

Abstract
Using Ebolavirus genomic and epidemiological data, we conducted the first joint analysis in which both data types were used to fit dynamic transmission models for an ongoing outbreak. Our results indicate that transmission is clustered, highlighting a potential bias in medical demand forecasts, and provide the first empirical estimate of underreporting.
Keywords: Ebola, West Africa, clustering, genome sequencing, epidemiology
The ongoing Ebola virus (EBOV) outbreak in West Africa is the largest ever recorded and shows little evidence of attenuation. As of 27 November 2014, the outbreak has spread mainly from Guinea to the neighboring nations of Liberia and Sierra Leone, with 15 935 cases reported as suspected, probable, or confirmed, and 5689 reported deaths in these 3 countries [1]. The high viral load in the blood and excreta, associated with late-stage infections and deceased patients, constitutes the highest risk for transmission [2]. Consequently, most transmission events occur in hospitals, households, and funeral settings, potentially contributing to an epidemic that is clustered in both space and time [3].
Typical mass-action models of disease transmission assume that infections occur between random pairs of individuals. In contrast, social clustering implies that transmission events will be correlated, occurring within mutually interconnected subgroups. Ignoring clustering can bias estimates of key epidemiological parameters [4–6], such as the basic reproduction number (R0, which indicates the early growth rate for an epidemic), can result in biased vaccine efficacy trials and can negatively impact assessments of necessary countermeasures, such as numbers of hospital beds, personal protective equipment, and staff. To overcome these biases, we investigated social clustering of the underlying contact patterns through which Ebola virus disease (EVD) spreads, parameterized by analysis of field-based infection contact and viral genome sequencing data. We provide the first quantitative estimate of clustered transmission and underreporting for an ongoing outbreak.
METHODS
We fit a transmission-oriented phylodynamic model [7] to 78 EBOV genome sequences collected from >70% of the confirmed cases arising in June 2014 of the current outbreak in Sierra Leone [8]. This model infers a time-based evolutionary reconstruction of the viral dynamics. We then used a Bayesian approach [9] on the same genomic data to reconstruct the transmission chains. We also fit a complementary susceptible exposed infectious removed (SEIR) network model that estimated clustering based on confirmed EVD cases and deaths [10, 11], inferring parameters for a clustered (φ > 0) and a nonclustered population (φ = 0). Parameters of these SEIR models were fitted to the cumulative numbers of laboratory-confirmed EBOV cases and laboratory-confirmed EBOV deaths obtained from the World Health Organization Global Alert and Response news from 27 May to 31 August 2014 (Supplementary Appendix), and the starting date for the SEIR model was sampled over the posterior distribution for the initial case supplied by our phylodynamic analysis.
RESULTS
The best-fit phylodynamic model for EBOV genomic data yielded an estimate of R0 = 1.4 (95% highest posterior density [HPD], 1.1–1.8). These results were robust to different prior and model specifications (Supplementary Methods). Using the genomic data, we also reconstructed transmission chains (Figure 1A). The estimated number of secondary infections was 1.35. Both of our genomic analyses estimated similar starting dates for the Sierra Leone outbreak, which were more recent than those proposed by Gire et al [8] but are still compatible with the initial Sierra Leone infections occurring at a known funeral in May 2014 [8].
Figure 1.
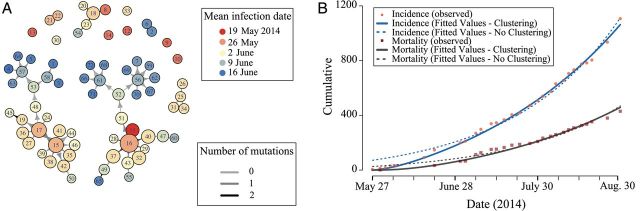
Evidence for clustered transmission and the reconstructed transmission chains in the 2014 West African Ebola virus outbreak in Sierra Leone. A, The inferred transmission tree between the patients whose Ebola virus genomes were sequenced. Colors indicate the inferred infection date, edge color indicates the number of mutations, and node sizes are proportional to the inferred number of infections. B, Cumulative incidence (pink circles) and mortality (red squares) of laboratory-confirmed Ebola virus disease cases in Sierra Leone, based on data from the World Health Organization Global Alert and Response news from 27 May to 30 August 2014. Support for a clustered model is demonstrated by its improved fit (solid lines) relative to the unclustered model (dashed lines), based both on incidence (blue lines) and mortality data (gray lines).
Our fit of the network-based SEIR model yielded an estimate of the proportion of potential high-risk contacts of an infected individual who are themselves contacts of φ = 0.71 (95% confidence interval [CI], .66–.72) and an estimate of R0 = 1.29 (95% CI, 1.27–1.37). Our comparison of epidemiological data analyzed by models with and without clustering provides strong evidence for clustering in the underlying contact network (Figure 1B; Supplementary Table 1).
Our analysis of EBOV genome sequences also provided an estimate of the proportion of cases sampled of 58% (20%–99% HPD). However, >70% of confirmed patients for the period of late May to mid-June in Sierra Leone were sequenced [8]. The discrepancy suggests that underreporting of cases is approximately 17%, with a maximum of 70%.
DISCUSSION
We have fitted both epidemiological and phylodynamic models to genomic and case-based data from the 2014 EVD outbreak in Sierra Leone, found evidence for clustered transmission, and estimated the underreporting rate. The evidence for clustered transmission was obtained from the epidemiological analysis of the case data. The phylodynamic inference provided an estimate of the start date of the outbreak, which improved the estimated SEIR parameters. Both the epidemiological and phylodynamic models produced consistent estimates of epidemiological parameters, supporting the consistency of our inference of clustered transmission. The clustering of transmission we infer has implications for the public health response such as the rate at which health resources, such as hospital beds, are required, for deriving realistic predictions about epidemic potential, and for the design of vaccine trials.
Our estimates of the reproductive number are lower than those from other modeling studies, all of which assumed that transmission occurs in a population without clustering and that infectious and susceptible individuals mix randomly [12–14]. The discrepancy between our results and earlier studies can be attributed to our relaxation of the strong assumption that susceptible and infectious individuals mix randomly. Our model demonstrates that as social clustering increases, the interactions between infected and susceptible individuals underlying transmission decrease, because contacts are shared among infectious individuals. In turn, this decrease in the contact rate between infectious and susceptible individuals requires an estimated 1.5- to 12.5-fold greater infectiousness to explain the spread of disease (Supplementary Table 1). This nonlinear interaction between higher infectiousness and decreased contact rates between susceptible and infectious individuals yields an R0 that is lower for a clustered disease such as EVD.
The primary transmission routes of EBOV are within households, at funerals, and in healthcare facilities. These transmission routes involve greater clustering than among random groups [15, 16]. For example, we estimated a clustering coefficient of φ = 0.21 (95% CI, .196–.223) from empirical contact tracing data obtained from the Liberian Ministry of Health and Social Welfare. In addition, by including clustering in the network, we were able to more accurately capture the early growth trends of the outbreak compared with an unclustered model (Figure 1B). Although the 2 genomic-based methods do not estimate clustering coefficients, they can still produce unbiased parameter estimates if clustered transmission occurs. Although the uncertainty in the phylodynamic model parameters might appear wide relative to our estimate, the confidence intervals exclude other published R0 estimates [12–14].
The agreement of our empirical, network, and genomic analyses supports a conclusion of significant social clustering, a conclusion that is robust to likely underreporting. Early estimates of this EVD outbreak suggested that for every case reported, >2 additional cases went unreported [17]. Any underreporting will lead our SEIR model to underestimate the clustering coefficient. In this respect, our estimate of the clustering coefficient will be conservative. Our phylodynamic estimate of underreporting is unbiased as long as underreporting is random. Although our estimate of underreporting has high uncertainty, our estimate that 17% of total actual cases go unreported is well below the early estimate of that 250% of reported cases equaling the actual total cases [17]. Underreporting could thus be far less prevalent than previous estimates implied.
The results of this analysis have important public health and clinical implications. First, the documentation of a high degree of clustering again highlights the importance of contact tracing. Specifically, if secondary cases are most likely to be close contacts of infected patients, identifying exposed individuals will be easier and quarantine procedures more effective. However, the discrepancy between the clustered and unclustered models decreases as the epidemic progresses (Figure 1). The improved fit of the unclustered model suggests that as EVD cases increase, moving out of the household and into the community, transmission may become less clustered. In turn, this implies that contact tracing may have a narrow window of effectiveness, highlighting the importance of rapid intervention [18]. Second, when predicting health resource demands, models must account for both clustered transmission and potentially lower R0 values. Accounting for these factors may reduce the total estimated outbreak size and decrease the rate of disease spread [6]. Third, genome sequences from other countries would allow for R0 comparisons and the quantification of viral circulation across borders, a task that is challenging with most other types of data that are readily available during an ongoing outbreak. Finally, vaccination clinical trial design for the current EBOV outbreak should incorporate complexities arising as a consequence of disease clustering [19]. Typically, in a more clustered population, an individual has a higher risk of multiple exposures, thereby increasing the individual's risk of infection. This observation makes highly clustered populations better candidates for vaccine efficacy trials than less clustered populations. However, natural immunity from previous exposures and asymptomatic infection [20], which might mitigate the vaccine's effect size, are also more prevalent in highly clustered populations. Further research into the clustered transmission of EVD, natural immunity, and potential asymptomatic infection is warranted.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online (http://cid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Financial support. This work was supported by the Santa Fe Institute and the Omidyar Group (to S. V. S.); the National Institutes of Health (grant numbers 2 U01 GM087719 and 5 U01 GM105627 to A. P. G., L. A. M., and J. P. T., and K24 DA017072 to F. L. A.); the National Science Foundation (RAPID grant 1514673 to A. P. G., M. N.-M. and J. P. T.); the Notsew Orm Sands Foundation (to A. P. G. and J. P. T.); and Fundacao de Amparo a Pesquisa de Sao Paulo (grant number 13/15144-8 to A. I.).
Potential conflicts of interest. All authors: No potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.World Health Organization. Ebola response roadmap situation report—26 November 2014. Available at: http://www.who.int/csr/disease/ebola/situation-reports/en/?m=20141126. Accessed 27 November 2014.
- 2.Ksiazek TG, Rollin PE, Williams AJ, et al. Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J Infect Dis. 1999;179(suppl 1):S177–87. doi: 10.1086/514321. [DOI] [PubMed] [Google Scholar]
- 3.Khan AS, Tshioko FK, Heymann DL, et al. The reemergence of Ebola hemorrhagic fever, Democratic Republic of the Congo, 1995. Commission de Lutte contre les Epidemies a Kikwit. J Infect Dis. 1999;179(suppl 1):S76–86. doi: 10.1086/514306. [DOI] [PubMed] [Google Scholar]
- 4.Meyers LA, Pourbohloul B, Newman MEJ, Skowronski DM, Brunham RC. Network theory and SARS: predicting outbreak diversity. J Theor Biol. 2005;232:71–81. doi: 10.1016/j.jtbi.2004.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smieszek T, Fiebig L, Scholz RW. Models of epidemics: when contact repetition and clustering should be included. Theor Biol Med Model. 2009;6:11. doi: 10.1186/1742-4682-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volz EM, Miller JC, Galvani A, Ancel Meyers L. Effects of heterogeneous and clustered contact patterns on infectious disease dynamics. PLoS Comput Biol. 2011;7:e1002042. doi: 10.1371/journal.pcbi.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouckaert R, Heled J, Kuhnert D, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gire SK, Goba A, Andersen KG, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 2014;345:1369–72. doi: 10.1126/science.1259657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jombart T, Cori A, Didelot X, Cauchemez S, Fraser C, Ferguson N. Bayesian reconstruction of disease outbreaks by combining epidemiologic and genomic data. PLoS Comput Biol. 2014;10:e1003457. doi: 10.1371/journal.pcbi.1003457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chowell G, Hengartner NW, Castillo-Chavez C, Fenimore PW, Hyman JM. The basic reproductive number of Ebola and the effects of public health measures: the cases of Congo and Uganda. J Theor Biol. 2004;229:119–26. doi: 10.1016/j.jtbi.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 11.White LF, Pagano M. A likelihood-based method for real-time estimation of the serial interval and reproductive number of an epidemic. Stat Med. 2008;27:2999–3016. doi: 10.1002/sim.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Althaus CL. Estimating the reproduction number of Ebola virus (EBOV) during the 2014 outbreak in West Africa. PLoS Curr. 2014 doi: 10.1371/currents.outbreaks.91afb5e0f279e7f29e7056095255b288. doi:10.1371/currents.outbreaks.91afb5e0f279e7f29e7056095255b288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisman D, Khoo E, Tuite A. Early epidemic dynamics of the West African 2014 Ebola outbreak: estimates derived with a simple two-parameter model. PLoS Curr. 2014 doi: 10.1371/currents.outbreaks.89c0d3783f36958d96ebbae97348d571. doi:10.1371/currents.outbreaks.89c0d3783f36958d96ebbae97348d571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.WHO Ebola Response Team. Ebola virus disease in West Africa—the first 9 months of the epidemic and forward projections. N Engl J Med. 2014;371:1481–95. doi: 10.1056/NEJMoa1411100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dowell SF, Mukunu R, Ksiazek TG, Khan AS, Rollin PE, Peters CJ. Transmission of Ebola hemorrhagic fever: a study of risk factors in family members, Kikwit, Democratic Republic of the Congo, 1995. Commission de Lutte contre les Epidemies a Kikwit. J Infect Dis. 1999;179(suppl 1):S87–91. doi: 10.1086/514284. [DOI] [PubMed] [Google Scholar]
- 16.Francesconi P, Yoti Z, Declich S, et al. Ebola hemorrhagic fever transmission and risk factors of contacts, Uganda. Emerg Infect Dis. 2003;9:1430–7. doi: 10.3201/eid0911.030339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meltzer MI, Atkins CY, Santibanez S, et al. Estimating the future number of cases in the Ebola epidemic—Liberia and Sierra Leone, 2014–2015. MMWR Surveill Summ. 2014;63(suppl 3):1–14. [PubMed] [Google Scholar]
- 18.Hawkes N. Ebola outbreak is a public health emergency of international concern, WHO warns. BMJ. 2014;349 doi: 10.1136/bmj.g5089. g5089. [DOI] [PubMed] [Google Scholar]
- 19.Cohen J, Kupferschmidt K. Ebola vaccine trials raise ethical issues. Science. 2014;346:289–90. doi: 10.1126/science.346.6207.289. [DOI] [PubMed] [Google Scholar]
- 20.Bellan SE, Pulliam JRC, Dushoff J, Meyers LA. Ebola control: effect of asymptomatic infection and acquired immunity. Lancet. 2014;384:1499–500. doi: 10.1016/S0140-6736(14)61839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.