ten Cate FJ, Stienen GJ, van der Velden J. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ Heart Fail. 2012;5:36–46. doi: 10.1161/CIRCHEARTFAILURE.111.963702. [DOI] [PubMed] [Google Scholar]
- 52.Bahrudin U, Morisaki H, Morisaki T, Ninomiya H, Higaki K, Nanba E, Igawa O, Takashima S, Mizuta E, Miake J, Yamamoto Y, Shirayoshi Y, Kitakaze M, Carrier L, Hisatome I. Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J Mol Biol. 2008;384:896–907. doi: 10.1016/j.jmb.2008.09.070. [DOI] [PubMed] [Google Scholar]
- 53.Sarikas A, Carrier L, Schenke C, Doll D, Flavigny J, Lindenberg KS, Eschenhagen T, Zolk O. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res. 2005;66:33–44. doi: 10.1016/j.cardiores.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Schlossarek S, Englmann DR, Sultan KR, Sauer M, Eschenhagen T, Carrier L. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res Cardiol. 2012;107:235. doi: 10.1007/s00395-011-0235-3. [DOI] [PubMed] [Google Scholar]
- 55.Schlossarek S, Frey N, Carrier L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J Mol Cell Cardiol. 2014;71:25–31. doi: 10.1016/j.yjmcc.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 56.Schlossarek S, Schuermann F, Geertz B, Mearini G, Eschenhagen T, Carrier L. Adrenergic stress reveals septal hypertrophy and proteasome impairment in heterozygous Mybpc3-targeted knock-in mice. J Muscle Res Cell Motil. 2012;33:5–15. doi: 10.1007/s10974-011-9273-6. [DOI] [PubMed] [Google Scholar]
- 57.Gedicke-Hornung C, Behrens-Gawlik V, Reischmann S, Geertz B, Stimpel D, Weinberger F, Schlossarek S, Precigout G, Braren I, Eschenhagen T, Mearini G, Lorain S, Voit T, Dreyfus PA, Garcia L, Carrier L. Rescue of cardiomyopathy through U7snRNA-mediated exon skipping in Mybpc3-targeted knock-in mice. EMBO Mol Med. 2013;5:1060–1077. doi: 10.1002/emmm.201202168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mearini G, Stimpel D, Kramer E, Geertz B, Braren I, Gedicke-Hornung C, Precigout G, Muller OJ, Katus HA, Eschenhagen T, Voit T, Garcia L, Lorain S, Carrier L. Repair of Mybpc3 mRNA by 5′-trans-splicing in a mouse model of hypertrophic cardiomyopathy. Mol Ther Nucleic Acids. 2013;2:e102. doi: 10.1038/mtna.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mearini G, Stimpel D, Geertz B, Weinberger F, Krämer E, Schlossarek S, Mourot-Filiatre J, Stöhr A, Dutshc A, Wijnker PJM, Braren I, Katus HA, Müller OJ, Voit T, Eschenhagen T, Carrier L. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat Commun. 2014;5:5515. doi: 10.1038/ncomms6515. [DOI] [PubMed] [Google Scholar]
- 60.Gulick J, Robbins J. Regulation of Transgene Expression Using Tetracycline. Curr Protoc Mol Biol. 2005. doi:10.1002/0471142727.mb2312s71. [DOI] [PubMed]
- 61.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 62.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 63.Reiser PJ, Portman MA, Ning XH, Schomisch Moravec C. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am J Physiol Heart Circ Physiol. 2001;280:H1814–H1820. doi: 10.1152/ajpheart.2001.280.4.H1814. [DOI] [PubMed] [Google Scholar]
- 64.Dillmann WH. Hormonal influences on cardiac myosin ATPase activity and myosin isoenzyme distribution. Mol Cell Endocrinol. 1984;34:169–181. doi: 10.1016/0303-7207(84)90173-4. [DOI] [PubMed] [Google Scholar]
- 65.Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, Anderson RL, Sueoka SH, Lee KH, Finer JT, Sakowicz R, Baliga R, Cox DR, Garard M, Godinez G, Kawas R, Kraynack E, Lenzi D, Lu PP, Muci A, Niu C, Qian X, Pierce DW, Pokrovskii M, Suehiro I, Sylvester S, Tochimoto T, Valdez C, Wang W, Katori T, Kass DA, Shen YT, Vatner SF, Morgans DJ. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331:1439–1443. doi: 10.1126/science.1200113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Du CK, Morimoto S, Nishii K, Minakami R, Ohta M, Tadano N, Lu QW, Wang YY, Zhan DY, Mochizuki M, Kita S, Miwa Y, Takahashi-Yanaga F, Iwamoto T, Ohtsuki I, Sasaguri T. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ Res. 2007;101:185–194. doi: 10.1161/CIRCRESAHA.106.146670. [DOI] [PubMed] [Google Scholar]
- 67.Charpentier E, Marraffini LA. Harnessing CRISPR-Cas9 immunity for genetic engineering. Curr Opin Microbiol. 2014;19C:114–119. doi: 10.1016/j.mib.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- 69.Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 70.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sehnert AJ, Huq A, Weinstein BM, Walker C, Fishman M, Stainier DY. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat Genet. 2002;31:106–110. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- 72.Xu X, Meiler SE, Zhong TP, Mohideen M, Crossley DA, Burggren WW, Fishman MC. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat Genet. 2002;30:205–209. doi: 10.1038/ng816. [DOI] [PubMed] [Google Scholar]
- 73.Bendig G, Grimmler M, Huttner IG, Wessels G, Dahme T, Just S, Trano N, Katus HA, Fishman MC, Rottbauer W. Integrin-linked kinase, a novel component of the cardiac mechanical stretch sensor, controls contractility in the zebrafish heart. Genes Dev. 2006;20:2361–2372. doi: 10.1101/gad.1448306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dhandapany PS, Razzaque MA, Muthusami U, Kunnoth S, Edwards JJ, Mulero-Navarro S, Riess I, Pardo S, Sheng J, Rani DS, Rani B, Govindaraj P, Flex E, Yokota T, Furutani M, Nishizawa T, Nakanishi T, Robbins J, Limongelli G, Hajjar RJ, Lebeche D, Bahl A, Khullar M, Rathinavel A, Sadler KC, Tartaglia M, Matsuoka R, Thangaraj K, Gelb BD. RAF1 mutations in childhood-onset dilated cardiomyopathy. Nat Genet. 2014;46:635–639. doi: 10.1038/ng.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang H, Li Z, Wang J, Sun K, Cui Q, Song L, Zou Y, Wang X, Liu X, Hui R, Fan Y. Mutations in NEXN, a Z-disc gene, are associated with hypertrophic cardiomyopathy. Am J Hum Genet. 2010;87:687–693. doi: 10.1016/j.ajhg.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, McLaren S, Sealy I, Caccamo M, Churcher C, Scott C, Barrett JC, Koch R, Rauch GJ, White S, Chow W, Kilian B, Quintais LT, Guerra-Assuncao JA, Zhou Y, Gu Y, Yen J, Vogel JH, Eyre T, Redmond S, Banerjee R, Chi J, Fu B, Langley E, Maguire SF, Laird GK, Lloyd D, Kenyon E, Donaldson S, Sehra H, Almeida-King J, Loveland J, Trevanion S, Jones M, Quail M, Willey D, Hunt A, Burton J, Sims S, McLay K, Plumb B, Davis J, Clee C, Oliver K, Clark R, Riddle C, Elliot D, Threadgold G, Harden G, Ware D, Begum S, Mortimore B, Kerry G, Heath P, Phillimore B, Tracey A, Corby N, Dunn M, Johnson C, Wood J, Clark S, Pelan S, Griffiths G, Smith M, Glithero R, Howden P, Barker N, Lloyd C, Stevens C, Harley J, Holt K, Panagiotidis G, Lovell J, Beasley H, Henderson C, Gordon D, Auger K, Wright D, Collins J, Raisen C, Dyer L, Leung K, Robertson L, Ambridge K, Leongamornlert D, McGuire S, Gilderthorp R, Griffiths C, Manthravadi D, Nichol S, Barker G, Whitehead S, Kay M, Brown J, Murnane C, Gray E, Humphries M, Sycamore N, Barker D, Saunders D, Wallis J, Babbage A, Hammond S, Mashreghi-Mohammadi M, Barr L, Martin S, Wray P, Ellington A, Matthews N, Ellwood M, Woodmansey R, Clark G, Cooper J, Tromans A, Grafham D, Skuce C, Pandian R, Andrews R, Harrison E, Kimberley A, Garnett J, Fosker N, Hall R, Garner P, Kelly D, Bird C, Palmer S, Gehring I, Berger A, Dooley CM, Ersan-Urun Z, Eser C, Geiger H, Geisler M, Karotki L, Kirn A, Konantz J, Konantz M, Oberlander M, Rudolph-Geiger S, Teucke M, Lanz C, Raddatz G, Osoegawa K, Zhu B, Rapp A, Widaa S, Langford C, Yang F, Schuster SC, Carter NP, Harrow J, Ning Z, Herrero J, Searle SM, Enright A, Geisler R, Plasterk RH, Lee C, Westerfield M, de Jong PJ, Zon LI, Postlethwait JH, Nusslein-Volhard C, Hubbard TJ, Roest Crollius H, Rogers J, Stemple DL. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stainier DY, Fouquet B, Chen JN, Warren KS, Weinstein BM, Meiler SE, Mohideen MA, Neuhauss SC, Solnica-Krezel L, Schier AF, Zwartkruis F, Stemple DL, Malicki J, Driever W, Fishman MC. Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development. 1996;123:285–292. doi: 10.1242/dev.123.1.285. [DOI] [PubMed] [Google Scholar]
- 78.Knoll R, Postel R, Wang J, Kratzner R, Hennecke G, Vacaru AM, Vakeel P, Schubert C, Murthy K, Rana BK, Kube D, Knoll G, Schafer K, Hayashi T, Holm T, Kimura A, Schork N, Toliat MR, Nurnberg P, Schultheiss HP, Schaper W, Schaper J, Bos E, Den Hertog J, van Eeden FJ, Peters PJ, Hasenfuss G, Chien KR, Bakkers J. Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation. 2007;116:515–525. doi: 10.1161/CIRCULATIONAHA.107.689984. [DOI] [PubMed] [Google Scholar]
- 79.Gu R, Bai J, Ling L, Ding L, Zhang N, Ye J, Ferro A, Xu B. Increased expression of integrin-linked kinase improves cardiac function and decreases mortality in dilated cardiomyopathy model of rats. PLoS ONE. 2012;7:e31279. doi: 10.1371/journal.pone.0031279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.White DE, Coutu P, Shi YF, Tardif JC, Nattel S, St Arnaud R, Dedhar S, Muller WJ. Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes Dev. 2006;20:2355–2360. doi: 10.1101/gad.1458906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 82.Eisen JS, Smith JC. Controlling morpholino experiments: don’t stop making antisense. Development. 2008;135:1735–1743. doi: 10.1242/dev.001115. [DOI] [PubMed] [Google Scholar]
- 83.Kettleborough RN, Busch-Nentwich EM, Harvey SA, Dooley CM, de Bruijn E, van Eeden F, Sealy I, White RJ, Herd C, Nijman IJ, Fenyes F, Mehroke S, Scahill C, Gibbons R, Wali N, Carruthers S, Hall A, Yen J, Cuppen E, Stemple DL. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature. 2013;496:494–497. doi: 10.1038/nature11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Holtzman NG, Schoenebeck JJ, Tsai HJ, Yelon D. Endocardium is necessary for cardiomyocyte movement during heart tube assembly. Development. 2007;134:2379–2386. doi: 10.1242/dev.02857. [DOI] [PubMed] [Google Scholar]
- 85.Smith KA, Chocron S, von der Hardt S, de Pater E, Soufan A, Bussmann J, Schulte-Merker S, Hammerschmidt M, Bakkers J. Rotation and asymmetric development of the zebrafish heart requires directed migration of cardiac progenitor cells. Dev Cell. 2008;14:287–297. doi: 10.1016/j.devcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 86.de Pater E, Clijsters L, Marques SR, Lin YF, Garavito-Aguilar ZV, Yelon D, Bakkers J. Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development. 2009;136:1633–1641. doi: 10.1242/dev.030924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Auman HJ, Coleman H, Riley HE, Olale F, Tsai HJ, Yelon D. Functional modulation of cardiac form through regionally confined cell shape changes. PLoS Biol. 2007;5:e53. doi: 10.1371/journal.pbio.0050053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang R, Han P, Yang H, Ouyang K, Lee D, Lin YF, Ocorr K, Kang G, Chen J, Stainier DY, Yelon D, Chi NC. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature. 2013;498:497–501. doi: 10.1038/nature12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Staudt DW, Liu J, Thorn KS, Stuurman N, Liebling M, Stainier DY. High-resolution imaging of cardiomyocyte behavior reveals two distinct steps in ventricular trabeculation. Development. 2014;141:585–593. doi: 10.1242/dev.098632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin KY, Chang WT, Lai YC, Liau I. Toward functional screening of cardioactive and cardiotoxic drugs with zebrafish in vivo using pseudodynamic three-dimensional imaging. Anal Chem. 2014;86:2213–2220. doi: 10.1021/ac403877h. [DOI] [PubMed] [Google Scholar]
- 91.Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG, II, Tan W, Penheiter SG, Ma AC, Leung AY, Fahrenkrug SC, Carlson DF, Voytas DF, Clark KJ, Essner JJ, Ekker SC. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–118. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ocorr K, Vogler G, Bodmer R. Methods to assess Drosophila heart development, function and aging. Methods. 2014;68:265–272. doi: 10.1016/j.ymeth.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bier E, Bodmer R. Drosophila: an emerging model for cardiac disease. Gene. 2004;342:1–11. doi: 10.1016/j.gene.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 95.Wolf MJ, Amrein H, Izatt JA, Choma MA, Reedy MC, Rockman HA. Drosophila as a model for the identification of genes causing adult human heart disease. Proc Natl Acad Sci USA. 2006;103:1394–1399. doi: 10.1073/pnas.0507359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Choma MA, Izatt SD, Wessells RJ, Bodmer R, Izatt JA. Images in cardiovascular medicine: in vivo imaging of the adult Drosophila melanogaster heart with real-time optical coherence tomography. Circulation. 2006;114:e35–e36. doi: 10.1161/CIRCULATIONAHA.105.593541. [DOI] [PubMed] [Google Scholar]
- 97.Ocorr KA, Crawley T, Gibson G, Bodmer R. Genetic variation for cardiac dysfunction in Drosophila. PLoS ONE. 2007;2:e601. doi: 10.1371/journal.pone.0000601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vu Manh TP, Mokrane M, Georgenthum E, Flavigny J, Carrier L, Semeriva M, Piovant M, Roder L. Expression of cardiac myosin-binding protein-C (cMyBP-C) in Drosophila as a model for the study of human cardiomyopathies. Hum Mol Genet. 2005;14:7–17. doi: 10.1093/hmg/ddi002. [DOI] [PubMed] [Google Scholar]
- 99.Zhang D, Meijering RAM, Sibon OC, Kampinga HH, Henning RH, Brundel B. Selective inhibition of histone deacetylase 6 (HDAC6) attenuates atrial fibrillation in HL-1 cardiomyocytes and Drosophila melanogaster. Heart Rhythm. 2011;8:1. [Google Scholar]
- 100.Hoogstra-Berends F, Meijering RA, Zhang D, Heeres A, Loen L, Seerden JP, Kuipers I, Kampinga HH, Henning RH, Brundel BJ. Heat shock protein-inducing compounds as therapeutics to restore proteostasis in atrial fibrillation. Trends Cardiovasc Med. 2012;22:62–68. doi: 10.1016/j.tcm.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 101.Zhang D, Wu CT, Qi X, Meijering RA, Hoogstra-Berends F, Tadevosyan A, Cubukcuoglu Deniz G, Durdu S, Akar AR, Sibon OC, Nattel S, Henning RH, Brundel BJ. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation. 2014;129:346–358. doi: 10.1161/CIRCULATIONAHA.113.005300. [DOI] [PubMed] [Google Scholar]
- 102.Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012;11:275–291. doi: 10.1038/nrd3682. [DOI] [PubMed] [Google Scholar]
- 103.Ke L, Qi XY, Dijkhuis AJ, Chartier D, Nattel S, Henning RH, Kampinga HH, Brundel BJ. Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J Mol Cell Cardiol. 2008;45:685–693. doi: 10.1016/j.yjmcc.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 104.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW American College of Cardiology Foundation/American Heart Association Task Force on Practice G. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2011;58:e212–e260. doi: 10.1016/j.jacc.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 105.Ho CY. Genetic considerations in hypertrophic cardiomyopathy. Prog Cardiovasc Dis. 2012;54:456–460. doi: 10.1016/j.pcad.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circ Res. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Puckelwartz MJ, McNally EM. Genetic profiling for risk reduction in human cardiovascular disease. Genes (Basel) 2014;5:214–234. doi: 10.3390/genes5010214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 110.Campbell SG, McCulloch AD. Multi-scale computational models of familial hypertrophic cardiomyopathy: genotype to phenotype. J R Soc Interface. 2011;8:1550–1561. doi: 10.1098/rsif.2011.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sheikh F, Ouyang K, Campbell SG, Lyon RC, Chuang J, Fitzsimons D, et al. Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease. J Clin Invest. 2012;122:1209–1221. doi: 10.1172/JCI61134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 113.Ertz-Berger BR, He H, Dowell C, Factor SM, Haim TE, Nunez S, et al. Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice. Proc Natl Acad Sci USA. 2005;102:18219–18224. doi: 10.1073/pnas.0509181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Manning EP, Guinto PJ, Tardiff JC. Correlation of molecular and functional effects of mutations in cardiac troponin T linked to familial hypertrophic cardiomyopathy: an integrative in silico/in vitro approach. J Biol Chem. 2012;287:14515–14523. doi: 10.1074/jbc.M111.257436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li XE, Suphamungmee W, Janco M, Geeves MA, Marston SB, Fischer S, Lehman W. The flexibility of two tropomyosin mutants, D175N and E180G, that cause hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2012;424:493–496. doi: 10.1016/j.bbrc.2012.06.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Manning EP, Tardiff JC, Schwartz SD. A model of calcium activation of the cardiac thin filament. Biochemistry. 2011;50:7405–7413. doi: 10.1021/bi200506k. [DOI] [PMC free article] [PubMed] [Google Scholar]