

Abstract
The clinical variability in patients with sarcomeric cardiomyopathies is striking: a mutation causes cardiomyopathy in one individual, while the identical mutation is harmless in a family member. Moreover, the clinical phenotype varies ranging from asymmetric hypertrophy to severe dilatation of the heart. Identification of a single phenotype-associated disease mechanism would facilitate the design of targeted treatments for patient groups with different clinical phenotypes. However, evidence from both the clinic and basic knowledge of functional and structural properties of the sarcomere argues against a ‘one size fits all’ therapy for treatment of one clinical phenotype. Meticulous clinical and basic studies are needed to unravel the initial and progressive changes initiated by sarcomere mutations to better understand why mutations in the same gene can lead to such opposing phenotypes. Ultimately, we need to design an ‘integrative physiology’ approach to fully realize patient/gene-tailored therapy. Expertise within different research fields (cardiology, genetics, cellular biology, physiology, and pharmacology) must be joined to link longitudinal clinical studies with mechanistic insights obtained from molecular and functional studies in novel cardiac muscle systems. New animal models, which reflect both initial and more advanced stages of sarcomeric cardiomyopathy, will also aid in achieving these goals. Here, we discuss current priorities in clinical and preclinical investigation aimed at increasing our understanding of pathophysiological mechanisms leading from mutation to disease. Such information will provide the basis to improve risk stratification and to develop therapies to prevent/rescue cardiac dysfunction and remodelling caused by sarcomere mutations.
Keywords: Sarcomere, Mutation, Cardiomyopathy
1. Health care problem
Sarcomeric cardiomyopathies, caused by mutations in genes encoding proteins of the sarcomere, constitute one of the most common causes of sudden cardiac death (SCD) in the young and represent major causes for cardiac transplantation. Disease onset typically ranges between 20 and 50 years of age, thus affecting patients in the prime of their life, and earlier onsets represent an important cause of childhood cardiomyopathy. Because of advances in cardiovascular genetics during the last three decades,1,2 a large number of mutations have been identified in patients with cardiomyopathies previously considered to be idiopathic. Consequently, an increasing number of mutation carriers are being followed. Furthermore, due to increased availability of genotype analysis, at-risk family members can be identified prior to the emergence of a clinical diagnosis. This type of genotype-directed family evaluation is a growing focus of clinical management. However, the use of genetic insights and genetic diagnosis in risk stratification and therapeutic strategies in the clinic remains limited.3,4 The recently developed model to calculate SCD risk in hypertrophic cardiomyopathy (HCM) includes many clinical variables (e.g. left ventricular wall thickness, left atrial dimension, left ventricular outflow tract gradient, age),5 but it does not yet include genetics. Current drug therapy is aimed at management of symptom palliation and ‘watching and waiting’ to see if at-risk mutation carriers develop clinical cardiomyopathy. There is a great need for proactive risk stratification, including genotype, and treatment strategies that will be able to delay and ultimately prevent disease development in mutation carriers. The existence of (rare) neonatal forms of sarcomeric cardiomyopathy that rapidly evolve into systolic heart failure and death within the first year of life6–13 reveals a need for molecular diagnosis early after birth or during pregnancy to be able to rapidly give the appropriate treatment. With such advances, genetic discoveries will meet their full potential to change medical practice.
There is impressive clinical variability between affected mutation carriers.14 Importantly, within the same family, a specific mutation may cause cardiomyopathy and SCD in one relative, while it may appear entirely harmless in another family member. Disease onset and severity differ widely; implying that additional determinants, including genetic variations, environmental and/or toxic disease triggers, and an age-related decline in protective mechanisms (protein quality control systems)15–18 may have substantial impact on disease susceptibility and outcomes. Mechanisms governing how mutations cause disease and how secondary disease modifiers impact outcomes are largely unknown. As a result, it remains challenging for cardiologists to perform accurate risk stratification or initiate appropriate preventive therapy for the individual patient, as risk may be underestimated and left undertreated in some individuals, but overestimated in others, who are unnecessarily exposed to drugs or devices with potential side effects and complications. Finally, understanding of human genetic variation is rapidly evolving. Investigation of normal populations has led to the recognition of the prevalence of non-pathogenic sequence variation in genes associated with inherited cardiovascular disease. For example, mutations in genes encoding transmembrane potassium and sodium channels, KCNQ1, KCNH2, and SCN5A, are known to cause long QT syndrome. However, sequence variation in these same genes can also be seen at a low rate in normal population.19 Similarly, rare sarcomere gene variants were also identified in ∼11% of the general population. Although almost none of these individuals had clinical manifestations of HCM, the presence of a rare sarcomere variant was associated with increased risk of adverse cardiovascular events.20 These observations suggest that existing mutation databases are likely ‘contaminated’ with polymorphisms with little or no clinical relevance, further challenging accurate interpretation of genetic testing.
2. Cardiomyopathy phenotypes
Sarcomeric cardiomyopathies are associated with abnormalities in myocardial structure and function. Sarcomere gene defects have emerged as a shared aetiologic element, but genotype–phenotype relations are complex. Sarcomere mutations have been associated with distinct, apparently divergent patterns of ventricular remodelling, including hypertrophy, dilation, non-compaction, and restrictive physiology. HCM is typically characterized by left ventricular hypertrophy (LVH), small LV cavity size, and vigorous LV systolic function. In contrast, dilated cardiomyopathy (DCM) is characterized by an enlarged left ventricle with diminished systolic function. Many attempts have been made to identify a unifying mechanism for the so-called HCM- and DCM-causing mutations, which would explain development of hypertrophy or dilation of the ventricle, respectively. The genotype–phenotype relation analyses became even more complex with the finding that sarcomere mutations also result in restrictive cardiomyopathy (RCM) or left ventricular non-compaction (LVNC). Sarcomeric DCM and LVNC do not have specific phenotypes allowing a clinical distinction from the non-sarcomeric forms. In the case of DCM, for example, forms associated with MHY7, TNNT2, TNNI3, and TPM1 mutations lack the ‘red flags’ which characterize other genetic aetiologies such as lamin A/C (conduction disturbances) or skeletal muscle involvement (dystrophin complex). If anything, sarcomeric DCM may be suspected based on age at presentation and long-term course. Specifically, presentation early in life, from infancy to adolescence, is not uncommon and associated with severe outcome including SCD and refractory heart failure leading to death or transplantation. This is in sharp contrast with the mild course observed in relatives diagnosed as adults. This clinical profile differs meaningfully from that seen in other genetic causes of DCM, such as that caused by lamin A/C or phospholamban mutations where manifestations typically do not develop until adulthood and are progressive.21–25 Of note, subtle abnormalities in systolic function are present in subclinical DCM mutation carriers, despite normal left ventricular size and systolic function. In contrast, impaired relaxation appears to be the predominant early manifestation of sarcomere mutations causing HCM. These findings support the theory that the mutation's intrinsic impact on sarcomere function influences whether a dilated or hypertrophic phenotype develops.13 The case of LVNC is even more complex, as even its nature as a distinct cardiomyopathy is object of debate. Mutations in MYH7, ACTC, and TNNT2 have been associated to date.26,27 The most evident gap in current understanding of the phenotypic spectrum of sarcomeric cardiomyopathies, including HCM, DCM, RCM, and LVNC, is that the reasons for development of such different phenotypes from defects in the same genes is largely unknown.
Moreover, the phenotypic spectrum of cardiomyopathies is not limited to modifications in LV morphology or function. For example, atrial dilatation and dysfunction leading to atrial fibrillation (AF) is exceedingly common in both HCM and DCM. AF in turn promotes further dilatation of the atria in a vicious Cycle that is more common and occurs much earlier than in the general population. Although atrial changes have been attributed to the haemodynamic abnormalities and elevated filling pressures intrinsic to cardiomyopathies, the concept of a primary atrial myopathy and its implications for management have not been addressed. Because AF is common and a prominent cause of morbidity in cardiomyopathies, this issue requires further understanding.28 As well elucidated for HCM, cells other than cardiomyocytes may be directly involved in the disease process, including the coronary arterioles, the interstitial fibrous tissue, and the mitral valve and subvalvar apparatus. Coronary microvascular function is markedly abnormal in both HCM and DCM, although likely due to different mechanisms.29 In HCM, microvascular dysfunction is largely the result of smooth muscle hyperplasia causing severe reduction in luminal area. Microvascular dysfunction is associated with severe blunting in coronary vascular reserve, the most important cause of ischaemia in patients with HCM, and an important predictor of outcome.14,30 Ischaemia occurring at the microvascular level is believed to represent the main cause of replacement-type fibrosis, a common finding in patients with cardiomyopathies, whose extent is directly related to the degree of systolic impairment.31 In the majority of patients with HCM, the mitral leaflets are markedly enlarged, although with normal histological appearance, and show anomalous chordae and papillary muscle insertions. These abnormalities are observed in patients of all ages, including young children, and are thought to be one of the main determinants of anterior systolic motion and dynamic outflow obstruction.32 The link between the genetic defect in sarcomeric proteins and these features is elusive and often dismissed as being secondary to the myocardial abnormalities. However, other mechanisms may be at play. For example, a single developmental pathway involving the proepicardial organ, i.e. the common progenitor of all these extramyocardial cell types, might provide a simple explanation to such apparent discrepancy and indicate novel targets for phenotype suppression or modification during the early stages of life.33
3. Clinical studies
A clear-cut separation of patient groups on the basis of type of remodelling turns out to be unrealistic, as current clinical classification lacks adequate precision and specificity. HCM patients presenting with heart failure may have evolved a phenotype that resembles DCM or may manifest profound restrictive physiology. As such, their classification may differ depending on the predominant stage of disease at the time of clinical evaluation. In addition, in-depth understanding about how cardiomyopathy progresses and how sarcomere mutations lead to disease is currently limited. The development of end-stage remodelled myocardium represents the final stage of a long complex pathway from the initial genetic and biophysical insult (Figure 1). Human studies have largely focused on the late stages of cardiomyopathy and therefore do not account for the confounding influence of disease itself on phenotypic progression or genotype–phenotype correlations. As such, prior studies have been importantly hindered in their efforts to identify primary disease mechanisms.
Figure 1.
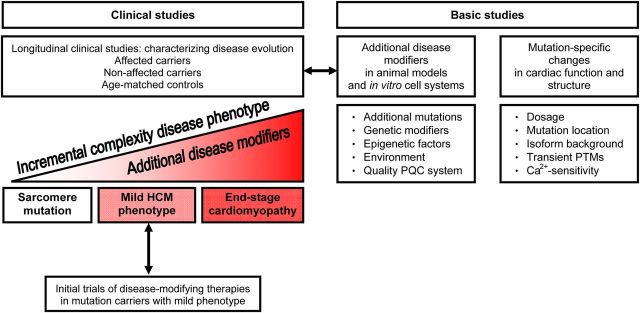
Schematic showing the progression of cardiac disease from the mutation to end-stage cardiomyopathy, which will involve secondary disease modifiers. Longitudinal clinical studies are needed to reveal secondary disease modifiers, which can be subsequently investigated in basic studies. To proof efficacy of novel therapies, initial trials should be performed in mutation carriers with a mild phenotype, which are expected to have a dynamic change in clinical phenotype within a relatively brief period of time. Basic studies should investigate myocardial effects of secondary disease modifiers and mutation-specific effects on the heart muscle. PQC, protein quality control; PTM, post-translational modification.
3.1. From mutation to cardiomyopathy: characterizing disease evolution
Identifying the genetic basis of disease is a prerequisite for developing ‘personalized’ medicine—rational, mechanism-based therapy. However, substantial challenges must be overcome to be able to initiate clinical trials and begin to realize this goal. We must first gain greater knowledge about the steps that lead from mutation to disease. Understanding the link between genotype and phenotype requires longitudinal clinical studies, starting with sarcomere mutation carriers early in life, before obvious disease has developed (Figure 1). The potential power of this approach was recently demonstrated by Ho and colleagues,34 who detected evidence for an early profibrotic state in HCM patients before the onset of hypertrophy or detectable fibrosis. Likewise, diastolic dysfunction has been reported in human HCM mutation carriers and transgenic mouse models before onset of the disease phenotype.2,35–37 Crilley et al.38 reported a reduction in the cardiac PCr-to-ATP ratio, a measure of energetic status, in mutation carriers with or without LVH. Reduced myocardial efficiency at the pre-hypertrophic stage of HCM was confirmed using PET and CMR studies to assess myocardial oxygen consumption and external work, respectively.39,40 Other changes in the early stage of cardiomyopathy include crypts41–44 and extracellular volume expansion assessed by cardiac MRI.45
At this early stage, the cardiac sarcomere mutation represents the single pathogenic constant. Presumably, during ageing, additional disease modifiers (Figure 1) initiate detrimental mutation-related changes in the heart. Longitudinal studies that begin early in life, anchored on genotype, are an ideal starting point for research on genotype–phenotype relations. Such studies will help to answer fundamental questions regarding penetrance, expressivity, and clinical outcomes. This is a highly ambitious effort that requires careful evaluation of a large number of families with affected and non-affected carriers as well as age-matched ‘control’ partners over long periods of time. Close collaboration is needed between all key investigators involved in sarcomeric cardiomyopathy, including geneticists, cardiologists, paediatrician cardiologists, and physiologists. Well-designed clinical studies will reveal disease mechanisms (epigenetic and environmental) that may modify the occurrence and progression of cardiomyopathy, which subsequently need to be investigated in basic studies (animal models, in vitro cell systems) to provide proof that these factors indeed enhance or delay onset and progression of disease (Figure 1).
In addition to longitudinal clinical studies, unbiased statistical approaches, utilizing techniques of machine learning or cluster analysis, may prove fruitful in trying to better identify previously unrecognized patterns in diseases with complex, overlapping phenotypes. Such studies may provide new insights to clinical outcomes and risk prediction, as well as genotype–phenotype correlations.
3.2. Clinical trials of disease modification and prevention
As our understanding of disease pathogenesis improves, we will be able to develop novel treatments intended to delay or attenuate phenotypic evolution and, ultimately, to prevent the emergence of disease all together. Such strategies will target key early steps in disease evolution, identified by basic investigation. This approach is highly appealing, as it seems both more feasible and more desirable to treat to delay or prevent disease, rather than trying to reverse or rescue severe changes associated with entrenched, late-stage disease. Indeed, animal studies have suggested that disease-modifying treatment was ineffective if started after a clinical phenotype developed.46,47
However, clinical trials to test these potential new therapies face unique and daunting challenges. For example, although the pathogenic sarcomere mutation is present at birth, overt disease may not develop for decades and in some cases may not develop at all or be associated with only very mild clinical consequences. Given the great variability in disease onset and course, who should be treated and when should treatment be started? Since mutations are present throughout life, will lifelong therapy be needed to inhibit phenotypic progression or emergence? Moreover, although sarcomeric cardiomyopathies can be associated with devastating outcomes, the event rate in the overall patient population is relatively low, especially if mutation carriers without clinical disease are included. As a result, trials will require treating large cohorts followed over long periods of time to demonstrate treatment benefit as reflected by hard outcomes, such as mortality or development of clinically overt disease. Achieving the scope and scale of such studies will require monumental effort given the relative rarity of sarcomeric cardiomyopathies.
Owing to these challenges, traditional approaches to clinical trial design may not be appropriate or successful. To allow initial test-of-concept trials to proceed over manageable time frames and with feasible cohort sizes, we will need to identify surrogate endpoints that accurately reflect disease progression and treatment benefit, and that are more likely to show dynamic change over shorter periods of time. Identifying additional robust, quantitative early phenotypes that reflect a continuum of disease progression from normal controls to patients with overt disease will be important to advance these efforts. Such phenotypes could function as surrogate endpoints in trials. Effective treatment would potentially show that early-stage mutation carriers become more ‘normal’ for the metrics under study.
Next, choosing which individuals to target and how long to treat them requires careful consideration. Although it is appealing to consider directing therapy towards the youngest mutation carriers without any phenotypic expression, such trials will be exceedingly difficult. At this stage, disease progression is likely to be extremely subtle and play out over many years. As a result, events are unlikely to accrue in the placebo-treated cohort, challenging the ability to demonstrate treatment benefit in the cohort receiving intervention during trials taking place over <10 years. It may be best to focus initial trials of disease modification on mutation carriers with mild phenotypic expression (Figure 1) and refine patient selection as more knowledge is obtained regarding both early phenotypes and pathways governing disease evolution.
Finally, more innovative and sophisticated statistical analysis plans will also be needed to be guard against false-negative trial results, without excessively increasing the risk for false-positive results.48 Experience gained from such initial trials of disease-modifying therapies will set the stage for more definitive trials to follow. Ideally, we will be able to determine accurate predictors of both adverse outcomes and imminent development of clinically overt disease. Therapy could then be targeted to those at highest risk who would derive greatest benefit from potentially lifelong treatment. Potential novel ways to address the challenges of trials to modify and prevent HCM are explored in the ongoing VANISH trial (Valsartan for Attenuating Disease Evolution in Early Sarcomeric HCM; NCT01912534).
4. Bench and preclinical studies
Apart from clinical studies, more advanced basic methodologies are crucial to establish the mechanistic links between mutation and the pathogenic molecular and cellular mechanisms underlying the cardiomyopathy phenotype. As a first step in understanding the pathophysiology of sarcomeric cardiomyopathies, it is essential to assess the functional effects of the mutant sarcomeric proteins per se. Until now, substantial data originated from experiments with engineered mouse models, gene transfer in cardiomyocytes or engineered heart tissue, or human recombinant proteins (reviewed in this issue).49–54 Based on these in vitro and transgenic rodent studies, it has been proposed that HCM gene mutations cause enhanced contractile function (hypercontractility), while DCM-associated mutations reduce function (hypocontractility). However, based on the complex functional and structural properties of the affected sarcomeric proteins, it is unlikely that mutations in different sarcomeric proteins would cause cardiomyopathy via a single disease mechanism. Myocardial dysfunction will depend on mutation expression, mutation location within the gene, age-related protein isoform background and secondary disease-related protein modifications. Below we highlight the mutation-related changes in cardiomyopathies which warrant future preclinical research (Figure 1).
4.1. Mutation expression and gene dosage
Sarcomeric cardiomyopathies are mainly inherited in an autosomal-dominant fashion, with the exception of some X-linked mutations, indicating that the presence of the mutation on one of the two alleles is sufficient to cause the disease phenotype. Therefore, expression of both wild-type and mutant alleles is basically expected to result in the incorporation of both wild-type and mutant proteins in the sarcomere. However, it is important to emphasize that the expression of the mutation is regulated at different levels by quality control mechanisms (such as the non-sense-mediated mRNA decay, ubiquitin-proteasome system, and/or the autophagy-lysosomal pathway)17,55,56 that could reduce or increase production of mutant proteins in comparison to wild-type. This is particularly the case for truncating MYBPC3 mutations, which result in reduced mutant mRNAs and/or proteins, leading to haploinsufficiency of the proteins in the sarcomere.57–59 A large variability in the level of wild-type cardiac myosin-binding protein-C (cMyBP-C) protein was found in tissue from HCM patients,59–61 which may correlate with the heterogeneity of the disease phenotype severity. In addition, heterozygous mutations could be subjected to allelic imbalance, which could affect disease severity. For example, higher levels of mutant mRNA appear to contribute to a more severe cardiac phenotype.62,63 Infants carrying homozygous or compound heterozygous truncating MYBPC3 mutations are expected to have low levels or complete absence of mutant cMyBP-C in the heart. As a consequence, these infants present with cardiomyopathy in the neonatal period and rapidly develop heart failure and die within the first year of life.6,8–12 Similarly, many transgenic rodent models exhibit gene dosage dependence on the phenotype severity. For example, homozygous Mybpc3-targeted knock-in mice developed an earlier and more severe disease phenotype than their heterozygous littermates.35,64,65 Similarly, a transgene dose-dependent increase in Ca2+ sensitivity of force development and concomitant decrease in relaxation parameters have been reported in transgenic mice expressing the HCM-associated E180G Tpm1 mutation.66 In addition, mice expressing high levels of the HCM-associated cTnTΔ160E Tnnt2 mutation showed a further decrease in relaxation capacity and exacerbated disturbances in Ca2+ transients compared with mice with low mutant cTnTΔ160E levels.67 These data are supported by recent molecular-based therapeutic approaches. Allele-specific RNA interference in heterozygous Myh6 mutant R403Q HCM mice showed that a reduction of ∼25% of the mutant allele expression was enough to prevent HCM development.68 Adeno-associated viral-mediated Mybpc3 gene transfer in Mybpc3-targeted knock-in mice showed replacement of the endogenous mutant by wild-type cMyBP-C protein and prevention of LVH and cardiac dysfunction in a dose-dependent manner.69 Thus, a body of evidence from the clinic and transgenic mouse models indicate that mutation dose contributes to the onset and severity of cardiomyopathy. However, little information is available on the dose needed to perturb sarcomere function and structure of the heart muscle as many in vitro studies focused on functional consequences of mutant protein in the absence of wild-type protein. A recent study based on troponin exchange experiments revealed that <50% of poison peptide is sufficient to perturb sarcomere function in human cardiomyocytes obtained from patients with a homozygous TNNT2 mutation or a heterozygous TNNI3 mutation.70
4.2. Mutation location
Apart from mutant protein dose, mutant protein conformation, which depends on location of the mutation in the gene, represents a likely pathomechanism underlying variable onset and phenotypic expression of cardiomyopathies. Maass et al.71 compared two HCM-associated Tnnt2 transgenic mouse models that express a missense or a truncating mutation, i.e. R92Q and cTnTtrunc, and found that remodelling was mutation dependent. Fibrosis was only seen in R92Q mice, which also showed a hypertrophic response to prolonged adrenergic stimulation, while cTnTtrunc mice did not. Likewise, S532P and F764L mutations in α-myosin heavy chain (Myh6) are both known to cause DCM, but their effect on hypertrophy and contractile function differs considerably.72 While hypertrophy was noted in F764L mice, the heart weights of S532P mice did not differ from non-transgenic mice. Moreover, cardiomyocytes from F764L mice produced significantly less force than those from S532P hearts.72 Recent data suggest mutation-specific perturbations in sarcomere function in human HCM samples.40,73–75 In particular, mutations in the head domain of myosin heavy chain (MyHC) reduce the contractile strength of sarcomeres.73,74 Moreover, a gene-specific increase in energetic cost of contraction was found both in vivo in HCM patients and in vitro in human HCM samples, being more severe in MYH7 than in MYBPC3 mutation carriers.40 The increase in tension cost in cardiac muscle strips of manifest HCM patients was explained primarily by a reduction in maximal force-generating capacity. Likewise, at the pre-hypertrophic phase of HCM, the decrease in myocardial efficiency was largely explained by a reduction in cardiac work rather than an increase in oxygen consumption. In contrast to the above-mentioned assumption that HCM mutations enhance contractile function, these recent studies in human HCM indicate that hypocontractile sarcomeres may represent a primary abnormality, in particular in cardiomyopathy caused by certain MYH7 mutations. The apparent difference in functional effects may be explained by the different HCM mutations investigated. The recent studies emphasize the need to establish the toxic effects of mutants on structure and function of the heart muscle with respect to mutation location and protein dose.
4.3. Protein isoform background
Effects of mutant proteins may be more or less severe depending on protein isoform background in the heart. The relevance of protein background for mutant-related effects has been demonstrated by the species-dependent MyHC isoform composition present in the heart. As indicated in the present issue,54 many studies in transgenic rodents have been performed on the Myh6 background, while human ventricles predominantly express the slow β-MyHC (MYH7). Functional and structural changes were different in mice expressing the first identified R403Q mutation in a Myh6 or Myh7 background.76,77 In addition, it has also been shown that a genetically engineered α- to β-MyHC shift mice in the contest of TNNT2 mutations could partially rescue observed energetic effects,78,79 further evidence that sarcomeric isoform composition (known to be altered in the context of pathogenic remodelling in humans) may play a greater role in modulating ventricular remodelling in sarcomeric cardiomyopathies. A recent study directly demonstrated the effects of switching troponin isoforms in an established transgenic mouse model of Tpm1-associated DCM.80 These results suggest that age-dependent isoform switching of sarcomeric proteins may directly modulate ventricular structure and function in a transient fashion.
4.4. Transient changes in post-translational protein modifications
Apart from the impact of isoform composition on disease outcome, the cardiomyopathic phenotype is not static and changes during the course of disease development as a result of secondary post-translational disease-modifying processes. An important post-translational modification (PTM) that changes during disease development is protein phosphorylation due to altered expression and activity changes of the myocardial kinases and phosphatases. An optimal balance between kinase and phosphatase activities is required to regulate cellular processes via targeted phosphorylation of proteins involved in cell signalling and contractility. During disease onset and progression, changes occur in protein phosphorylation due to an imbalance between kinase and phosphatase activities, which subsequently underlie structural and functional changes of the heart muscle. One of the most described perturbations is the down-regulation and desensitization of the β-adrenergic receptors in heart failure. At the level of the sarcomeres, reduced β-adrenergic-mediated phosphorylation by protein kinase A (PKA) is associated with reduced phosphorylation of myofilament proteins such as cardiac troponin I and cMyBP-C. A reduction in myofilament protein phosphorylation has been associated with an increased myofilament Ca2+ sensitivity. Increased Ca2+ sensitivity compared with healthy sarcomeres has been proposed as a hallmark of HCM, opposite to reduced myofilament Ca2+ sensitivity in DCM. Recent studies in human HCM samples with mutations in thick filament proteins, MyHC and cMyBP-C, indicate that high myofilament Ca2+ sensitivity is mostly due to reduced myofilament protein phosphorylation compared with non-failing myocardium rather than the mutation itself.70 The secondary disease-related high myofilament Ca2+ sensitivity may be a target of treatment in patients with manifest HCM, but most likely is not the initial trigger of cardiomyopathy onset. Likewise, PTMs induced by increased oxidative stress81 may contribute to cardiac dysfunction and disease progression.
4.5. Myofilament Ca2+ sensitivity, disease pathogenesis, and arrhythmias
Myofilament Ca2+ sensitivity is universally increased in sarcomeric cardiomyopathies associated with cardiac hypertrophy. As discussed above, the Ca2+ sensitization is often due to altered phosphorylation patterns of sarcomeres, but can also be a primary consequence as shown for Tnnt2 mutations. Upon myofilament Ca2+ sensitization, altered intracellular Ca2+ homeostasis and slowed muscle relaxation are expected, which will contribute to diastolic dysfunction regardless of the underlying molecular mechanism.82 Although the molecular mechanisms of how increased Ca2+ sensitivity can cause cardiac hypertrophy and contributes to HCM pathogenesis remains unclear, proof-of-concept studies showed that hypertrophy was prevented and relaxation enhanced when myofilaments were genetically desensitized in mice expressing the HCM-associated Tm180 Tpm1 mutation.83 Even in the absence of cardiac hypertrophy, increased myofilament Ca2+ sensitivity generated acutely by drugs or chronically by Tnnt2 mutations has been shown to cause susceptibility to ventricular arrhythmias in mice.84 Two independent arrhythmia mechanisms have been proposed: Ca2+-sensitizing Tnnt2 mutations were shown to increase the cytosolic Ca2+-binding affinity (Ca2+ buffering), Ca2+-dependent action potential remodelling and promote pause-dependent Ca2+-triggered arrhythmias.85,86 Myofilament Ca2+ sensitization was also linked to focal energy deprivation and re-entry arrhythmias during stress, but the underlying cause is unclear and is currently investigated.87 Although remains to be tested if primary or acquired myofilament Ca2+-sensitization differ, normalizing myofilament Ca2+ sensitivity may be beneficial on several levels: (i) normalizing it early may prevent disease progression and (ii) if that opportunity has been missed, it may still be effective to prevent lethal ventricular arrhythmias. Whether Ca2+-desensitizing drugs will be useful therapeutically needs to be explored. In addition, studies are warranted to investigate the underlying mechanisms in large animal models and humans to validate the proposed mechanisms and identify therapeutic targets.88
On the other hand, thin filament mutations that reduce myofilament Ca2+ sensitivity have been associated with a primary DCM phenotype in humans,88,90 which has been reproduced in mouse models.90–92 Normalizing reduced Ca2+ sensitivity either using transgenic approaches or with the Ca2+ sensitizer levosimendan was effective in preventing the development of DCM in mice expressing a human TNNT2 mutation. Hence, future basic and clinical studies are warranted to better identify how and to what extent abnormal myofilament Ca2+ sensitivity contributes to disease pathogenesis and arrhythmia risk (i.e. using iPSC models53 and large animal models54), and whether it can be used to risk stratify mutation carriers (e.g. to test whether human carriers of Ca2+-sensitizing mutations exhibit an increased risk for pause-triggered ectopic ventricular beats that are predicted by studies in mice84). If confirmed by such studies, targeting altered myofilament Ca2+ sensitivity, although possibly only a downstream consequence of the disease mutation, would nevertheless be an attractive, mutation-independent treatment strategy in sarcomeric cardiomyopathy.
5. Future
The critical gap in our knowledge is insight in the way a mutation becomes toxic. The large clinical variability implies that additional factors eventually determine whether a mutation becomes effective and injures the heart. Disease severity will depend on expression of the mutant allele, disease-modifying genes, cardiac load, and additional non-genetic disease modifiers including epigenetic modifications, miRNA, PTMs of proteins, an age-related decline in protein quality control, and environmental disease triggers. This complex interaction makes it difficult to determine the main pathophysiological mechanism. Previous paradigms need to be tested properly in disease model systems, which take into account expression levels of mutant proteins, disease progression, and, as indicated in the paper by Duncker and colleagues,54 the protein isoform content as present in the human heart. Observations from rodent studies need to be tested properly in a human-like model systems (iPSC-derived disease modelling in cardiac myocytes and engineered heart tissue) as described by Eschenhagen et al.53 to test applicability for the human situation. Ultimately, identification of disease modifiers that underlie the complex pathophysiology of sarcomeric cardiomyopathy may serve as rational therapeutic targets.
Acknowledgements
L.C. is supported by grants from the DZHK (German Centre for Cardiovascular Research) and the German Ministry of Research and Education (BMBF), the Leducq Foundation (Research grant Nr. 11, CVD 04), and Association Institut de Myologie (Paris). J.v.d.V. is supported by the Netherlands organization for scientific research (NWO; VIDI grant 91711344). We further acknowledge support from the 7th Framework Program of the European Union (“BIG-HEART”, grant agreement 241577).
Conflict of interest: none declared.
References
- 1.Ho CY, Charron P, Richard P, Girolami F, van Spaendonck-Zwarts KY, Pinto YM. Genetic causes: state of the art. Cardiovasc Res. 2015;105:397–408. doi: 10.1093/cvr/cvv025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ho CY, Carlsen C, Thune JJ, Havndrup O, Bundgaard H, Farrohi F, Rivero J, Cirino AL, Andersen PS, Christiansen M, Maron BJ, Orav EJ, Kober L. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:314–321. doi: 10.1161/CIRCGENETICS.109.862128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H Authors/Task Force members. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC) Eur Heart J. 2014;35:2733–2779. doi: 10.1093/eurheartj/ehu284. [DOI] [PubMed] [Google Scholar]
- 4.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Anesthesiologists, Interventions, Society of Thoracic Surgeons. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:2761–2796. doi: 10.1161/CIR.0b013e318223e230. [DOI] [PubMed] [Google Scholar]
- 5.O'Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, Biagini E, Gimeno JR, Limongelli G, McKenna WJ, Omar RZ, Elliott PM Hypertrophic Cardiomyopathy Outcomes I. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD) Eur Heart J. 2014;35:2010–2020. doi: 10.1093/eurheartj/eht439. [DOI] [PubMed] [Google Scholar]
- 6.Marziliano N, Merlini PA, Vignati G, Orsini F, Motta V, Bandiera L, Intrieri M, Veronese S. A case of compound mutations in the MYBPC3 gene associated with biventricular hypertrophy and neonatal death. Neonatology. 2012;102:254–258. doi: 10.1159/000339847. [DOI] [PubMed] [Google Scholar]
- 7.El-Saiedi SA, Seliem ZS, Esmail RI. Hypertrophic cardiomyopathy: prognostic factors and survival analysis in 128 Egyptian patients. Cardiol Young. 2014;24:702–708. doi: 10.1017/S1047951113001030. [DOI] [PubMed] [Google Scholar]
- 8.Dellefave LM, Pytel P, Mewborn S, Mora B, Guris DL, Fedson S, Waggoner D, Moskowitz I, McNally EM. Sarcomere mutations in cardiomyopathy with left ventricular hypertrabeculation. Circ Cardiovasc Genet. 2009;2:442–449. doi: 10.1161/CIRCGENETICS.109.861955. [DOI] [PubMed] [Google Scholar]
- 9.Lekanne Deprez RH, Muurling-Vlietman JJ, Hruda J, Baars MJ, Wijnaendts LC, Stolte-Dijkstra I, Alders M, van Hagen JM. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J Med Genet. 2006;43:829–832. doi: 10.1136/jmg.2005.040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xin B, Puffenberger E, Tumbush J, Bockoven JR, Wang H. Homozygosity for a novel splice site mutation in the cardiac myosin-binding protein C gene causes severe neonatal hypertrophic cardiomyopathy. Am J Med Genet Part A. 2007;143A:2662–2667. doi: 10.1002/ajmg.a.31981. [DOI] [PubMed] [Google Scholar]
- 11.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M. Hypertrophic Cardiomyopathy: distribution of disease genes, spectrum of mutations and implications for molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 12.Schaefer E, Helms P, Marcellin L, Desprez P, Billaud P, Chanavat V, Rousson R, Millat G. Next-generation sequencing (NGS) as a fast molecular diagnosis tool for left ventricular noncompaction in an infant with compound mutations in the MYBPC3 gene. Eur J Med Genet. 2014;57:129–132. doi: 10.1016/j.ejmg.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 13.Lakdawala NK, Thune JJ, Colan SD, Cirino AL, Farrohi F, Rivero J, McDonough B, Sparks E, Orav EJ, Seidman JG, Seidman CE, Ho CY. Subtle abnormalities in contractile function are an early manifestation of sarcomere mutations in dilated cardiomyopathy. Circ Cardiovasc Genet. 2012;5:503–510. doi: 10.1161/CIRCGENETICS.112.962761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olivotto I, d'Amati G, Basso C, van Rossum A, Patten M, Emdin M, Pinto Y, Tomberli B, Camici PG, Michels M. Defining phenotypes and disease progression in sarcomeric cardiomyopathies: contemporary role of clinical investigations. Cardiovasc Res. 2015;105:409–423. doi: 10.1093/cvr/cvv024. [DOI] [PubMed] [Google Scholar]
- 15.Schlossarek S, Schuermann F, Geertz B, Mearini G, Eschenhagen T, Carrier L. Adrenergic stress reveals septal hypertrophy and proteasome impairment in heterozygous Mybpc3-targeted knock-in mice. J Muscle Res Cell Motil. 2012;33:5–15. doi: 10.1007/s10974-011-9273-6. [DOI] [PubMed] [Google Scholar]
- 16.Schlossarek S, Englmann DR, Sultan KR, Sauer M, Eschenhagen T, Carrier L. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Bas Res Cardiol. 2012;107:235. doi: 10.1007/s00395-011-0235-3. [DOI] [PubMed] [Google Scholar]
- 17.Schlossarek S, Frey N, Carrier L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J Mol Cell Cardiol. 2014;71:25–31. doi: 10.1016/j.yjmcc.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Robbins J. Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol. 2014;71:16–24. doi: 10.1016/j.yjmcc.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AA, Ackerman MJ. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120:1752–1760. doi: 10.1161/CIRCULATIONAHA.109.863076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bick AG, Flannick J, Ito K, Cheng S, Vasan RS, Parfenov MG, Herman DS, DePalma SR, Gupta N, Gabriel SB, Funke BH, Rehm HL, Benjamin EJ, Aragam J, Taylor HA, Jr, Fox ER, Newton-Cheh C, Kathiresan S, O'Donnell CJ, Wilson JG, Altshuler DM, Hirschhorn JN, Seidman JG, Seidman C. Burden of rare sarcomere gene variants in the Framingham and Jackson Heart Study cohorts. Am J Human Genet. 2012;91:513–519. doi: 10.1016/j.ajhg.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakdawala N, Dellefave L, Redwood CS, Sparks E, Cirino AL, Depalma S, Colan SD, Funke B, Zimmerman RS, Robinson P, Watkins H, Seidman CE, Seidman JG, McNally EM, Ho CY. Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: the distinctive natural history of sarcomeric DCM. J Am Coll Cardiol. 2010;55:320–329. doi: 10.1016/j.jacc.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 23.Mogensen J, Murphy RT, Bahl A, Redwood C, Watkins H, Burke M, Elliott PM, McKenna WJ. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2033–2040. doi: 10.1016/j.jacc.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 24.Villard E, Duboscq-Bidot L, Charron P, Benaiche A, Conraads V, Sylvius N, Komajda M. Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur Heart J. 2005;26:794–803. doi: 10.1093/eurheartj/ehi193. [DOI] [PubMed] [Google Scholar]
- 25.Carballo S, Robinson P, Otway R, Fatkin D, Jongbloed JD, de Jonge N, Blair E, van Tintelen JP, Redwood C, Watkins H. Identification and functional characterization of cardiac troponin I as a novel disease gene in autosomal dominant dilated cardiomyopathy. Circ Res. 2009;105:375–382. doi: 10.1161/CIRCRESAHA.109.196055. [DOI] [PubMed] [Google Scholar]
- 26.Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, Greutmann M, Hürlimann D, Yegitbasi M, Pons L, Gramlich M, Drenckhahn JD, Heuser A, Berger F, Jenni R, Thierfelder L. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117:2893–2901. doi: 10.1161/CIRCULATIONAHA.107.746164. [DOI] [PubMed] [Google Scholar]
- 27.Hoedemaekers Y, Caliskan K, Majoor-Krakauer D, van de Laar I, Michels M, Witsenburg M, ten Cate FJ, Simoons ML, Dooijes D. Cardiac b-myosin heavy chain defects in two families with non-compaction cardiomyopathy: linking non-compaction to hypertrophic, restrictive, and dilated cardiomyopathies. Eur Heart J. 2007;28:2732–2737. doi: 10.1093/eurheartj/ehm429. [DOI] [PubMed] [Google Scholar]
- 28.Di Donna P, Olivotto I, Delcre SD, Caponi D, Scaglione M, Nault I, Montefusco A, Girolami F, Cecchi F, Haissaguerre M, Gaita F. Efficacy of catheter ablation for atrial fibrillation in hypertrophic cardiomyopathy: impact of age, atrial remodelling, and disease progression. Europace. 2010;12:347–355. doi: 10.1093/europace/euq013. [DOI] [PubMed] [Google Scholar]
- 29.Camici PG, Crea F. Coronary microvascular dysfunction. New Engl J Med. 2007;356:830–840. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- 30.Camici PG, Olivotto I, Rimoldi OE. The coronary circulation and blood flow in left ventricular hypertrophy. J Mol Cell Cardiol. 2012;52:857–864. doi: 10.1016/j.yjmcc.2011.08.028. [DOI] [PubMed] [Google Scholar]
- 31.Olivotto I, Maron BJ, Appelbaum E, Harrigan CJ, Salton C, Gibson CM, Udelson JE, O'Donnell C, Lesser JR, Manning WJ, Maron MS. Spectrum and clinical significance of systolic function and myocardial fibrosis assessed by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. Am J Cardiol. 2010;106:261–267. doi: 10.1016/j.amjcard.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 32.Maron MS, Olivotto I, Harrigan C, Appelbaum E, Gibson CM, Lesser JR, Haas TS, Udelson JE, Manning WJ, Maron BJ. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation. 2011;124:40–47. doi: 10.1161/CIRCULATIONAHA.110.985812. [DOI] [PubMed] [Google Scholar]
- 33.Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Developmental origins of hypertrophic cardiomyopathy phenotypes: a unifying hypothesis. Nature Reviews Cardiol. 2009;6:317–321. doi: 10.1038/nrcardio.2009.9. [DOI] [PubMed] [Google Scholar]
- 34.Ho CY, Lopez B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, Gonzalez A, Colan SD, Seidman JG, Diez J, Seidman CE. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. New Engl J Med. 2010;363:552–563. doi: 10.1056/NEJMoa1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fraysse B, Weinberger F, Bardswell SC, Cuello F, Vignier N, Geertz B, Starbatty J, Kramer E, Coirault C, Eschenhagen T, Kentish JC, Avkiran M, Carrier L. Increased myofilament Ca2+ sensitivity and diastolic dysfunction as early consequences of Mybpc3 mutation in heterozygous knock-in mice. J Mol Cell Cardiol. 2012;52:1299–1307. doi: 10.1016/j.yjmcc.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Germans T, Russel IK, Gotte MJ, Spreeuwenberg MD, Doevendans PA, Pinto YM, van der Geest RJ, van der Velden J, Wilde AA, van Rossum AC. How do hypertrophic cardiomyopathy mutations affect myocardial function in carriers with normal wall thickness? Assessment with cardiovascular magnetic resonance. J Cardiovasc Magnetic Resonance. 2010;12:13. doi: 10.1186/1532-429X-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michels M, Soliman OI, Kofflard MJ, Hoedemaekers YM, Dooijes D, Majoor-Krakauer D, ten Cate FJ. Diastolic abnormalities as the first feature of hypertrophic cardiomyopathy in Dutch myosin-binding protein C founder mutations. JACC Cardiovasc Imag. 2009;2:58–64. doi: 10.1016/j.jcmg.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, McKenna WJ, Ostman-Smith I, Clarke K, Watkins H. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. 2003;41:1776–1782. doi: 10.1016/s0735-1097(02)03009-7. [DOI] [PubMed] [Google Scholar]
- 39.Timmer SA, Germans T, Brouwer WP, Lubberink M, van der Velden J, Wilde AA, Christiaans I, Lammertsma AA, Knaapen P, van Rossum AC. Carriers of the hypertrophic cardiomyopathy MYBPC3 mutation are characterized by reduced myocardial efficiency in the absence of hypertrophy and microvascular dysfunction. Eur J Heart Fail. 2011;13:1283–1289. doi: 10.1093/eurjhf/hfr135. [DOI] [PubMed] [Google Scholar]
- 40.Witjas-Paalberends ER, Güçlü A, Germans T, Knaapen P, Harms HJ, Vermeer AM, Christiaans I, Wilde AA, Dos Remedios C, Lammertsma AA, van Rossum AC, Stienen GJ, van Slegtenhorst M, Schinkel AF, Michels M, Ho CY, Poggesi C, van der Velden J. Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovasc Res. 2014;103:248–257. doi: 10.1093/cvr/cvu127. [DOI] [PubMed] [Google Scholar]
- 41.Germans T, Wilde AA, Dijkmans PA, Chai W, Kamp O, Pinto YM, van Rossum AC. Structural abnormalities of the inferoseptal left ventricular wall detected by cardiac magnetic resonance imaging in carriers of hypertrophic cardiomyopathy mutations. J Am Coll Cardiol. 2006;48:2518–2523. doi: 10.1016/j.jacc.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 42.Brouwer WP, Germans T, Head MC, van der Velden J, Heymans MW, Christiaans I, Houweling AC, Wilde AA, van Rossum AC. Multiple myocardial crypts on modified long-axis view are a specific finding in pre-hypertrophic HCM mutation carriers. Eur Heart J Cardiovasc Imag. 2012;13:292–297. doi: 10.1093/ehjci/jes005. [DOI] [PubMed] [Google Scholar]
- 43.Maron MS, Rowin EJ, Lin D, Appelbaum E, Chan RH, Gibson CM, Lesser JR, Lindberg J, Haas TS, Udelson JE, Manning WJ, Maron BJ. Prevalence and clinical profile of myocardial crypts in hypertrophic cardiomyopathy. Circulation Cardiovasc Imag. 2012;5:441–447. doi: 10.1161/CIRCIMAGING.112.972760. [DOI] [PubMed] [Google Scholar]
- 44.Captur G, Lopes LR, Mohun TJ, Patel V, Li C, Bassett P, Finocchiaro G, Ferreira VM, Esteban MT, Muthurangu V, Sherrid MV, Day SM, Canter CE, McKenna WJ, Seidman CE, Bluemke DA, Elliott PM, Ho CY, Moon JC. Prediction of sarcomere mutations in subclinical hypertrophic cardiomyopathy. Circulation Cardiovasc Imag. 2014;7:863–871. doi: 10.1161/CIRCIMAGING.114.002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho CY, Abbasi SA, Neilan TG, Shah RV, Chen Y, Heydari B, Cirino AL, Lakdawala NK, Orav EJ, Gonzalez A, Lopez B, Diez J, Jerosch-Herold M, Kwong RY. T1 measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circulation Cardiovasc Imag. 2013;6:415–422. doi: 10.1161/CIRCIMAGING.112.000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, Seidman CE, Seidman JG. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–1020. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP, Molkentin JD, Norris RA, Tager AM, Hoffman SR, Markwald RR, Seidman CE, Seidman JG. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun H, Davison BA, Cotter G, Pencina MJ, Koch GG. Evaluating treatment efficacy by multiple end points in phase II acute heart failure clinical trials: analyzing data using a global method. Circ Heart Fail. 2012;5:742–749. doi: 10.1161/CIRCHEARTFAILURE.112.969154. [DOI] [PubMed] [Google Scholar]
- 49.Friedrich FW, Wilding BR, Reischmann S, Crocini C, Lang P, Charron P, Müller OJ, McGrath MJ, Vollert I, Hansen A, Linke WA, Hengstenberg C, Bonne G, Morner S, Wichter T, Madeira H, Arbustini E, Eschenhagen T, Mitchell CA, Isnard R, Carrier L. Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum Mol Genet. 2012;21:3237–3254. doi: 10.1093/hmg/dds157. [DOI] [PubMed] [Google Scholar]
- 50.Crocini C, Arimura T, Reischmann S, Eder A, Braren I, Naito H, Hansen A, Eschenhagen T, Kimura A, Carrier L. Impact of ANKRD1 mutations associated with hypertrophic cardiomyopathy on cardiac contraction. Bas Res Cardiol. 2013;108:349–360. doi: 10.1007/s00395-013-0349-x. [DOI] [PubMed] [Google Scholar]
- 51.Stöhr A, Friedrich FW, Flenner F, Geertz B, Eder A, Schaaf S, Hirt MN, Uebeler J, Schlossarek S, Carrier L, Hansen A, Eschenhagen T. Contractile abnormalities and altered drug response in engineered heart tissue from Mybpc3-targeted HCM mouse models. J Mol Cell Cardiol. 2013;63:189–198. doi: 10.1016/j.yjmcc.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 52.Friedrich FW, Reischmann S, Schwalm A, Unger A, Ramanujam D, Münch J, Müller OM, Hengstenberg C, Galve E, Charron P, Linke WA, Engelhardt S, Patten M, Richard P, van der Velden J, Eschenhagen T, Isnard R, Carrier L. FHL2 expression and variants in hypertrophic cardiomyopathy. Bas Res Cardiol. 2014;109:451–458. doi: 10.1007/s00395-014-0451-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eschenhagen T, Mummery C, Knollmann BC. Modeling sarcomeric cardiomyopathies in the dish – from human heart samples to iPSC cardiomyocytes. Cardiovasc Res. 2015;105:424–438. doi: 10.1093/cvr/cvv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duncker DJ, Bakker J, Brundel BJ, Robbins J, Tardiff JC, Carrier L. Animal – and in silico models for the study of sarcomeric cardiomyopathies. Cardiovasc Res. 2015;105:439–448. doi: 10.1093/cvr/cvv006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. 2010;121:997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carrier L, Schlossarek S, Willis MS, Eschenhagen T. The ubiquitin-proteasome system and nonsense-mediated mRNA decay in hypertrophic cardiomyopathy. Cardiovasc Res. 2010;85:330–338. doi: 10.1093/cvr/cvp247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marston S, Copeland O, Gehmlich K, Schlossarek S, Carrier L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J Muscle Res Cell Motil. 2012;33:75–80. doi: 10.1007/s10974-011-9268-3. [DOI] [PubMed] [Google Scholar]
- 58.Marston S, Copeland ON, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 59.van Dijk SJ, Dooijes D, Dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, Ten Cate FJ, Stienen GJ, van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy. Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 60.van Dijk SJ, Paalberends ER, Najafi A, Michels M, Sadayappan S, Carrier L, Boontje NM, Kuster DW, van Slegtenhorst M, Dooijes D, Dos Remedios C, Ten Cate FJ, Stienen GJ, van der Velden J. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ Heart Fail. 2012;5:36–46. doi: 10.1161/CIRCHEARTFAILURE.111.963702. [DOI] [PubMed] [Google Scholar]
- 61.Helms AS, Davis F, Coleman D, Bartolone S, Glazier AA, Pagani F, Yob JM, Sadayappan S, Pedersen E, Lyons R, Westfall MV, Jones R, Russell MW, Day SM. Sarcomere mutation-specific expression patterns in human hypertrophic cardiomyopathy. Circ Cardiovasc Gen. 2014;7:434–443. doi: 10.1161/CIRCGENETICS.113.000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tripathi S, Schultz I, Becker E, Montag J, Borchert B, Francino A, Navarro-Lopez F, Perrot A, Ozcelik C, Osterziel KJ, McKenna WJ, Brenner B, Kraft T. Unequal allelic expression of wild-type and mutated beta-myosin in familial hypertrophic cardiomyopathy. Bas Res Cardiol. 2011;106:1041–1055. doi: 10.1007/s00395-011-0205-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Montgomery DE, Tardiff JC, Chandra M. Cardiac troponin T mutations: correlation between the type of mutation and the nature of myofilament dysfunction in transgenic mice. J Physiol. 2001;536:583–592. doi: 10.1111/j.1469-7793.2001.0583c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1235–1244. doi: 10.1172/JCI7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vignier N, Schlossarek S, Fraysse B, Mearini G, Kramer E, Pointu H, Mougenot N, Guiard J, Reimer R, Hohenberg H, Schwartz K, Vernet M, Eschenhagen T, Carrier L. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ Res. 2009;105:239–248. doi: 10.1161/CIRCRESAHA.109.201251. [DOI] [PubMed] [Google Scholar]
- 66.Michele DE, Gomez CA, Hong KE, Westfall MV, Metzger JM. Cardiac dysfunction in hypertrophic cardiomyopathy mutant tropomyosin mice is transgene-dependent, hypertrophy-independent, and improved by beta-blockade. Circ Res. 2002;91:255–262. doi: 10.1161/01.res.0000027530.58419.82. [DOI] [PubMed] [Google Scholar]
- 67.Moore RK, Grinspan LT, Jimenez J, Guinto PJ, Ertz-Berger B, Tardiff JC. HCM-linked 160E cardiac troponin T mutation causes unique progressive structural and molecular ventricular remodeling in transgenic mice. J Mol Cell Cardiol. 2013;58:188–198. doi: 10.1016/j.yjmcc.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang J, Wakimoto H, Seidman JG, Seidman CE. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342:111–114. doi: 10.1126/science.1236921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mearini G, Stimpel D, Geertz B, Weinberger F, Kramer E, Schlossarek S, Mourot-Filiatre J, Stoehr A, Dutsch A, Wijnker PJ, Braren I, Katus HA, Muller OJ, Voit T, Eschenhagen T, Carrier L. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat Commun. 2014;5:5515. doi: 10.1038/ncomms6515. [DOI] [PubMed] [Google Scholar]
- 70.Sequeira V, Wijnker PJ, Nijenkamp LL, Kuster DW, Najafi A, Witjas-Paalberends ER, Regan JA, Boontje N, Ten Cate FJ, Germans T, Carrier L, Sadayappan S, van Slegtenhorst MA, Zaremba R, Foster DB, Murphy AM, Poggesi C, Dos Remedios C, Stienen GJ, Ho CY, Michels M, van der Velden J. Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ Res. 2013;112:1491–1505. doi: 10.1161/CIRCRESAHA.111.300436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maass AH, Ikeda K, Oberdorf-Maass S, Maier SK, Leinwand LA. Hypertrophy, fibrosis, and sudden cardiac death in response to pathological stimuli in mice with mutations in cardiac troponin T. Circulation. 2004;110:2102–2109. doi: 10.1161/01.CIR.0000144460.84795.E3. [DOI] [PubMed] [Google Scholar]
- 72.Palmer BM, Schmitt JP, Seidman CE, Seidman JG, Wang Y, Bell SP, Lewinter MM, Maughan DW. Elevated rates of force development and MgATP binding in F764L and S532P myosin mutations causing dilated cardiomyopathy. J Mol Cell Cardiol. 2013;57:23–31. doi: 10.1016/j.yjmcc.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Witjas-Paalberends ER, Piroddi N, Stam K, van Dijk SJ, Sequeira Oliviera V, Ferrara C, Scellini B, Hazebroek M, ten Cate FJ, van Slegtenhorst M, dos Remedios C, Niessen HWM, Tesi C, Stienen GJM, Heymans S, Michels M, Poggesi C, van der Velden J. Mutations in MYH7 reduce the force generating capacity of sarcomeres in human familial hypertrophic cardiomyopathy. Cardiovasc Res. 2013;99:432–441. doi: 10.1093/cvr/cvt119. [DOI] [PubMed] [Google Scholar]
- 74.Kraft T, Witjas-Paalberends ER, Boontje NM, Tripathi S, Brandis A, Montag J, Hodgkinson JL, Francino A, Navarro-Lopez F, Brenner B, Stienen GJ, van der Velden J. Familial hypertrophic cardiomyopathy: functional effects of myosin mutation R723G in cardiomyocytes. J Mol Cell Cardiol. 2013;57:13–22. doi: 10.1016/j.yjmcc.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 75.Witjas-Paalberends E, Ferrara C, Scellini B, Piroddi N, Montag J, Tesi C, Stienen G, Michels M, Ho C, Kraft T, Poggesi C, van der Velden J. Faster cross-bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. J Physiol. 2014;592:3257–3272. doi: 10.1113/jphysiol.2014.274571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lowey S, Bretton V, Gulick J, Robbins J, Trybus KM. Transgenic mouse alpha- and beta-cardiac myosins containing the R403Q mutation show isoform-dependent transient kinetic differences. J Biol Chem. 2013;288:14780–14787. doi: 10.1074/jbc.M113.450668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, Rincon M, Gulick J, Robbins J. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. J Biol Chem. 2008;283:20579–20589. doi: 10.1074/jbc.M800554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He H, Hoyer K, Tao H, Rice R, Jimenez J, Tardiff JC, Ingwall JS. Myosin-driven rescue of contractile reserve and energetics in mouse hearts bearing familial hypertrophic cardiomyopathy-associated mutant troponin T is mutation-specific. J Physiol (Lond) 2012;590:5371–5388. doi: 10.1113/jphysiol.2012.234252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ford SJ, Mamidi R, Jimenez J, Tardiff JC, Chandra M. Effects of R92 mutations in mouse cardiac troponin T are influenced by changes in myosin heavy chain isoform. J Mol Cell Cardiol. 2012;53:542–551. doi: 10.1016/j.yjmcc.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McConnell M, Jayasundar J, Grinspan L, Fass O, Schwartz B, Tardiff J. Dynamic effects of tropomyosin D230N mutation and fetal troponin T on the tropomyosin overlap region. Biophys J. 2014;106:p32a. (abstract) [Google Scholar]
- 81.Canton M, Menazza S, Sheeran FL, Polverino de Laureto P, Di Lisa F, Pepe S. Oxidation of myofibrillar proteins in human heart failure. J Am Coll Cardiol. 2011;57:300–309. doi: 10.1016/j.jacc.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 82.Huke S, Knollmann BC. Increased myofilament Ca2+-sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol. 2010;48:824–833. doi: 10.1016/j.yjmcc.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alves ML, Gaffin RD, Wolska BM. Rescue of familial cardiomyopathies by modifications at the level of sarcomere and Ca2+ fluxes. J Mol Cell Cardiol. 2010;48:834–842. doi: 10.1016/j.yjmcc.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118:3893–3903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, Mackow JC, Fabritz L, Potter JD, Morad M. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ Res. 2003;92:428–436. doi: 10.1161/01.RES.0000059562.91384.1A. [DOI] [PubMed] [Google Scholar]
- 86.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ Res. 2012;111:170–179. doi: 10.1161/CIRCRESAHA.112.270041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huke S, Venkataraman R, Faggioni M, Bennuri S, Hwang HS, Baudenbacher F, Knollmann BC. Focal energy deprivation underlies arrhythmia susceptibility in mice with calcium-sensitized myofilaments. Circ Res. 2013;112:1334–1344. doi: 10.1161/CIRCRESAHA.113.301055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tardiff JC, Carrier L, Bers DM, Poggesi C, Ferrantini C, Coppini R, Maier LS, Ashrafian H, Huke S, van der Velden J. Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc Res. 2015;105:457–470. doi: 10.1093/cvr/cvv023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robinson P, Mirza M, Knott A, Abdulrazzak H, Willott R, Marston S, Watkins H, Redwood C. Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. J Biol Chem. 2002;277:40710–40716. doi: 10.1074/jbc.M203446200. [DOI] [PubMed] [Google Scholar]
- 90.Robinson P, Lipscomb S, Preston LC, Altin E, Watkins H, Ashley CC, Redwood CS. Mutations in fast skeletal troponin I, troponin T, and beta-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007;21:896–905. doi: 10.1096/fj.06-6899com. [DOI] [PubMed] [Google Scholar]
- 91.Lombardi R, Bell A, Senthil V, Sidhu J, Noseda M, Roberts R, Marian AJ. Differential interactions of thin filament proteins in two cardiac troponin T mouse models of hypertrophic and dilated cardiomyopathies. Cardiovasc Res. 2008;79:109–117. doi: 10.1093/cvr/cvn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rajan S, Ahmed RP, Jagatheesan G, Petrashevskaya N, Boivin GP, Urboniene D, Arteaga GM, Wolska BM, Solaro RJ, Liggett SB, Wieczorek DF. Dilated cardiomyopathy mutant tropomyosin mice develop cardiac dysfunction with significantly decreased fractional shortening and myofilament calcium sensitivity. Circ Res. 2007;101:205–214. doi: 10.1161/CIRCRESAHA.107.148379. [DOI] [PubMed] [Google Scholar]